
Whole Blood Metabolite Profiles Reflect Changes in Energy Metabolism in Heart Failure

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
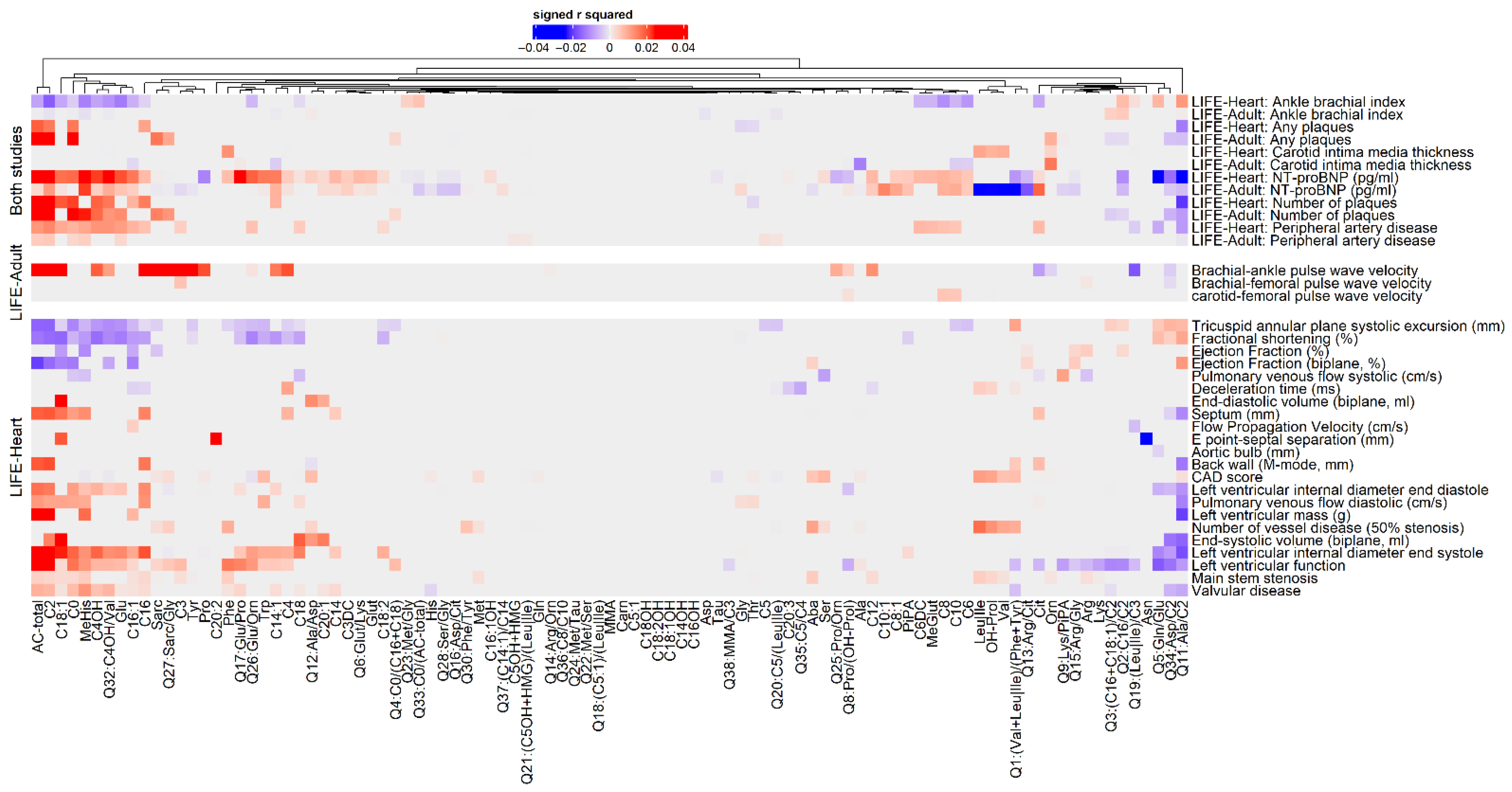
2.1. Association Analysis of Metabolites and ASCVD Phenotypes
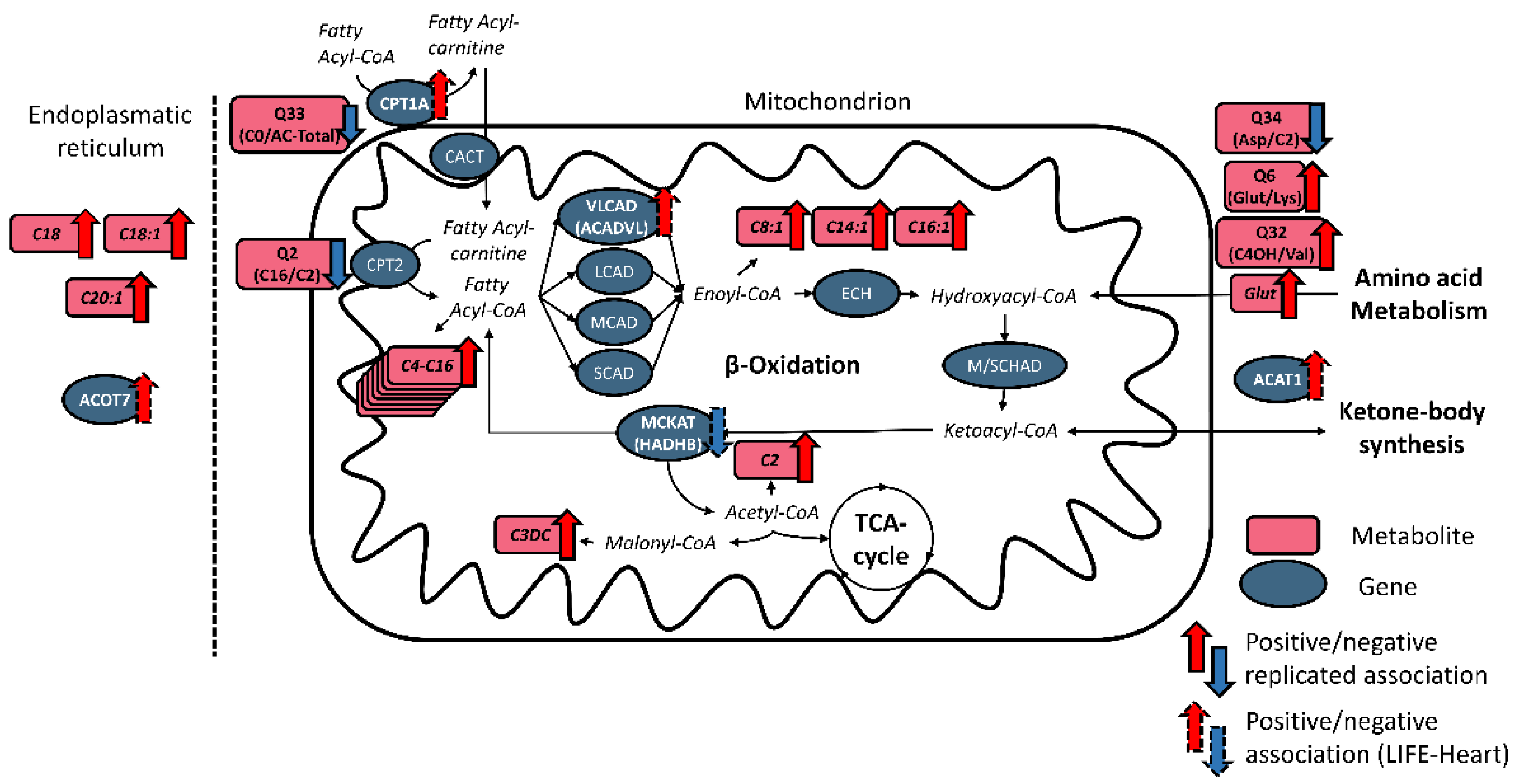
2.2. NT-proBNP Associated with Metabolites and Genes Involved in Fatty Acid Metabolism
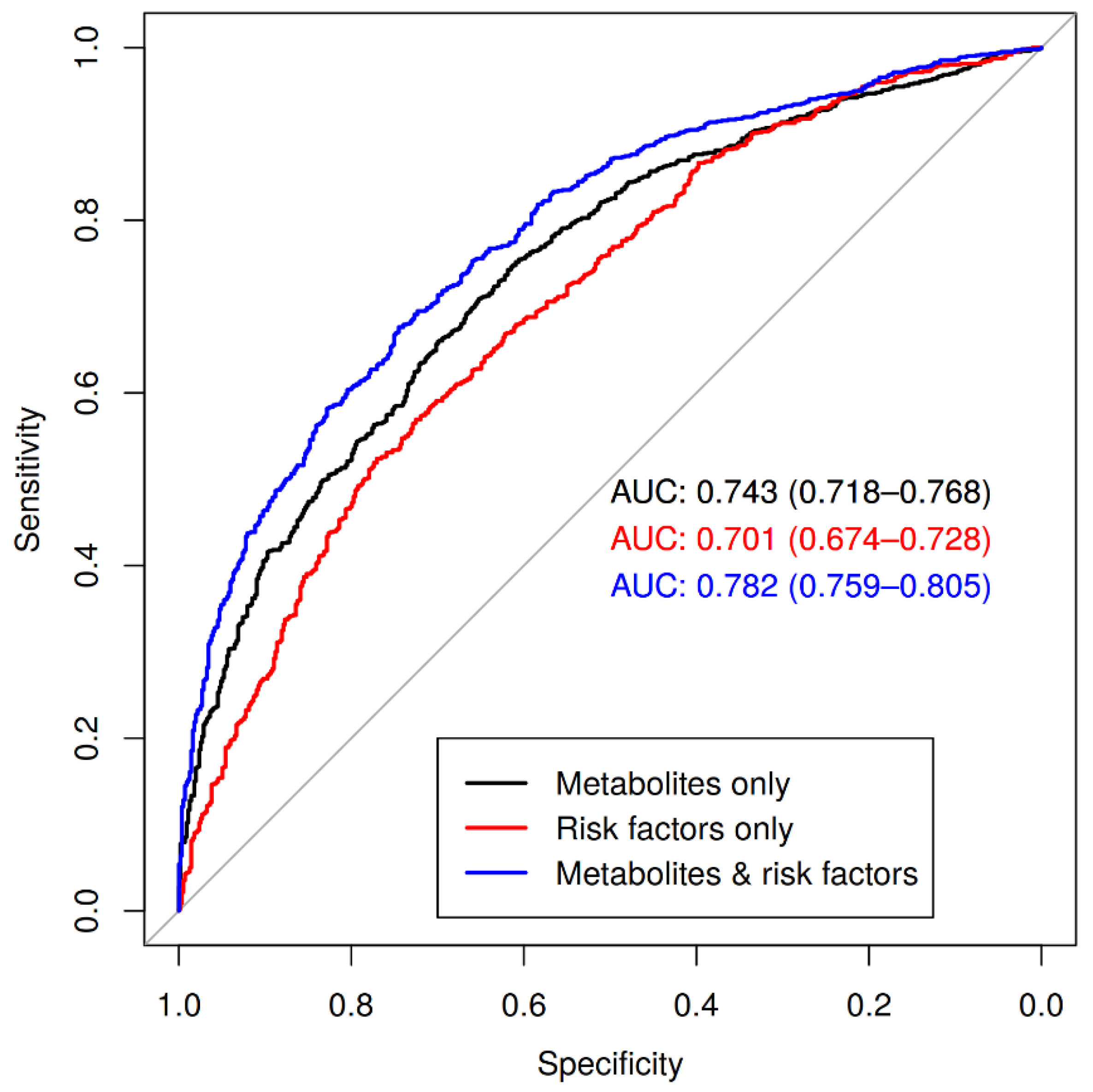
2.3. Metabolic Profile as Predictor of Coronary Artery Disease
3. Discussion
3.1. Metabolites of Fatty Acid Oxidation Reliably Associate with ASCVD Phenotypes
3.2. PBMC Expression of Genes Involved in Oxidation of Fatty Acids Associated with NT-proBNP in LIFE-Heart
3.3. Full Metabolite Profile Is Predictive for Coronary Artery Disease
3.4. Limitations
3.5. Conclusions
4. Materials and Methods
4.1. Study Characteristics and Design
4.2. Metabolite Measurement and Pre-Processing
4.3. Gene-Expression Measurement and Pre-Processing
4.4. Analysis of Cofactors
4.5. Discovery and Replication of Metabolite Associations with ASCVD Phenotypes
4.6. Discovery and Replication of Associations of ASCVD Phenotypes with Blood Gene Expression
4.7. Classification/Risk-Prediction Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wende, A.R.; Brahma, M.K.; McGinnis, G.R.; Young, M.E. Metabolic Origins of Heart Failure. JACC Basic Transl. Sci. 2017, 2, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceglarek, U.; Leichtle, A.; Brügel, M.; Kortz, L.; Brauer, R.; Bresler, K.; Thiery, J.; Fiedler, G.M. Challenges and developments in tandem mass spectrometry based clinical metabolomics. Mol. Cell. Endocrinol. 2009, 301, 266–271. [Google Scholar] [CrossRef]
- Brauer, R.; Leichtle, A.B.; Fiedler, G.M.; Thiery, J.; Ceglarek, U. Preanalytical standardization of amino acid and acylcarnitine metabolite profiling in human blood using tandem mass spectrometry. Metabolomics 2011, 7, 344–352. [Google Scholar] [CrossRef]
- Ceglarek, U.; Müller, P.; Stach, B.; Bührdel, P.; Thiery, J.; Kiess, W. Validation of the phenylalanine/tyrosine ratio determined by tandem mass spectrometry: Sensitive newborn screening for phenylketonuria. Clin. Chem. Lab. Med. 2002, 40, 693–697. [Google Scholar] [CrossRef]
- Yin, P.; Lehmann, R.; Xu, G. Effects of pre-analytical processes on blood samples used in metabolomics studies. Anal. Bioanal. Chem. 2015, 407, 4879–4892. [Google Scholar] [CrossRef] [Green Version]
- Zukunft, S.; Sorgenfrei, M.; Prehn, C.; Möller, G.; Adamski, J. Targeted Metabolomics of Dried Blood Spot Extracts. Chromatographia 2013, 76, 1295–1305. [Google Scholar] [CrossRef]
- McCann, M.R.; La George De Rosa, M.V.; Rosania, G.R.; Stringer, K.A. L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine. Metabolites 2021, 11, 51. [Google Scholar] [CrossRef]
- McGarrah, R.W.; Crown, S.B.; Zhang, G.-F.; Shah, S.H.; Newgard, C.B. Cardiovascular Metabolomics. Circ. Res. 2018, 122, 1238–1258. [Google Scholar] [CrossRef] [PubMed]
- Albert, C.L.; Tang, W.H.W. Metabolic Biomarkers in Heart Failure. Heart Fail. Clin. 2018, 14, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Labarthe, F.; Fortier, A.; Bouchard, B.; Thompson Legault, J.; Bolduc, V.; Rigal, O.; Chen, J.; Ducharme, A.; Crawford, P.A.; et al. Circulating acylcarnitine profile in human heart failure: A surrogate of fatty acid metabolic dysregulation in mitochondria and beyond. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H768–H781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.-L.; Wang, C.-H.; Shiao, M.-S.; Liu, M.-H.; Huang, Y.-Y.; Huang, C.-Y.; Mao, C.-T.; Lin, J.-F.; Ho, H.-Y.; Yang, N.-I. Metabolic disturbances identified in plasma are associated with outcomes in patients with heart failure: Diagnostic and prognostic value of metabolomics. J. Am. Coll. Cardiol. 2015, 65, 1509–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poorthuis, M.H.F.; Halliday, A.; Massa, M.S.; Sherliker, P.; Clack, R.; Morris, D.R.; Clarke, R.; de Borst, G.J.; Bulbulia, R.; Lewington, S. Validation of Risk Prediction Models to Detect Asymptomatic Carotid Stenosis. J. Am. Heart Assoc. 2020, 9, e014766. [Google Scholar] [CrossRef] [PubMed]
- Tajima, G.; Hara, K.; Yuasa, M. Carnitine palmitoyltransferase II deficiency with a focus on newborn screening. J Hum Genet 2019, 64, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Tajima, G.; Hara, K.; Tsumura, M.; Kagawa, R.; Okada, S.; Sakura, N.; Maruyama, S.; Noguchi, A.; Awaya, T.; Ishige, M.; et al. Newborn screening for carnitine palmitoyltransferase II deficiency using (C16+C18:1)/C2: Evaluation of additional indices for adequate sensitivity and lower false-positivity. Mol. Genet. Metab. 2017, 122, 67–75. [Google Scholar] [CrossRef]
- Pereyra, A.S.; Hasek, L.Y.; Harris, K.L.; Berman, A.G.; Damen, F.W.; Goergen, C.J.; Ellis, J.M. Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J. Biol. Chem. 2017, 292, 18443–18456. [Google Scholar] [CrossRef] [Green Version]
- Bedi, K.C.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Kelly, J.P.; McGarrah, R.W.; Hellkamp, A.S.; Fiuzat, M.; Testani, J.M.; Wang, T.S.; Verma, A.; Samsky, M.D.; Donahue, M.P.; et al. Prognostic Implications of Long-Chain Acylcarnitines in Heart Failure and Reversibility with Mechanical Circulatory Support. J. Am. Coll. Cardiol. 2016, 67, 291–299. [Google Scholar] [CrossRef]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W.; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Metabolomic Profiling Identifies Novel Circulating Biomarkers of Mitochondrial Dysfunction Differentially Elevated in Heart Failure with Preserved Versus Reduced Ejection Fraction: Evidence for Shared Metabolic Impairments in Clinical Heart Failure. JAHA 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cluntun, A.A.; Badolia, R.; Lettlova, S.; Parnell, K.M.; Shankar, T.S.; Diakos, N.A.; Olson, K.A.; Taleb, I.; Tatum, S.M.; Berg, J.A.; et al. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab. 2021, 33, 629–648.e10. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [Green Version]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R.; et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: A multisystems approach. Circ. Heart Fail. 2014, 7, 1022–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bay, M.; Kirk, V.; Parner, J.; Hassager, C.; Nielsen, H.; Krogsgaard, K.; Trawinski, J.; Boesgaard, S.; Aldershvile, J. NT-proBNP: A new diagnostic screening tool to differentiate between patients with normal and reduced left ventricular systolic function. Heart 2003, 89, 150–154. [Google Scholar] [CrossRef]
- Britton, C.H.; Mackey, D.W.; Esser, V.; Foster, D.W.; Burns, D.K.; Yarnall, D.P.; Froguel, P.; McGarry, J.D. Fine chromosome mapping of the genes for human liver and muscle carnitine palmitoyltransferase I (CPT1A and CPT1B). Genomics 1997, 40, 209–211. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161. [Google Scholar] [CrossRef]
- Lewandowski, E.D.; Fischer, S.K.; Fasano, M.; Banke, N.H.; Walker, L.A.; Huqi, A.; Wang, X.; Lopaschuk, G.D.; O’Donnell, J.M. Acute liver carnitine palmitoyltransferase I overexpression recapitulates reduced palmitate oxidation of cardiac hypertrophy. Circ. Res. 2013, 112, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Miyata, K.; Nakayama, M.; Mizuta, S.; Hokimoto, S.; Sugamura, K.; Oshima, S.; Oike, Y.; Sugiyama, S.; Ogawa, H.; Yamamura, K. Elevated mature macrophage expression of human ABHD2 gene in vulnerable plaque. Biochem. Biophys. Res. Commun. 2008, 365, 207–213. [Google Scholar] [CrossRef]
- Lord, C.C.; Thomas, G.; Brown, J.M. Mammalian alpha beta hydrolase domain (ABHD) proteins: Lipid metabolizing enzymes at the interface of cell signaling and energy metabolism. Biochim. Biophys. Acta 2013, 1831, 792–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, M.; Henger, S.; Beutner, F.; Teren, A.; Baber, R.; Willenberg, A.; Ceglarek, U.; Pott, J.; Burkhardt, R.; Thiery, J. Cohort Profile: The Leipzig Research Center for Civilization Diseases-Heart Study (LIFE-Heart). Int. J. Epidemiol. 2020, 49, 1439–1440h. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Engel, C.; Ahnert, P.; Alfermann, D.; Arelin, K.; Baber, R.; Beutner, F.; Binder, H.; Brähler, E.; Burkhardt, R.; et al. The LIFE-Adult-Study: Objectives and design of a population-based cohort study with 10,000 deeply phenotyped adults in Germany. BMC Public Health 2015, 15, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuchel, C.; Kirsten, H.; Ceglarek, U.; Scholz, M. Metabolite-Investigator: An integrated user-friendly workflow for metabolomics multi-study analysis. Bioinformatics 2021, 37, 2218–2220. [Google Scholar] [CrossRef]
- Beuchel, C.; Becker, S.; Dittrich, J.; Kirsten, H.; Toenjes, A.; Stumvoll, M.; Loeffler, M.; Thiele, H.; Beutner, F.; Thiery, J.; et al. Clinical and lifestyle related factors influencing whole blood metabolite levels–A comparative analysis of three large cohorts. Mol. Metab. 2019, 29, 76–85. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Burkhardt, R.; Kirsten, H.; Beutner, F.; Holdt, L.M.; Gross, A.; Teren, A.; Tönjes, A.; Becker, S.; Krohn, K.; Kovacs, P.; et al. Integration of Genome-Wide SNP Data and Gene-Expression Profiles Reveals Six Novel Loci and Regulatory Mechanisms for Amino Acids and Acylcarnitines in Whole Blood. PLoS Genet. 2015, 11, e1005510. [Google Scholar] [CrossRef] [Green Version]
- Holdt, L.M.; Hoffmann, S.; Sass, K.; Langenberger, D.; Scholz, M.; Krohn, K.; Finstermeier, K.; Stahringer, A.; Wilfert, W.; Beutner, F.; et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 2013, 9, e1003588. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, H.; Al-Hasani, H.; Holdt, L.; Gross, A.; Beutner, F.; Krohn, K.; Horn, K.; Ahnert, P.; Burkhardt, R.; Reiche, K.; et al. Dissecting the genetics of the human transcriptome identifies novel trait-related trans-eQTLs and corroborates the regulatory relevance of non-protein coding loci†. Hum. Mol. Genet. 2015, 24, 4746–4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Aboyans, V.; Criqui, M.H.; Abraham, P.; Allison, M.A.; Creager, M.A.; Diehm, C.; Fowkes, F.G.R.; Hiatt, W.R.; Jönsson, B.; Lacroix, P.; et al. Measurement and interpretation of the ankle-brachial index: A scientific statement from the American Heart Association. Circulation 2012, 126, 2890–2909. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 30 June 2021).
- Nagelkerke, N.J.D. A note on a general definition of the coefficient of determination. Biometrika 1991, 78, 691–692. [Google Scholar] [CrossRef]
- Huang, Q.Q.; Ritchie, S.C.; Brozynska, M.; Inouye, M. Power, false discovery rate and Winner’s Curse in eQTL studies. Nucleic Acids Res. 2018, 46, e133. [Google Scholar] [CrossRef]
- Peterson, C.B.; Bogomolov, M.; Benjamini, Y.; Sabatti, C. Many Phenotypes Without Many False Discoveries: Error Controlling Strategies for Multitrait Association Studies. Genet. Epidemiol. 2016, 40, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Di, W.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.S.; Reitsma, J.B.; Altman, D.G.; Moons, K.G.M. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD): The TRIPOD Statement. BMC Med. 2015, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28. [Google Scholar] [CrossRef] [Green Version]
- Harrell, F.E.; Lee, K.L.; Mark, D.B. Multivariable Prognostic Models: Issues in Developing Models, Evaluating Assumptions and Adequacy, and Measuring and Reducing Errors. Statist. Med. 1996, 15, 361–387. [Google Scholar] [CrossRef]
- DeLong, E.R.; DeLong, D.M.; Clarke-Pearson, D.L. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics 1988, 44, 837–845. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Van Calster, B.; Nieboer, D.; Vergouwe, Y.; de Cock, B.; Pencina, M.J.; Steyerberg, E.W. A calibration hierarchy for risk models was defined: From utopia to empirical data. J. Clin. Epidemiol. 2016, 74, 167–176. [Google Scholar] [CrossRef]
- De Cock, B.; Nieboer, D.; Van Calster, B.; Steyerberg, E.; Vergouwe, Y. CalibrationCurves: Calibration Performance. 2016. Available online: https://github.com/BravoDC/CalibrationCurves/ (accessed on 30 June 2021).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beuchel, C.; Dittrich, J.; Pott, J.; Henger, S.; Beutner, F.; Isermann, B.; Loeffler, M.; Thiery, J.; Ceglarek, U.; Scholz, M. Whole Blood Metabolite Profiles Reflect Changes in Energy Metabolism in Heart Failure. Metabolites 2022, 12, 216. https://doi.org/10.3390/metabo12030216
Beuchel C, Dittrich J, Pott J, Henger S, Beutner F, Isermann B, Loeffler M, Thiery J, Ceglarek U, Scholz M. Whole Blood Metabolite Profiles Reflect Changes in Energy Metabolism in Heart Failure. Metabolites. 2022; 12(3):216. https://doi.org/10.3390/metabo12030216
Chicago/Turabian StyleBeuchel, Carl, Julia Dittrich, Janne Pott, Sylvia Henger, Frank Beutner, Berend Isermann, Markus Loeffler, Joachim Thiery, Uta Ceglarek, and Markus Scholz. 2022. "Whole Blood Metabolite Profiles Reflect Changes in Energy Metabolism in Heart Failure" Metabolites 12, no. 3: 216. https://doi.org/10.3390/metabo12030216
APA StyleBeuchel, C., Dittrich, J., Pott, J., Henger, S., Beutner, F., Isermann, B., Loeffler, M., Thiery, J., Ceglarek, U., & Scholz, M. (2022). Whole Blood Metabolite Profiles Reflect Changes in Energy Metabolism in Heart Failure. Metabolites, 12(3), 216. https://doi.org/10.3390/metabo12030216