
The MicroRNA miR-277 Controls Physiology and Pathology of the Adult Drosophila Midgut by Regulating the Expression of Fatty Acid β-Oxidation-Related Genes in Intestinal Stem Cells

 , and
, and
Abstract
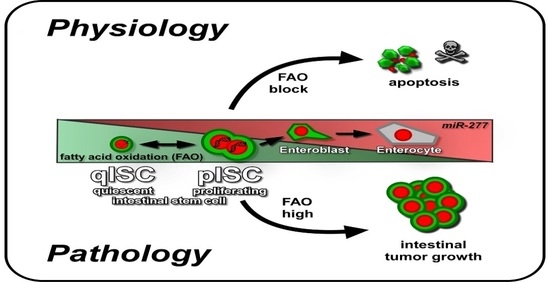
:
1. Introduction
2. Results
2.1. Identification of miR-277 as a Regulator of Cellular Metabolism
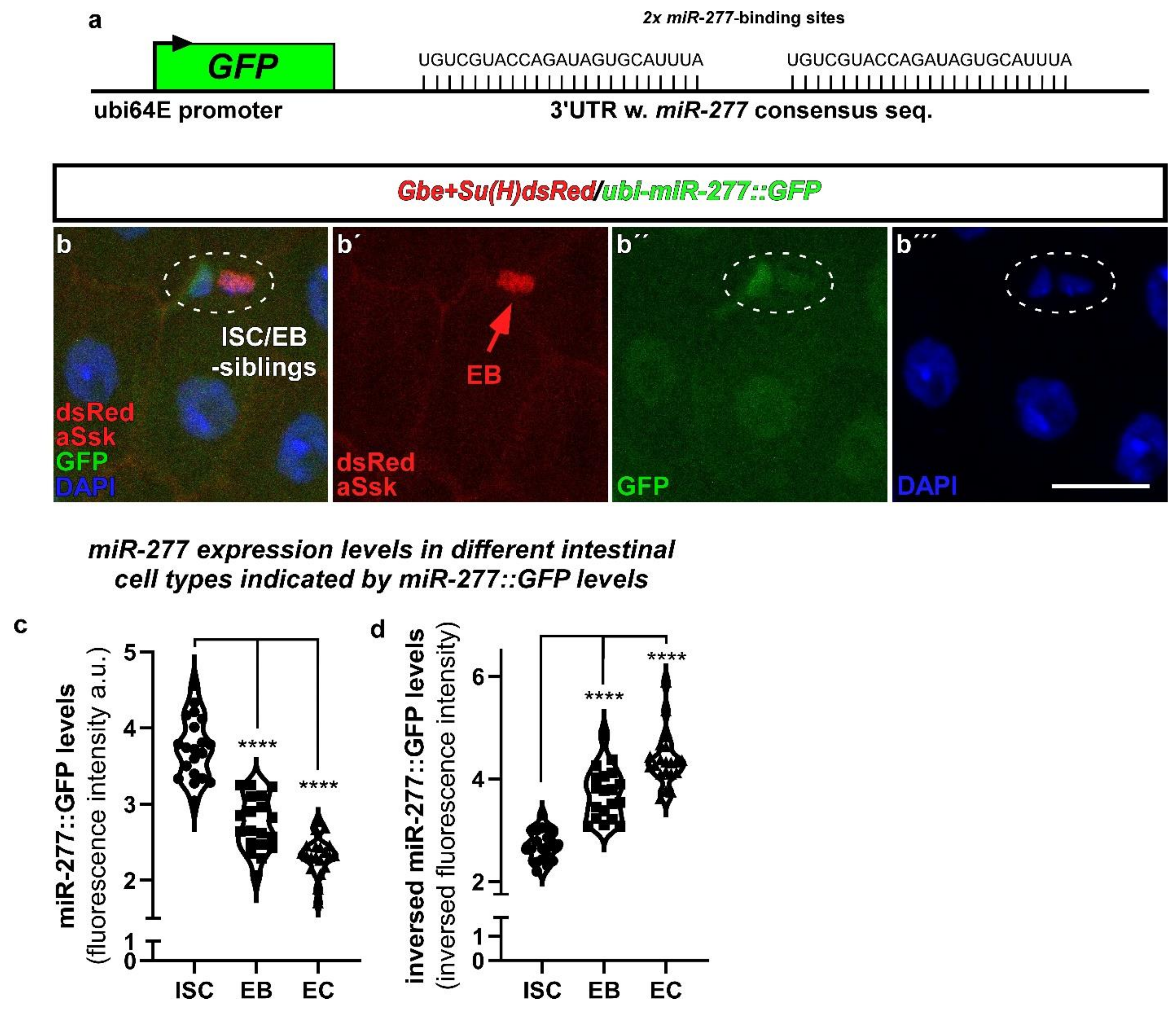
2.2. miR-277 Is Expressed in Differentiating EB and EC in the Adult Drosophila Intestine
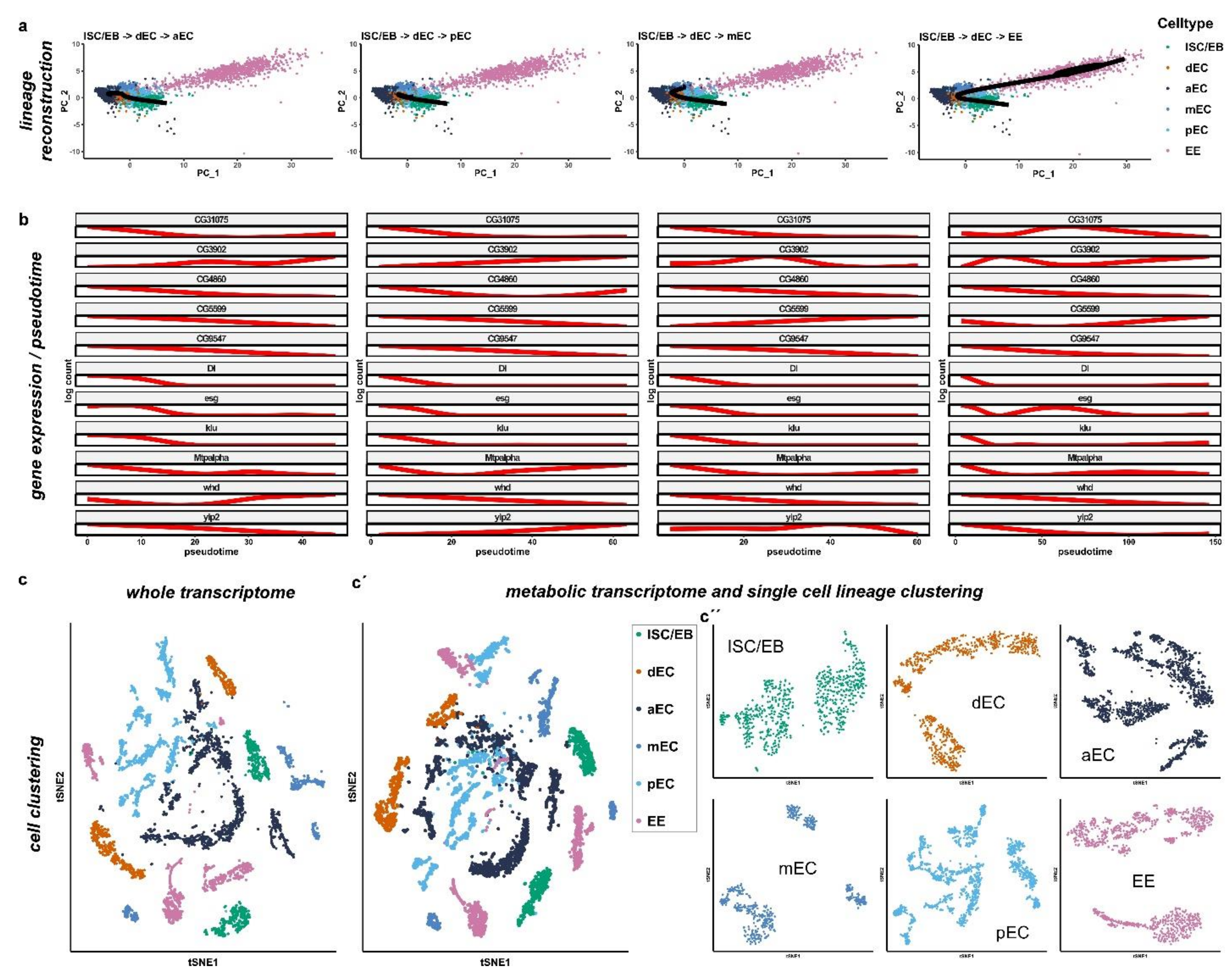
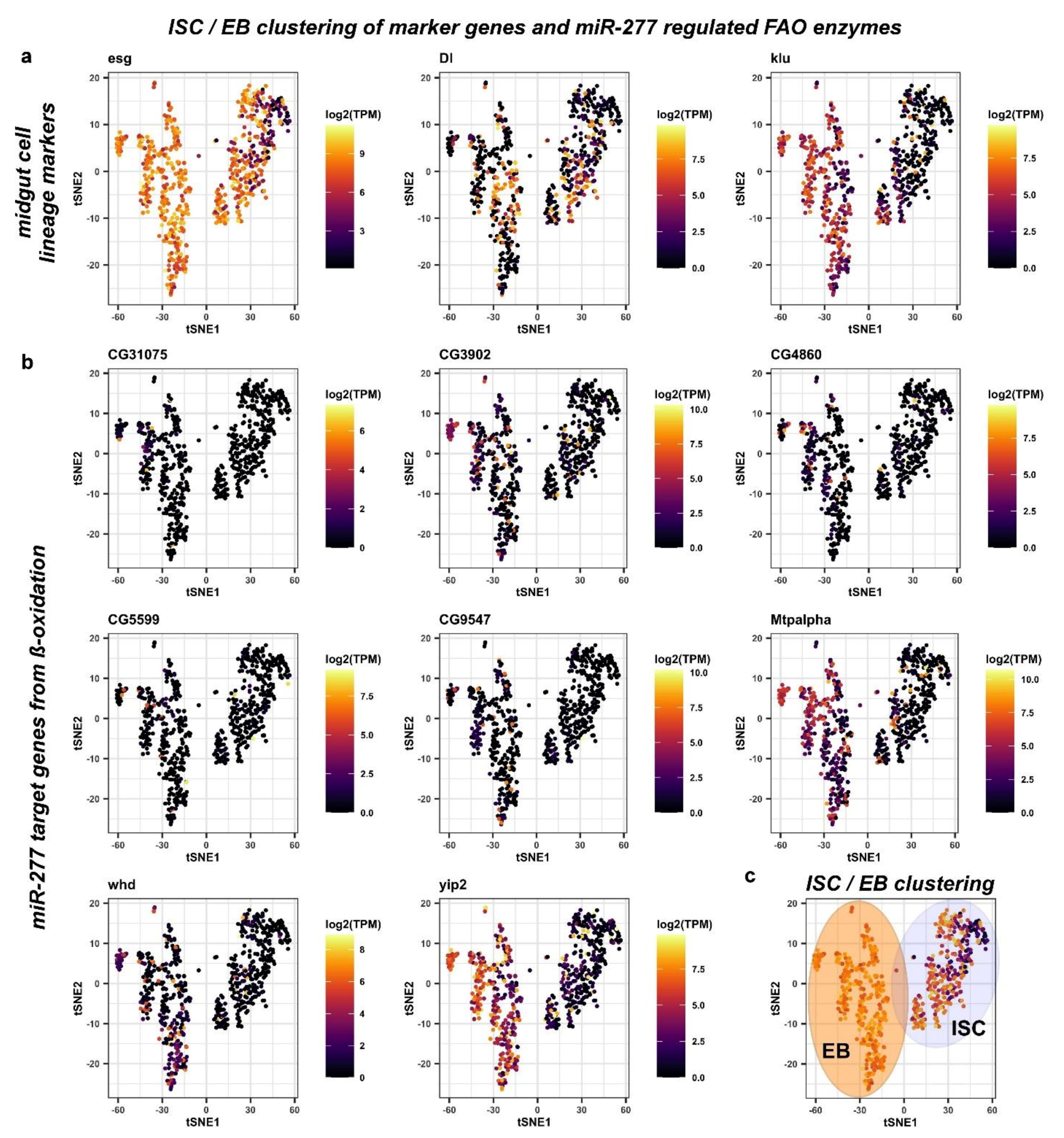
2.3. miR-277 Target Gene Expression in Reconstructed Intestinal Lineage Trajectories from scRNAseq
2.4. FAO defines Differences among Intestinal Stem Cells and Enteroblasts
2.5. miR-277 Levels affect Midgut Homeostasis and Progenitor Survival
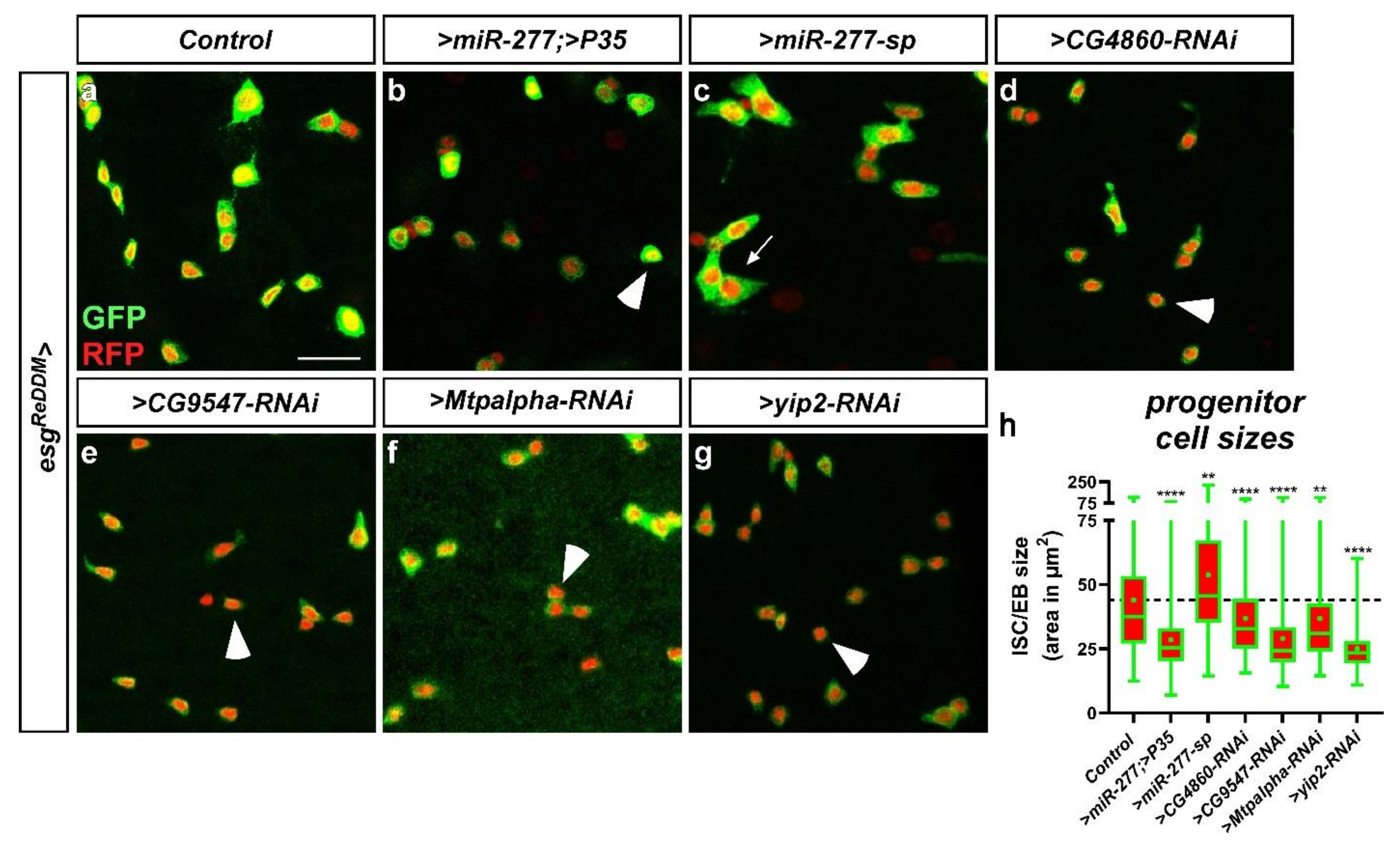
2.6. miR-277 and FAO Deficiency affect ISC Morphology and Subsequently Survival in Physiology and Pathology
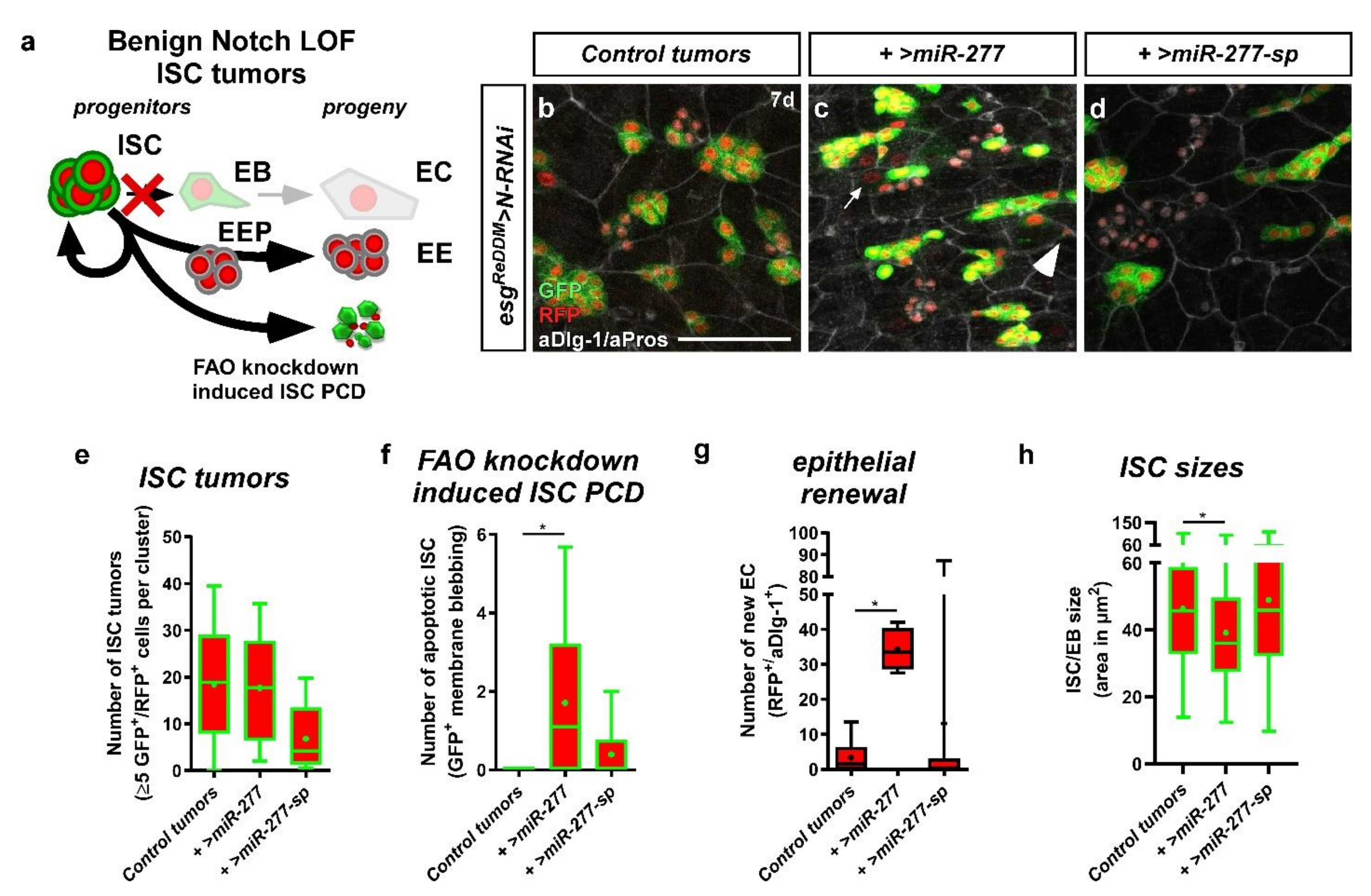
2.7. miR-277 in a Benign ISC Tumor Model
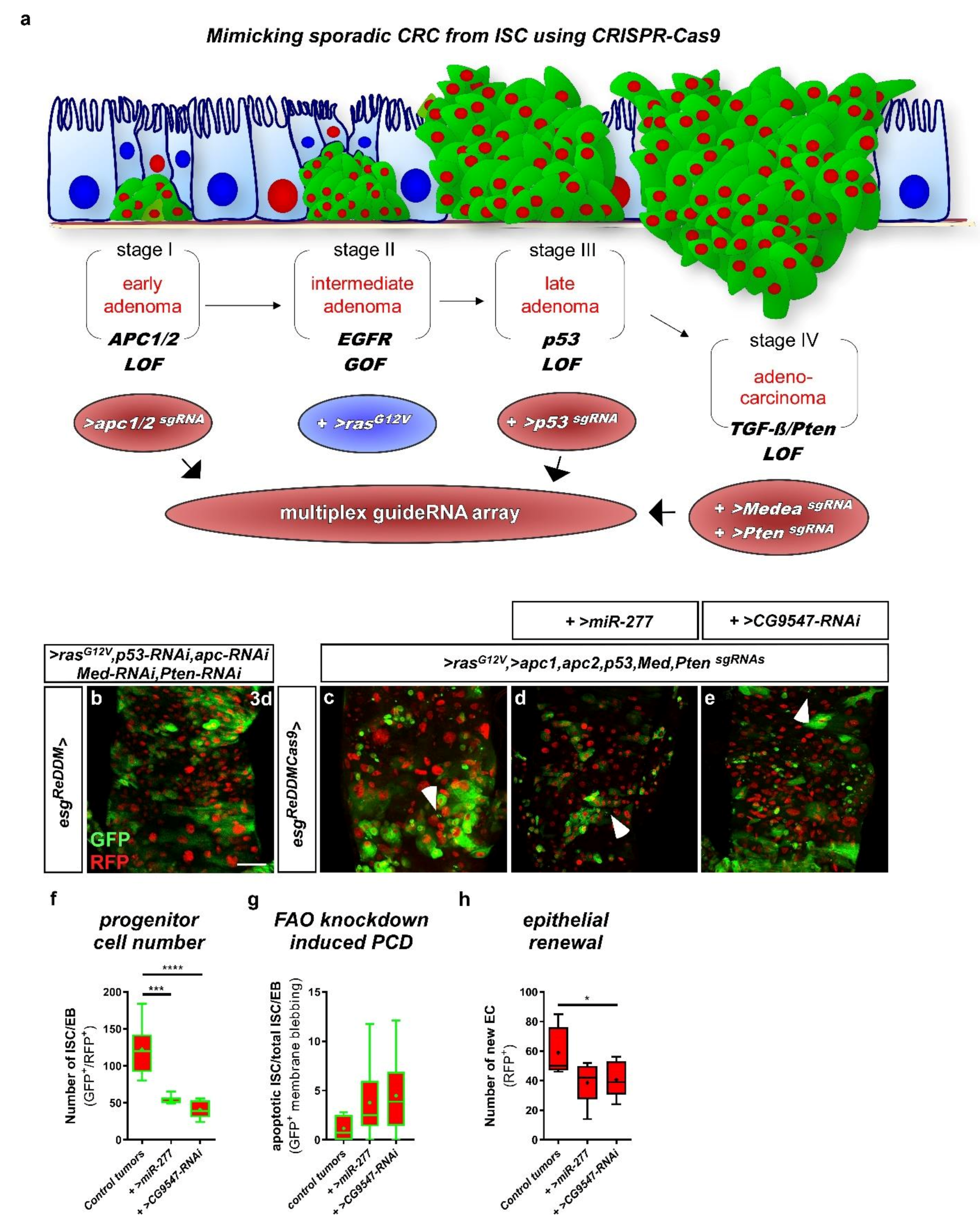
2.8. miR-277 and Colorectal Tumorigenesis
3. Discussion
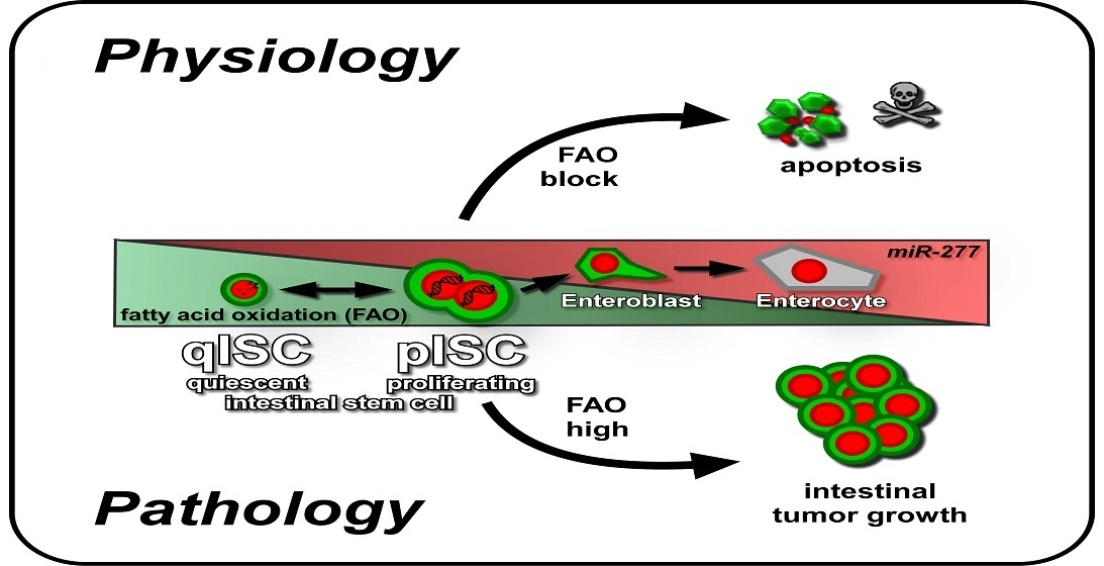
3.1. A Putative Role of Fatty Acid β-Oxidation in Controlling Quiescence in Stem Cells and their Lineage
3.2. miR-277, Fatty Acid Oxidation and ISC Apoptosis
3.3. The Role of miR-277 and FAO Genes in a CRC Model
4. Materials and Methods
4.1. In Silico microRNA Target Prediction
4.2. Genetics and fly Husbandry/Fly Strains
4.3. Food Composition and Fly Keeping
4.4. RNA Isolation and cDNA Synthesis
4.5. Real-Time qPCR and Conventional PCR
4.6. Plasmid Cloning
4.7. gRNA Design
4.8. Immunohistochemistry
4.9. Image Acquisition
4.10. Quantification of Proliferation, Cell Size and Fluorophore Intensity Measurements
4.11. Statistical Analysis
4.12. Metabolic Landscape of Adult Drosophila Midgut at Single Cell Level
4.12.1. Preprocessing
4.12.2. Normalization
4.12.3. Pathway Activity Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lai, X.; Wolkenhauer, O.; Vera, J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res. 2016, 44, 6019–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Subramanyam, D.; Blelloch, R.; Derynck, R. Regulation of epithelial–mesenchymal and mesenchymal–epithelial transitions by microRNAs. Curr. Opin. Cell Biol. 2013, 25, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Antonello, Z.; Reiff, T.; Ballesta-Illan, E.; Dominguez, M. Robust intestinal homeostasis relies on cellular plasticity in enteroblasts mediated by miR-8–Escargot switch. EMBO J. 2015, 34, 2025–2041. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Jiang, Q.; Luo, D.; Zhao, L.; Fu, X.; Chen, Y.; Song, X.; Li, L.; Zhao, H.; He, Y.; et al. miR-200b is a key regulator of tumor progression and metabolism targeting lactate dehydrogenase A in human malignant glioma. Oncotarget 2016, 7, 48423–48431. [Google Scholar] [CrossRef]
- Zipper, L.; Jassmann, D.; Burgmer, S.; Görlich, B.; Reiff, T. Ecdysone steroid hormone remote controls intestinal stem cell fate decisions via the PPARγ-homolog Eip75B in Drosophila. eLife 2020, 9, e55795. [Google Scholar] [CrossRef]
- Reiff, T.; Jacobson, J.; Cognigni, P.; Antonello, Z.; Ballesta, E.; Tan, K.J.; Yew, J.Y.; Dominguez, M.; Miguel-Aliaga, I. Endocrine remodelling of the adult intestine sustains reproduction in Drosophila. eLife 2015, 4, e06930. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.H.; Maldera, J.A.; Krunic, D.; Paiva-Silva, G.O.; Pénalva, C.; Teleman, A.A.; Edgar, B.A. Fitness trade-offs incurred by ovary-to-gut steroid signalling in Drosophila. Nature 2020, 584, 415–419. [Google Scholar] [CrossRef]
- Dutta, D.; Dobson, A.; Houtz, P.L.; Gläßer, C.; Revah, J.; Korzelius, J.; Patel, P.; Edgar, B.A.; Buchon, N. Regional Cell-Specific Transcriptome Mapping Reveals Regulatory Complexity in the Adult Drosophila Midgut. Cell Rep. 2015, 12, 346–358. [Google Scholar] [CrossRef] [Green Version]
- Buchon, N.; Osman, D.; David, F.P.; Fang, H.Y.; Boquete, J.P.; Deplancke, B.; Lemaitre, B. Morphological and molecular characterization of adult midgut compartmentalization in Drosophila. Cell Rep. 2013, 3, 1725–1738. [Google Scholar] [CrossRef] [Green Version]
- Stine, R.R.; Sakers, A.P.; TeSlaa, T.; Kissig, M.; Stine, Z.E.; Kwon, C.W.; Cheng, L.; Lim, H.-W.; Kaestner, K.H.; Rabinowitz, J.D.; et al. PRDM16 Maintains Homeostasis of the Intestinal Epithelium by Controlling Region-Specific Metabolism. Cell Stem. Cell 2019, 25, 830–845.e8. [Google Scholar] [CrossRef]
- Ohlstein, B.; Spradling, A. The adult Drosophila posterior midgut is maintained by pluripotent stem cells. Nature 2006, 439, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Micchelli, C.A.; Perrimon, N. Evidence that stem cells reside in the adult Drosophila midgut epithelium. Nature 2006, 439, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Hung, R.J.; Hu, Y.; Kirchner, R.; Liu, Y.; Xu, C.; Comjean, A.; Tattikota, S.G.; Li, F.; Song, W.; Sui, S.H.; et al. A cell atlas of the adult Drosophila midgut. Proc. Natl. Acad. Sci. USA 2020, 117, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, N.; Wang, C.; Huang, P.; Huang, H.; Jin, Z.; Yu, Z.; Cai, T.; Jiao, R.; Xi, R. Transient Scute activation via a self-stimulatory loop directs enteroendocrine cell pair specification from self-renewing intestinal stem cells. Nat. Cell Biol. 2018, 20, 152–161. [Google Scholar] [CrossRef]
- Hu, C.; Fan, L.; Cen, P.; Chen, E.; Jiang, Z.; Li, L. Energy Metabolism Plays a Critical Role in Stem Cell Maintenance and Differentiation. Int. J. Mol. Sci. 2016, 17, 253. [Google Scholar] [CrossRef] [Green Version]
- Shyh-Chang, N.; Daley, G.Q.; Cantley, L.C. Stem cell metabolism in tissue development and aging. Development 2013, 140, 2535–2547. [Google Scholar] [CrossRef] [Green Version]
- Biteau, B.; Karpac, J.; Supoyo, S.; DeGennaro, M.; Lehmann, R.; Jasper, H. Lifespan Extension by Preserving Proliferative Homeostasis in Drosophila. PLoS Genet. 2010, 6, e1001159. [Google Scholar] [CrossRef] [Green Version]
- Mattila, J.; Kokki, K.; Hietakangas, V.; Boutros, M. Stem Cell Intrinsic Hexosamine Metabolism Regulates Intestinal Adaptation to Nutrient Content. Dev. Cell 2018, 47, 112–121.e3. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Takashima, S.; Paul, M.; Guo, M.; Hartenstein, V. Mitochondrial dynamics regulates Drosophila intestinal stem cell differentiation. Cell Death Discov. 2018, 4, 81. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Zhang, C.; Marchetti, M.; Hammouda, O.; Edgar, B. EGFR Signaling Activates Intestinal Stem Cells by Promoting Mitochondrial Biogenesis. SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- Reiff, T.; Antonello, Z.A.; Ballesta-Illán, E.; Mira, L.; Sala, S.; Navarro, M.; Martinez, L.M.; Dominguez, M. Notch and EGFR regulate apoptosis in progenitor cells to ensure gut homeostasis in Drosophila. EMBO J. 2019, 38, e101346. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.C.; Wisidagama, D.R.; Bensard, C.; Zhao, H.; Wei, P.; Tanner, J.; Flores, A.; Mohlman, J.; Sorensen, L.K.; Earl, C.S.; et al. Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 2017, 19, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008, 79, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esslinger, S.M.; Schwalb, B.; Helfer, S.; Michalik, K.M.; Witte, H.; Maier, K.C.; Martin, D.; Michalke, B.; Tresch, A.; Cramer, P.; et al. Drosophila miR-277 controls branched-chain amino acid catabolism and affects lifespan. RNA Biol. 2013, 10, 1042–1056. [Google Scholar] [CrossRef]
- Ling, L.; Kokoza, V.A.; Zhang, C.; Aksoy, E.; Raikhel, A.S. MicroRNA-277 targets insulin-like peptides 7 and 8 to control lipid metabolism and reproduction in Aedes aegypti mosquitoes. Proc. Natl. Acad. Sci. USA 2017, 114, E8017–E8024. [Google Scholar] [CrossRef] [Green Version]
- Schertel, C.; Rutishauser, T.; Förstemann, K.; Basler, K. Functional Characterization of Drosophila microRNAs by a Novel in Vivo Library. Genetics 2012, 192, 1543–1552. [Google Scholar] [CrossRef] [Green Version]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. Bantam Encodes a Developmentally Regulated microRNA that Controls Cell Proliferation and Regulates the Proapoptotic Gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Ohlstein, B.; Spradling, A. Multipotent Drosophila Intestinal Stem Cells Specify Daughter Cell Fates by Differential Notch Signaling. Science 2007, 315, 988–992. [Google Scholar] [CrossRef] [Green Version]
- Antonello, Z.A.; Reiff, T.; Dominguez, M. Mesenchymal to epithelial transition during tissue homeostasis and regeneration: Patching up the Drosophila midgut epithelium. Fly 2015, 9, 132–137. [Google Scholar] [CrossRef]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Horwich, M.D.; Zamore, P.D. Design and delivery of antisense oligonucleotides to block microRNA function in cultured Drosophila and human cells. Nat. Protoc. 2008, 3, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Leader, D.P.; A Krause, S.; Pandit, A.; A Davies, S.; Dow, J.A.T. FlyAtlas 2: A new version of the Drosophila melanogaster expression atlas with RNA-Seq, miRNA-Seq and sex-specific data. Nucleic Acids Res. 2017, 46, D809–D815. [Google Scholar] [CrossRef] [PubMed]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Yin, C.; Yang, F.; Zhang, Y.; Huang, H.; Wang, J.; Deng, B.; Cai, T.; Rao, Y.; Xi, R. The Cellular Diversity and Transcription Factor Code of Drosophila Enteroendocrine Cells. Cell Rep. 2019, 29, 4172–4185.e5. [Google Scholar] [CrossRef] [Green Version]
- Korzelius, J.; Azami, S.; Ronnen-Oron, T.; Koch, P.; Baldauf, M.; Meier, E.; A Rodriguez-Fernandez, I.; Groth, M.; Sousa-Victor, P.; Jasper, H. The WT1-like transcription factor Klumpfuss maintains lineage commitment of enterocyte progenitors in the Drosophila intestine. Nat. Commun. 2019, 10, 4123. [Google Scholar] [CrossRef] [Green Version]
- Korzelius, J.; Naumann, S.K.; A Loza-Coll, M.; Chan, J.S.; Dutta, D.; Oberheim, J.; Gläßer, C.; Southall, T.D.; Brand, A.; Jones, D.L.; et al. Escargot maintains stemness and suppresses differentiation in Drosophila intestinal stem cells. EMBO J. 2014, 33, 2967–2982. [Google Scholar] [CrossRef]
- A Loza-Coll, M.; Southall, T.D.; Sandall, S.L.; Brand, A.; Jones, D.L. Regulation of Drosophila intestinal stem cell maintenance and differentiation by the transcription factor Escargot. EMBO J. 2014, 33, 2983–2996. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Pang, Z.; Huang, H.; Wang, C.; Cai, T.; Xi, R. Transcription Factor Antagonism Controls Enteroendocrine Cell Specification from Intestinal Stem Cells. Sci. Rep. 2017, 7, 988. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zhao, Y.; Buchon, N.; Engström, Y. The POU/Oct Transcription Factor Nubbin Controls the Balance of Intestinal Stem Cell Maintenance and Differentiation by Isoform-Specific Regulation. Stem Cell Rep. 2018, 10, 1565–1578. [Google Scholar] [CrossRef]
- Tsuji, T.; Hasegawa, E.; Isshiki, T. Neuroblast entry into quiescence is regulated intrinsically by the combined action of spatial Hox proteins and temporal identity factors. Development 2008, 135, 3859–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchet, E.; Annicotte, J.-S.; Lagarrigue, S.; Aguilar, V.; Clapé, C.; Chavey, C.; Fritz, V.; Casas, F.; Apparailly, F.; Auwerx, J.; et al. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. 2011, 13, 1146–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-L.; Kumar, D.B.U.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2015, 23, 206–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, J.; Bandura, J.; Zhang, P.; Jin, Y.; Reuter, H.; Edgar, B.A. EGFR-dependent TOR-independent endocycles support Drosophila gut epithelial regeneration. Nat. Commun. 2017, 8, 15125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Hay, B.; Wolff, T.; Rubin, G.M. Expression of baculovirus P35 prevents cell death in Drosophila. Development 1994, 120, 2121–2129. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; Van Es, J.H.; Van De Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; Van Den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Bangi, E.; Murgia, C.; Teague, A.G.; Sansom, O.J.; Cagan, R.L. Functional exploration of colorectal cancer genomes using Drosophila. Nat. Commun. 2016, 7, 13615. [Google Scholar] [CrossRef]
- Xing, Y.; Su, T.T.; Ruohola-Baker, H. Tie-mediated signal from apoptotic cells protects stem cells in Drosophila melanogaster. Nat. Commun. 2015, 6, 7058. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.H.; Dutta, D.; Edgar, B.A. Niche Appropriation by Drosophila Intestinal Stem Cell Tumors. Nat. Cell Biol. 2015, 17, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Bahuguna, S.; Redhai, S.; Zhou, J.; Wang, T.; Port, F.; Boutros, M. Conditional CRISPR-Cas Genome Editing in Drosophila to Generate Intestinal Tumors. Cells 2021, 10, 3156. [Google Scholar] [CrossRef] [PubMed]
- Port, F.; Strein, C.; Stricker, M.; Rauscher, B.; Heigwer, F.; Zhou, J.; Beyersdörffer, C.; Frei, J.; Hess, A.; Kern, K.; et al. A large-scale resource for tissue-specific CRISPR mutagenesis in Drosophila. eLife 2020, 9, e53865. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2019, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.R.; Zeng, X.; Zhao, J.; Liu, Y.; Hou, G.; Liu, H.; Hou, S.X. The lipolysis pathway sustains normal and transformed stem cells in adult Drosophila. Nature 2016, 538, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.K.; Toshniwal, A.G.; Mandal, S.; Mandal, L. Fatty acid β-oxidation is required for the differentiation of larval hematopoietic progenitors in Drosophila. eLife 2020, 9, e53247. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA Induces Cell Growth and Proliferation by Promoting the Acetylation of Histones at Growth Genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Tu, B.P. Acetyl-CoA and the Regulation of Metabolism: Mechanisms and Consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Grenley, M.O.; Bravo, M.-J.; Blumhagen, R.Z.; Edgar, B.A. EGFR/Ras/MAPK Signaling Mediates Adult Midgut Epithelial Homeostasis and Regeneration in Drosophila. Cell Stem. Cell 2011, 8, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Demarco, R.S.; Uyemura, B.S.; D’Alterio, C.; Jones, D.L. Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat. Cell Biol. 2019, 21, 710–720. [Google Scholar] [CrossRef]
- Torroja, L.; Ortuño-Sahagún, D.; Ferrús, A.; Hämmerle, B.; Barbas, J.A. scully, an Essential Gene of Drosophila, is Homologous to Mammalian Mitochondrial Type II l-3-hydroxyacyl-CoA Dehydrogenase/Amyloid-β Peptide-binding Protein. J. Cell Biol. 1998, 141, 1009–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Endow, S.; E Miller, S.; Ly, P.T. Mitochondria-enriched protrusions are associated with brain and intestinal stem cells in Drosophila. Commun. Biol. 2019, 2, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Balachandra, S.; Ngo, S.; O’Brien, L.E. Feedback regulation of steady-state epithelial turnover and organ size. Nature 2017, 548, 588–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Cheng, C.-W.; Cao, A.; Tripathi, S.; Mana, M.D.; Bauer-Rowe, K.E.; Abu-Remaileh, M.; Clavain, L.; Erdemir, A.; Lewis, C.A.; et al. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem. Cell 2018, 22, 769–778.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boren, J.; Brindle, K.M. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 2012, 19, 1561–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudero, S. Direct Regulation of Mitochondrial Fatty Acid Oxidation by Anti-Apoptotic MCL-1. Ph.D. Thesis, Harvard University, Cambridge, MA, USA, 2017. [Google Scholar]
- Iuchi, K.; Ema, M.; Suzuki, M.; Yokoyama, C.; Hisatomi, H. Oxidized unsaturated fatty acids induce apoptotic cell death in cultured cells. Mol. Med. Rep. 2019, 19, 2767–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strub, B.R.; Parkes, T.L.; Mukai, S.T.; Bahadorani, S.; Coulthard, A.B.; Hall, N.; Phillips, J.P.; Hilliker, A.J. Mutations of the withered (whd) gene in Drosophila melanogaster confer hypersensitivity to oxidative stress and are lesions of the carnitine palmitoyltransferase I (CPT I) gene. Genome 2008, 51, 409–420. [Google Scholar] [CrossRef]
- Cao, W.; Liu, N.; Tang, S.; Bao, L.; Shen, L.; Yuan, H.; Zhao, X.; Lu, H. Acetyl-Coenzyme A acyltransferase 2 attenuates the apoptotic effects of BNIP3 in two human cell lines. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 873–880. [Google Scholar] [CrossRef]
- Montgomery, R.K.; Carlone, D.L.; Richmond, C.A.; Farilla, L.; Kranendonk, M.E.G.; Henderson, D.E.; Baffour-Awuah, N.Y.; Ambruzs, D.M.; Fogli, L.K.; Algra, S.; et al. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc. Natl. Acad. Sci. USA 2010, 108, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.S.; Chia, L.A.; Li, X.; Ootani, A.; Su, J.; Lee, J.Y.; Su, N.; Luo, Y.; Heilshorn, S.C.; Amieva, M.R.; et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc. Natl. Acad. Sci. USA 2011, 109, 466–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, A.E.; Wang, Y.; Li, Y.; Poulin, E.J.; Means, A.L.; Washington, M.K.; Higginbotham, J.N.; Juchheim, A.; Prasad, N.; Levy, S.E.; et al. The Pan-ErbB Negative Regulator Lrig1 Is an Intestinal Stem Cell Marker that Functions as a Tumor Suppressor. Cell 2012, 149, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; van Oudenaarden, A.; Clevers, H. Identifying the Stem Cell of the Intestinal Crypt: Strategies and Pitfalls. Cell Stem. Cell 2012, 11, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, B.; Chen, E.X. Anti-EGFR Monoclonal Antibodies for Treatment of Colorectal Cancers: Development of Cetuximab and Panitumumab. J. Clin. Pharmacol. 2012, 52, 128–155. [Google Scholar] [CrossRef] [PubMed]
- Basak, O.; Beumer, J.; Wiebrands, K.; Seno, H.; van Oudenaarden, A.; Clevers, H. Induced Quiescence of Lgr5+ Stem Cells in Intestinal Organoids Enables Differentiation of Hormone-Producing Enteroendocrine Cells. Cell Stem. Cell 2017, 20, 177–190.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [Green Version]
- Knobloch, M.; Braun, S.; Zurkirchen, L.; Von Schoultz, C.; Zamboni, N.; Araúzo-Bravo, M.J.; Kovacs, W.; Karalay, Ö.; Suter, U.; Machado, R.A.M.; et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 2012, 493, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Stark, A.; Brennecke, J.; Russell, R.B.; Cohen, S.M. Identification of Drosophila MicroRNA Targets. PLOS Biol. 2003, 1, e60. [Google Scholar] [CrossRef]
- Housden, B.; Millen, K.; Bray, S.J. Drosophila Reporter Vectors Compatible with ΦC31 Integrase Transgenesis Techniques and Their Use to Generate New Notch Reporter Fly Lines. G3 Genes|Genomes|Genetics 2012, 2, 79–82. [Google Scholar] [CrossRef] [Green Version]
- Port, F.; Bullock, S.L. Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nat. Methods 2016, 13, 852–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratz, S.J.; Ukken, F.P.; Rubinstein, C.D.; Thiede, G.; Donohue, L.K.; Cummings, A.M.; O’Connor-Giles, K.M. Highly Specific and Efficient CRISPR/Cas9-Catalyzed Homology-Directed Repair in Drosophila. Genetics 2014, 196, 961–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, Y.; Motoishi, M.; Furuse, K.; Furuse, M. A tetraspanin regulates septate junction formation in Drosophila midgut. J. Cell Sci. 2016, 129, 1155–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.V.; Li, J.J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 2018, 9, 997. [Google Scholar] [CrossRef] [Green Version]
- Van Der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2625. [Google Scholar]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- LLun, A.T.; Bach, K.; Marioni, J.C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 2016, 17, 75. [Google Scholar] [CrossRef]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019, 10, 3763. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| Rp49 | TGGTTTCCGGCAAGCTTCAA | TGTTGTCGATACCCTTGGGC |
| miR-277 | GCGTGTCAGGAGTGCATTTG | GATTGTACGTTCTGGAATGTCGT |
| CG31075 | TCCGAGGGAGATAAGGCTGA | GAATGCCTTGTCCCGATCCA |
| CG3902 | CTTCTCCCTGAAGACCGTCG | GGATGGCTACCGTGGCATTA |
| CG4860 | CGACCGGGAGGAGCTTTATC | TCCAATCCGGAACCACCATAC |
| CG5599 | TCGATGACGGAATCCCTGAAAA | TCTCCTTGGCCACTAACTGC |
| CG9547 | CAAGCTGATTGGTGCCTTTGG | GCGCACTAGTAATCCACGTCT |
| MtpAlpha | CCAGTCCTTCGTCATGGACA | CACGGATCACATCGAGAATCTTCA |
| whd | AACTTCTACGGCACGGATGC | TGCCCTGAACCATGATAGGC |
| Yip2 | CATGAGTTGCAGCGCAAGAAG | GCTGTAGGATTAGACAGCCTCG |
| Targeted Gene | Target Sequence | Number of Off-Targets |
|---|---|---|
| apc1 | GGGCATCGCCGAGCTCAGTC | 3 |
| apc2 | GGAGAGACGATCCGCTCAGA | 5 |
| cic | GGCTTGCCCGGGGAGCTTAG | 4 |
| p53 | GGCTATTACGTGCCCCAATA | 5 |
| Med | GGTGAAGGACGAATACTCAG | 1 |
| Pten | GACGGTTTCTGAATAGGCCC | 4 |
| Primer | Sequence (5’-3’) |
|---|---|
| BbsI_apc1_for | ATAAGAAGACCTTGCAGGGCATCGCCGAGCTCAGTCGTTTCAGAGCTATGCTGGAAAC |
| SapI_apc2_for | ATAAGCTCTTCCTGCAGGAGAGACGATCCGCTCAGAGTTTCAGAGCTATGCTGGAAAC |
| BbsI_cic_for | ATAAGAAGACCTTGCAGGCTTGCCCGGGGAGCTTAGGTTTCAGAGCTATGCTGGAAAC |
| SapI_p53_for | ATAAGCTCTTCCTGCAGGCTATTACGTGCCCCAATAGTTTCAGAGCTATGCTGGAAAC |
| BbsI_Med_for | ATAAGAAGACCTTGCAGGTGAAGGACGAATACTCAGGTTTCAGAGCTATGCTGGAAAC |
| final_rev_Pten_BbsI | ATAAGAAGACCCAAACGACGGTTTCTGAATAGGCCCTGCACCAGCCGGGAATCGAACC |
| universal_rev_2xSapI_BbsI | ATAAGAAGACCCAAACTGAAGAGCTGAACGGCTCTTCTGCACCAGCCGGGAATCGAACC |
| universal_rev_2xBbsI_SapI | ATAAGCTCTTCAAACTGGTCTTCTGAAGGGAAGACTATGCACCAGCCGGGAATCGAACC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zipper, L.; Batchu, S.; Kaya, N.H.; Antonello, Z.A.; Reiff, T. The MicroRNA miR-277 Controls Physiology and Pathology of the Adult Drosophila Midgut by Regulating the Expression of Fatty Acid β-Oxidation-Related Genes in Intestinal Stem Cells. Metabolites 2022, 12, 315. https://doi.org/10.3390/metabo12040315
Zipper L, Batchu S, Kaya NH, Antonello ZA, Reiff T. The MicroRNA miR-277 Controls Physiology and Pathology of the Adult Drosophila Midgut by Regulating the Expression of Fatty Acid β-Oxidation-Related Genes in Intestinal Stem Cells. Metabolites. 2022; 12(4):315. https://doi.org/10.3390/metabo12040315
Chicago/Turabian StyleZipper, Lisa, Sai Batchu, Nida Hatice Kaya, Zeus Andrea Antonello, and Tobias Reiff. 2022. "The MicroRNA miR-277 Controls Physiology and Pathology of the Adult Drosophila Midgut by Regulating the Expression of Fatty Acid β-Oxidation-Related Genes in Intestinal Stem Cells" Metabolites 12, no. 4: 315. https://doi.org/10.3390/metabo12040315
APA StyleZipper, L., Batchu, S., Kaya, N. H., Antonello, Z. A., & Reiff, T. (2022). The MicroRNA miR-277 Controls Physiology and Pathology of the Adult Drosophila Midgut by Regulating the Expression of Fatty Acid β-Oxidation-Related Genes in Intestinal Stem Cells. Metabolites, 12(4), 315. https://doi.org/10.3390/metabo12040315