
Analogues of Natural Chalcones as Efficient Inhibitors of AKR1C3

 , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
2. Results
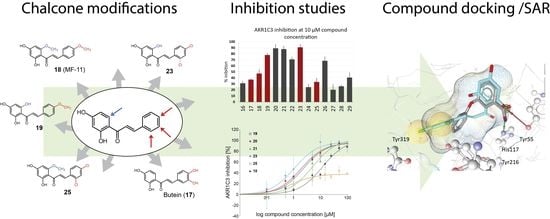
2.1. Synthesis of Chalcones
2.2. Bioactivity of Chalcones with AKR1C3 and Related Targets
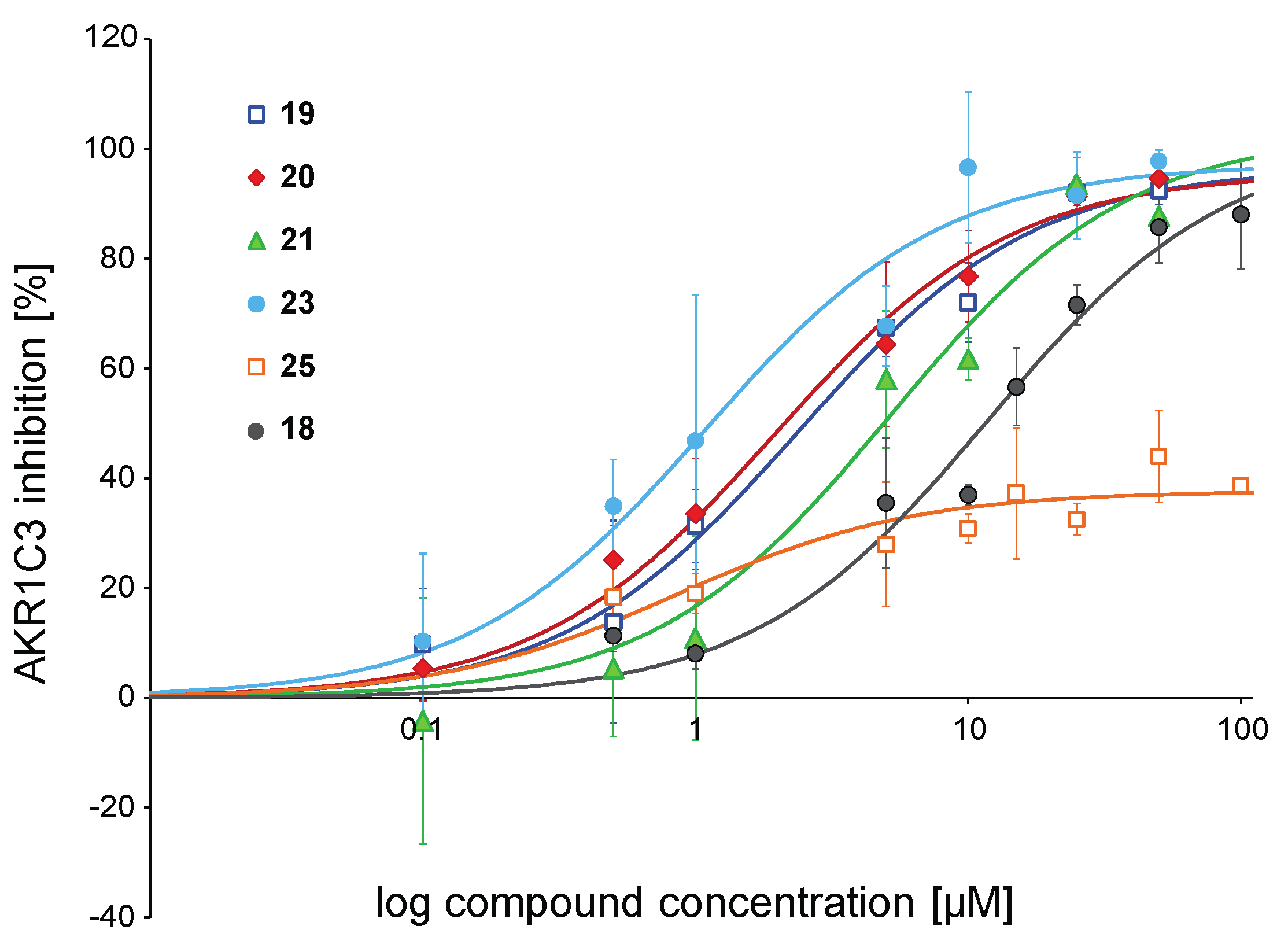
2.2.1. Inhibition of the Enzymatic Activity of AKR1C3
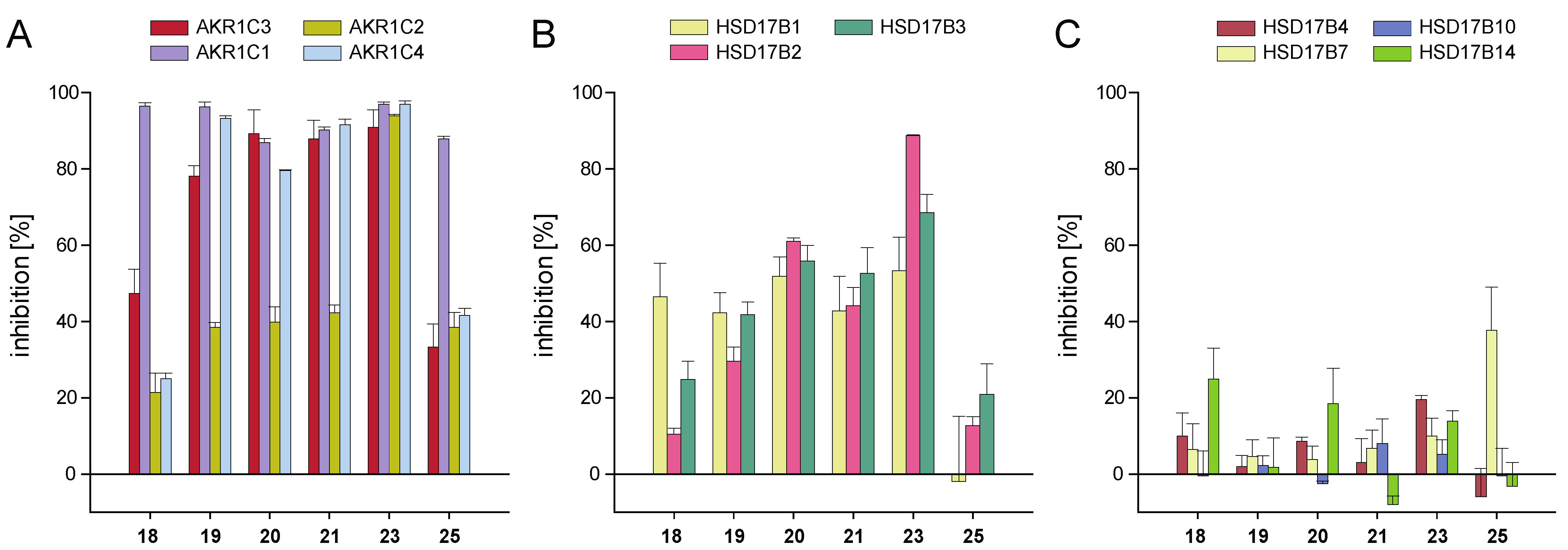
2.2.2. Selectivity of Selected AKR1C3-Inhibiting Chalcones
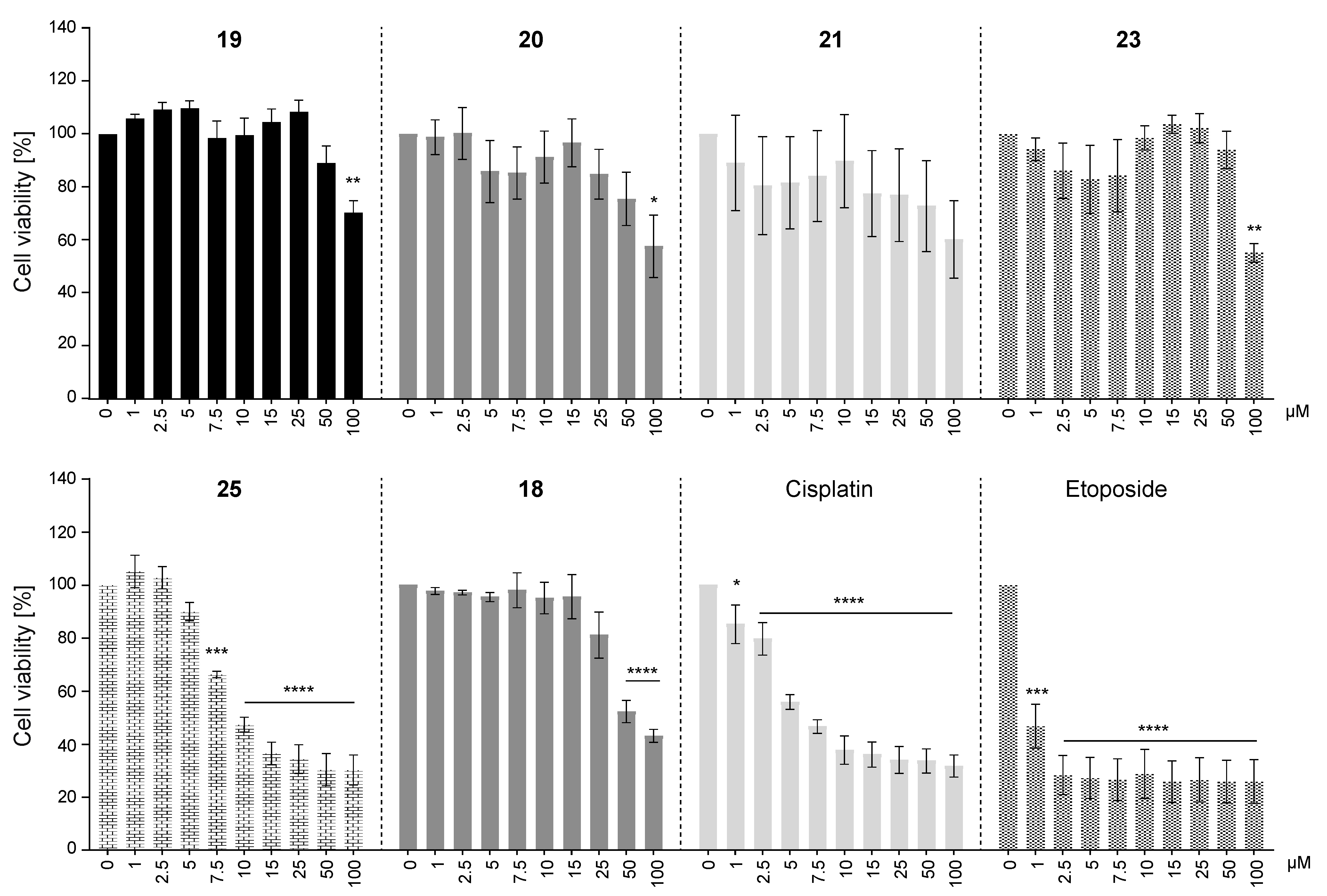
2.2.3. Cytotoxicity of Selected AKR1C3-Inhibiting Chalcones
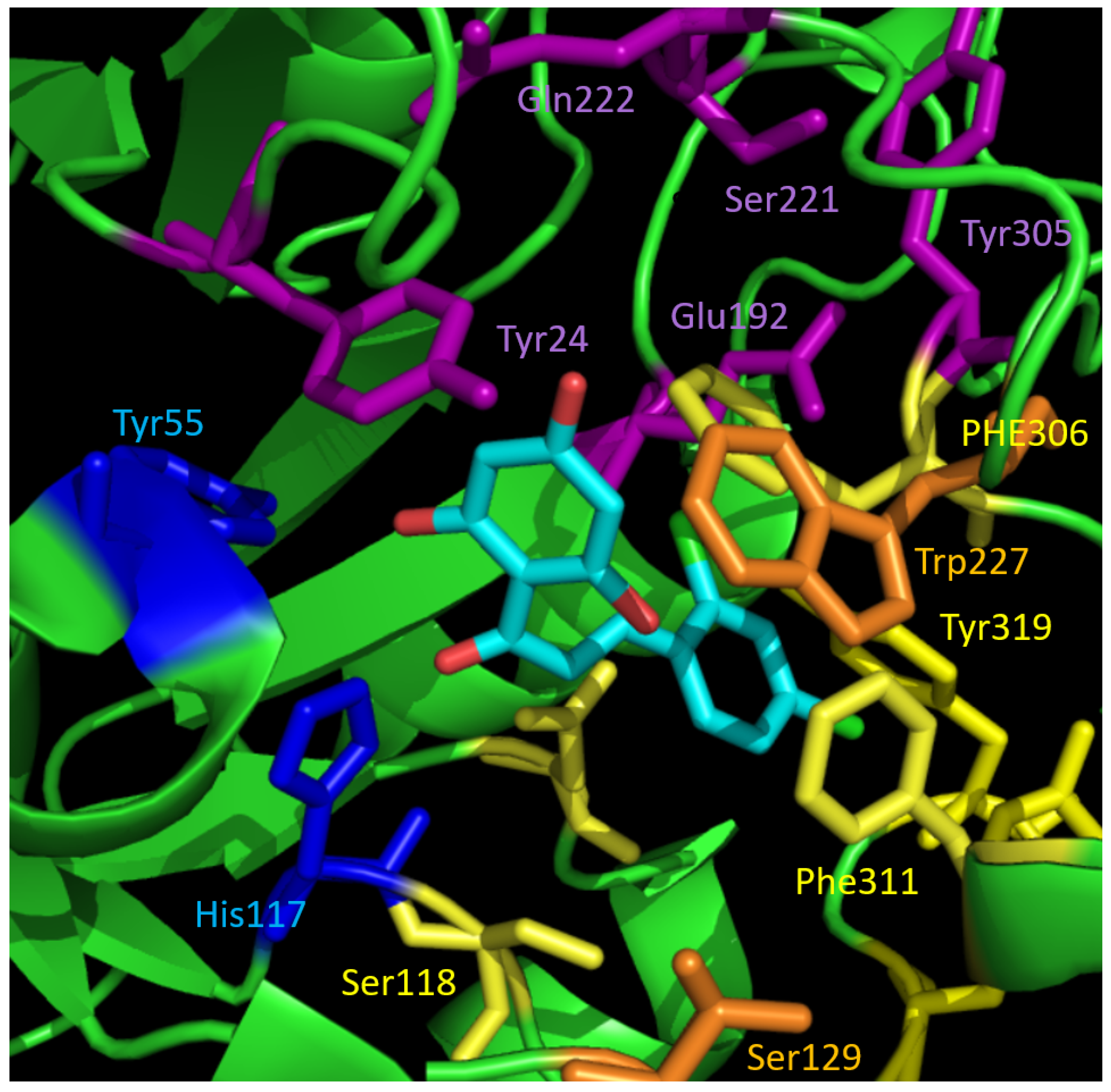
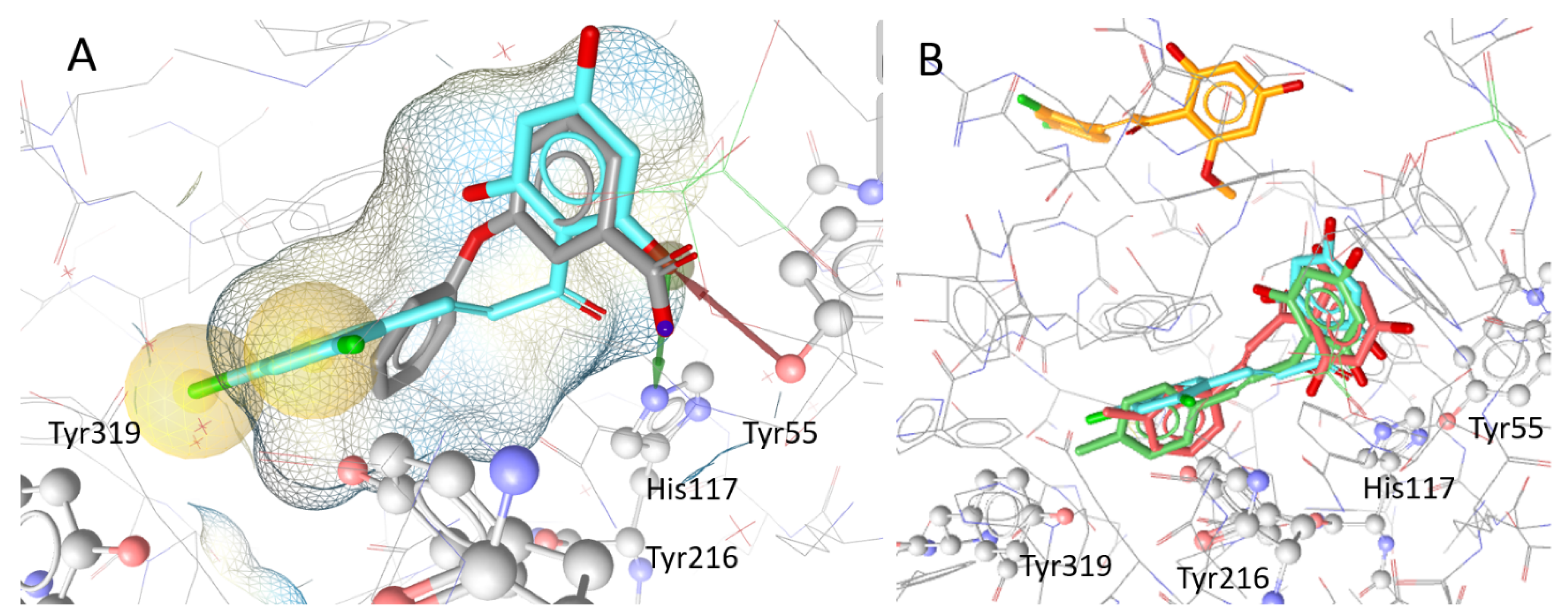
2.3. Docking and SAR Analysis for Selected AKR1C3-Inhibiting Chalcones
3. Discussion
4. Materials and Methods
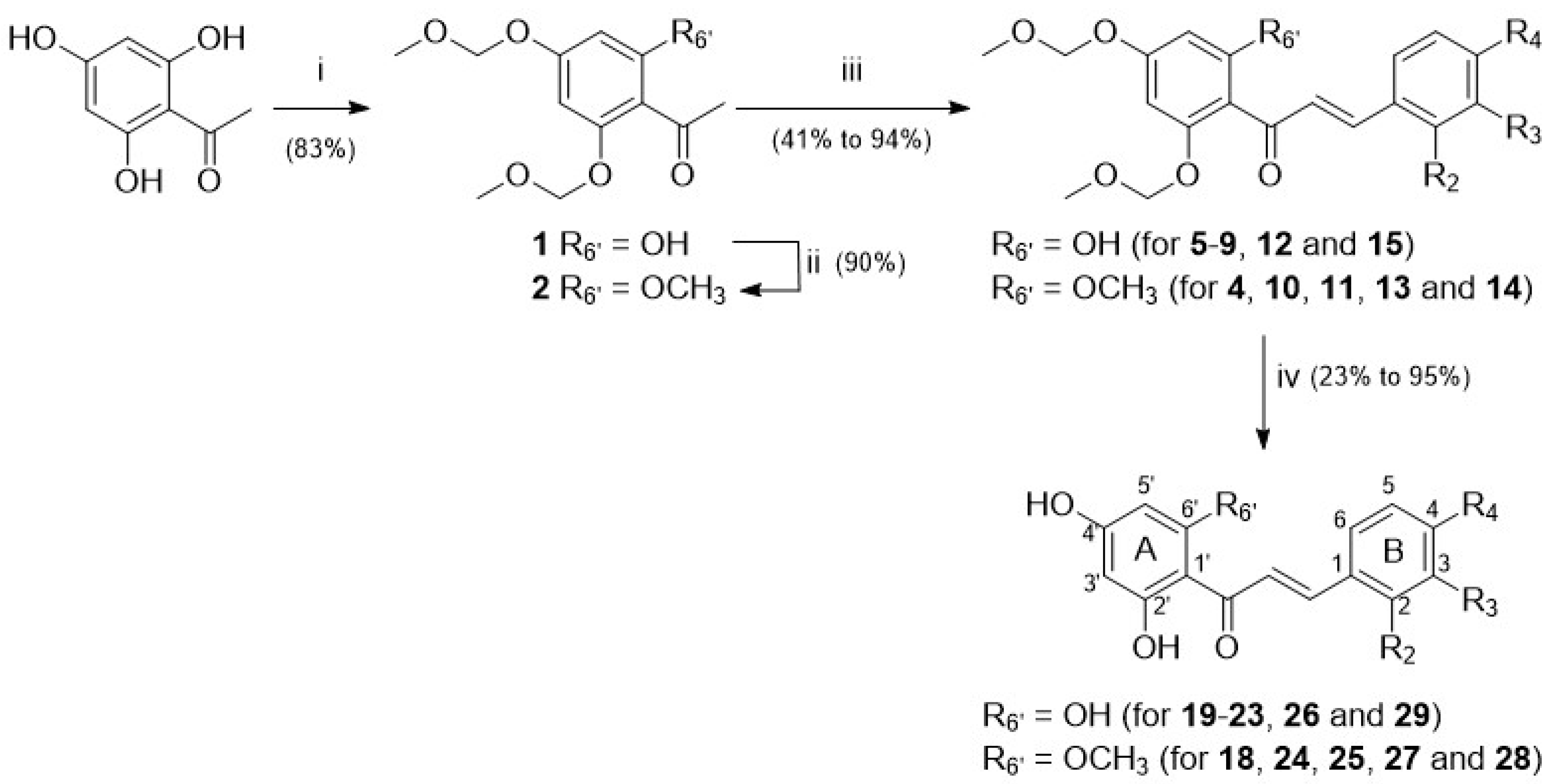
4.1. Synthesis of Chalcones
4.1.1. General Procedures
General Procedure for Chalcone Formation
General Procedure for Phenol Deprotection
4.2. Enzymatic Inhibition Assays
4.2.1. Enzymes
4.2.2. Inhibition Assays
4.3. Cytotoxicity Assays
4.3.1. Cell Culture
4.3.2. Cytotoxicity Assays
4.4. Compound Docking and SAR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qiu, W.; Zhou, M.; Labrie, F.; Lin, S.-X. Crystal Structures of the Multispecific 17β-Hydroxysteroid Dehydrogenase Type 5: Critical Androgen Regulation in Human Peripheral Tissues. Mol. Endocrinol. 2004, 18, 1798–1807. [Google Scholar] [CrossRef]
- Moeller, G.; Adamski, J. Integrated view on 17beta-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2009, 301, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Wangtrakuldee, P.; Auchus, R.J. Structural and Functional Biology of Aldo-Keto Reductase Steroid-Transforming Enzymes. Endocr. Rev. 2019, 40, 447–475. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, G.; Luu-The, V.; Tětu, B.; Labrie, F. Immunocytochemical Localization of Type 5 17 β-Hydroxysteroid Dehydrogenase in Human Reproductive Tissues. J. Histochem. Cytochem. 1999, 47, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-K.; Steckelbroeck, S.; Fung, K.-M.; Jones, A.N.; Penning, T.M. Characterization of a monoclonal antibody for human aldo-keto reductase AKR1C3 (type 2 3α-hydroxysteroid dehydrogenase/type 5 17β-hydroxysteroid dehydrogenase); immunohistochemical detection in breast and prostate. Steroids 2004, 69, 795–801. [Google Scholar] [CrossRef]
- Penning, T.M.; Burczynski, M.E.; Jez, J.M.; Hung, C.-F.; Lin, H.-K.; Ma, H.; Moore, M.; Palackal, N.; Ratnam, K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351, 67–77. [Google Scholar] [CrossRef]
- Byrns, M.C.; Duan, L.; Lee, S.H.; Blair, I.A.; Penning, T.M. Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prostaglandin metabolism that may explain its over-expression in breast cancer. J. Steroid Biochem. Mol. Biol. 2010, 118, 177–187. [Google Scholar] [CrossRef]
- Dufort, I.; Rheault, P.; Huang, X.-F.; Soucy, P.; Luu-The, V. Characteristics of a Highly Labile Human Type 5 17β-Hydroxysteroid Dehydrogenase. Endocrinology 1999, 140, 568–574. [Google Scholar] [CrossRef]
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem.-Biol. Interact. 2015, 234, 236–246. [Google Scholar] [CrossRef]
- Nakamura, Y.; Hornsby, P.J.; Casson, P.; Morimoto, R.; Satoh, F.; Xing, Y.; Kennedy, M.R.; Sasano, H.; Rainey, W.E. Type 5 17β-Hydroxysteroid Dehydrogenase (AKR1C3) Contributes to Testosterone Production in the Adrenal Reticularis. J. Clin. Endocrinol. Metab. 2009, 94, 2192–2198. [Google Scholar] [CrossRef]
- Labrie, F.; Luu-The, V.; Lin, S.-X.; Simard, J.; Labrie, C. Role of 17β-Hydroxysteroid Dehydrogenases in Sex Steroid Formation in Peripheral Intracrine Tissues. Trends Endocrinol. Metab. 2000, 11, 421–427. [Google Scholar] [CrossRef]
- Schiffer, L.; Arlt, W.; Storbeck, K.-H. Intracrine androgen biosynthesis, metabolism and action revisited. Mol. Cell. Endocrinol. 2018, 465, 4–26. [Google Scholar] [CrossRef]
- Yepuru, M.; Wu, Z.; Kulkarni, A.; Yin, F.; Barrett, C.M.; Kim, J.; Steiner, M.S.; Miller, D.D.; Dalton, J.T.; Narayanan, R. Steroidogenic Enzyme AKR1C3 Is a Novel Androgen Receptor-Selective Coactivator that Promotes Prostate Cancer Growth. Clin. Cancer Res. 2013, 19, 5613–5625. [Google Scholar] [CrossRef]
- Komoto, J.; Yamada, T.; Watanabe, K.; Takusagawa, F. Crystal Structure of Human Prostaglandin F Synthase (AKR1C3). Biochemistry 2004, 43, 2188–2198. [Google Scholar] [CrossRef]
- Matsuura, K.; Shiraishi, H.; Hara, A.; Sato, K.; Deyashiki, Y.; Ninomiya, M.; Sakai, S. Identification of a Principal mRNA Species for Human 3α-Hydroxysteroid Dehydrogenase Isoform (AKR1C3) That Exhibits High Prostaglandin D2 11-Ketoreductase Activity. J. Biochem. 1998, 124, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. Aldo-Keto Reductase (AKR) 1C3 inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 1329–1340. [Google Scholar] [CrossRef]
- Penning, T.M. AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol. Cell. Endocrinol. 2019, 489, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Adeniji, A.O.; Chen, M.; Penning, T.M. AKR1C3 as a target in castrate resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2013, 137, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Rižner, T.L.; Penning, T.M. Aldo-keto reductase 1C3—Assessment as a new target for the treatment of endometriosis. Pharmacol. Res. 2020, 152, 104446. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.K.; Acosta-Jimenez, L.; Coates, P.J.; McMahon, M.; Carey, F.A.; Honda, T.; Hayes, J.D.; Henderson, C.J.; Wolf, C.R. Aldo-keto reductases are biomarkers of NRF2 activity and are co-ordinately overexpressed in non-small cell lung cancer. Br. J. Cancer 2016, 115, 1530–1539, Erratum in Br. J. Cancer 2017, 117, e1. [Google Scholar] [CrossRef]
- Nakarai, C.; Osawa, K.; Akiyama, M.; Matsubara, N.; Ikeuchi, H.; Yamano, T.; Hirota, S.; Tomita, N.; Usami, M.; Kido, Y. Expression of AKR1C3 and CNN3 as markers for detection of lymph node metastases in colorectal cancer. Clin. Exp. Med. 2015, 15, 333–341. [Google Scholar] [CrossRef]
- Peraldo-Neia, C.; Ostano, P.; Mello-Grand, M.; Guana, F.; Gregnanin, I.; Boschi, D.; Oliaro-Bosso, S.; Pippione, A.C.; Carenzo, A.; De Cecco, L.; et al. AKR1C3 is a biomarker and druggable target for oropharyngeal tumors. Cell. Oncol. 2021, 44, 357–372. [Google Scholar] [CrossRef]
- Manesh, D.M.; El-Hoss, J.; Evans, K.; Richmond, J.; Toscan, C.E.; Bracken, L.S.; Hedrick, A.; Sutton, R.; Marshall, G.M.; Wilson, W.R.; et al. AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood 2015, 126, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Frycz, B.A.; Murawa, D.; Borejsza-Wysocki, M.; Wichtowski, M.; Spychała, A.; Marciniak, R.; Murawa, P.; Drews, M.; Jagodziński, P.P. Transcript level of AKR1C3 is down-regulated in gastric cancer. Biochem. Cell Biol. 2016, 94, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, S.; Chen, Y.; Liu, Y.; Feng, F.; Liu, W.; Guo, Q.; Zhao, L.; Sun, H. Overview of AKR1C3: Inhibitor Achievements and Disease Insights. J. Med. Chem. 2020, 63, 11305–11329. [Google Scholar] [CrossRef] [PubMed]
- Novotna, R.; Wsol, V.; Xiong, G.; Maser, E. Inactivation of the anticancer drugs doxorubicin and oracin by aldo–keto reductase (AKR) 1C3. Toxicol. Lett. 2008, 181, 1–6. [Google Scholar] [CrossRef]
- Hofman, J.; Malcekova, B.; Skarka, A.; Novotna, E.; Wsol, V. Aldo-Keto Reductase 1c3 Induces Anthracycline Resistance in Cancer Cells by the Reduction of Daunorubicin and Idarubicin. Drug Metab. Rev. 2014, 45, 193. [Google Scholar]
- Penning, T.M.; Jonnalagadda, S.; Trippier, P.C.; Rižner, T.L. Aldo-Keto Reductases and Cancer Drug Resistance. Pharmacol. Rev. 2021, 73, 1150–1171. [Google Scholar] [CrossRef]
- Penning, T.M.; Asangani, I.A.; Sprenger, C.; Plymate, S. Intracrine androgen biosynthesis and drug resistance. Cancer Drug Resist. 2020, 3, 912–929. [Google Scholar] [CrossRef]
- Chen, M.; Adeniji, A.O.; Twenter, B.M.; Winkler, J.D.; Christianson, D.W.; Penning, T.M. Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg. Med. Chem. Lett. 2012, 22, 3492–3497. [Google Scholar] [CrossRef]
- Kafka, M.; Mayr, F.; Temml, V.; Möller, G.; Adamski, J.; Höfer, J.; Schwaiger, S.; Heidegger, I.; Matuszczak, B.; Schuster, D.; et al. Dual Inhibitory Action of a Novel AKR1C3 Inhibitor on Both Full-Length AR and the Variant AR-V7 in Enzalutamide Resistant Metastatic Castration Resistant Prostate Cancer. Cancers 2020, 12, 2092. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Sun, M.; Zhou, Y.; Zhuo, X.; Wang, S.; Jiang, S.; Peng, Z.; Kang, K.; Zheng, X.; Sun, M. Design, Synthesis and Cytotoxicity Evaluation of Novel Indole Derivatives Containing Benzoic Acid Group as Potential AKR1C3 Inhibitors. Chem. Biodivers. 2020, 17, e2000519. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.-M.; Chang, L.-L.; Ying, M.-D.; Cao, J.; He, Q.-J.; Zhu, H.; Yang, B. Aldo-Keto Reductase AKR1C1-AKR1C4: Functions, Regulation, and Intervention for Anti-cancer Therapy. Front. Pharmacol. 2017, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Zhu, D.; You, J.; Dong, X.; Yang, B.; Zhu, H.; He, Q. Liquiritin, as a Natural Inhibitor of AKR1C1, Could Interfere With the Progesterone Metabolism. Front. Physiol. 2019, 10, 833. [Google Scholar] [CrossRef]
- Jasim, H.A.; Nahar, L.; Jasim, M.A.; Moore, S.A.; Ritchie, K.J.; Sarker, S.D. Chalcones: Synthetic Chemistry follows Where Nature Leads. Biomolecules 2021, 11, 1203. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; Satomi, Y.; Mizutani, Y.; Ukimura, O.; Kawauchi, A.; Sakai, T.; Baba, M.; Okuyama, T.; Nishino, H.; Miki, T. Isoliquiritigenin Inhibits the Growth of Prostate Cancer. Eur. Urol. 2003, 43, 580–586. [Google Scholar] [CrossRef]
- Shukla, S.; Sood, A.K.; Goyal, K.; Singh, A.; Sharma, V.; Guliya, N.; Gulati, S.; Kumar, S. Chalcone Scaffolds as Anticancer Drugs: A Review on Molecular Insight in Action of Mechanisms and Anticancer Properties. Anti-Cancer Agents Med. Chem. 2021, 21, 1650–1670. [Google Scholar] [CrossRef]
- Jandial, D.; Blair, C.; Zhang, S.; Krill, L.; Zhang, Y.-B.; Zi, X. Molecular targeted approaches to cancer therapy and prevention using chalcones. Curr. Cancer Drug Targets 2014, 14, 181–200. [Google Scholar] [CrossRef]
- Brožič, P.; Golob, B.; Gomboc, N.; Rižner, T.L.; Gobec, S. Cinnamic acids as new inhibitors of 17β-hydroxysteroid dehydrogenase type 5 (AKR1C3). Mol. Cell. Endocrinol. 2006, 248, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Zang, T.; Verma, K.; Chen, M.; Jin, Y.; Trippier, P.C.; Penning, T.M. Screening baccharin analogs as selective inhibitors against type 5 17β-hydroxysteroid dehydrogenase (AKR1C3). Chem.-Biol. Interact. 2015, 234, 339–348. [Google Scholar] [CrossRef]
- Gazvoda, M.; Beranič, N.; Turk, S.; Burja, B.; Kočevar, M.; Rižner, T.L.; Gobec, S.; Polanc, S. 2,3-Diarylpropenoic acids as selective non-steroidal inhibitors of type-5 17β-hydroxysteroid dehydrogenase (AKR1C3). Eur. J. Med. Chem. 2013, 62, 89–97. [Google Scholar] [CrossRef]
- Krazeisen, A.; Breitling, R.; Möller, G.; Adamski, J. Phytoestrogens inhibit human 17β-hydroxysteroid dehydrogenase type 5. Mol. Cell. Endocrinol. 2001, 171, 151–162. [Google Scholar] [CrossRef]
- Škarydová, L.; Živná, L.; Xiong, G.; Maser, E.; Wsól, V. AKR1C3 as a potential target for the inhibitory effect of dietary flavonoids. Chem.-Biol. Interact. 2009, 178, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Skarydova, L.; Hofman, J.; Chlebek, J.; Havrankova, J.; Kosanova, K.; Skarka, A.; Hostalkova, A.; Plucha, T.; Cahlikova, L.; Wsol, V. Isoquinoline alkaloids as a novel type of AKR1C3 inhibitors. J. Steroid Biochem. Mol. Biol. 2014, 143, 250–258. [Google Scholar] [CrossRef]
- Le Bail, J.-C.; Pouget, C.; Fagnere, C.; Basly, J.-P.; Chulia, A.-J.; Habrioux, G. Chalcones are potent inhibitors of aromatase and 17β-hydroxysteroid dehydrogenase activities. Life Sci. 2001, 68, 751–761. [Google Scholar] [CrossRef]
- Thévenin, M.; Mouray, E.; Grellier, P.; Dubois, J. Facile Formation of Methylenebis(chalcone)s through Unprecedented Methylenation Reaction. Application to Antiparasitic and Natural Product Synthesis. Eur. J. Org. Chem. 2014, 2014, 2986–2992. [Google Scholar] [CrossRef]
- Boehlow, T.R.; Harburn, J.J.; Spilling, C.D. Approaches to the Synthesis of Some Tyrosine-Derived Marine Sponge Metabolites: Synthesis of Verongamine and Purealidin N. J. Org. Chem. 2001, 66, 3111–3118. [Google Scholar] [CrossRef]
- Urgaonkar, S.; La Pierre, H.S.; Meir, I.; Lund, H.; RayChaudhuri, D.; Shaw, J.T. Synthesis of Antimicrobial Natural Products Targeting FtsZ: (±)-Dichamanetin and (±)-2‘ ‘‘-Hydroxy-5‘ ‘-benzylisouvarinol-B. Org. Lett. 2005, 7, 5609–5612. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, D.P.; Kiyota, T.; Dong, Y.; Ikezu, T.; Vennerstrom, J.L. Phenolic Bis-styrylbenzenes as β-Amyloid Binding Ligands and Free Radical Scavengers. J. Med. Chem. 2010, 53, 7992–7999. [Google Scholar] [CrossRef]
- Zhang, B.; Duan, D.; Ge, C.; Yao, J.; Liu, Y.; Li, X.; Fang, J. Synthesis of Xanthohumol Analogues and Discovery of Potent Thioredoxin Reductase Inhibitor as Potential Anticancer Agent. J. Med. Chem. 2015, 58, 1795–1805. [Google Scholar] [CrossRef]
- Sui, X.; Quan, Y.-C.; Chang, Y.; Zhang, R.-P.; Xu, Y.-F.; Guan, L.-P. Synthesis and studies on antidepressant activity of 2′,4′,6′-trihydroxychalcone derivatives. Med. Chem. Res. 2012, 21, 1290–1296. [Google Scholar] [CrossRef]
- Vogel, S.; Ohmayer, S.; Brunner, G.; Heilmann, J. Natural and non-natural prenylated chalcones: Synthesis, cytotoxicity and anti-oxidative activity. Bioorg. Med. Chem. 2008, 16, 4286–4293. [Google Scholar] [CrossRef]
- Jin, Y.L.; Jin, X.Y.; Jin, F.; Sohn, D.H.; Kim, H.S. Structure activity relationship studies of anti-inflammatory TMMC derivatives: 4-Dimethylamino group on the B ring responsible for lowering the potency. Arch. Pharmacal Res. 2008, 31, 1145–1152. [Google Scholar] [CrossRef]
- Nguyen, V.-S.; Dong, L.-P.; Wang, S.-C.; Wang, Q. The First Total Synthesis of Sophoflavescenol, Flavenochromane C, and Citrusinol. Eur. J. Org. Chem. 2015, 2015, 2297–2302. [Google Scholar] [CrossRef]
- Jeong, S.; Lee, S.; Kim, K.; Lee, Y.; Lee, J.; Oh, S.; Choi, J.-W.; Kim, S.W.; Hwang, K.-C.; Lim, S. Isoliquiritigenin Derivatives Inhibit RANKL-Induced Osteoclastogenesis by Regulating p38 and NF-κB Activation in RAW 264.7 Cells. Molecules 2020, 25, 3908. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.U.; Yosaatmadja, Y.; Teague, R.M.; Chai, M.Z.L.; Turnbull, A.P.; Squire, C.J. Crystal Structures of Three Classes of Non-Steroidal Anti-Inflammatory Drugs in Complex with Aldo-Keto Reductase 1C3. PLoS ONE 2012, 7, e43965. [Google Scholar] [CrossRef]
- Saito, Y.; Mizokami, A.; Tsurimoto, H.; Izumi, K.; Goto, M.; Nakagawa-Goto, K. 5′-Chloro-2,2′-dihydroxychalcone and related flavanoids as treatments for prostate cancer. Eur. J. Med. Chem. 2018, 157, 1143–1152. [Google Scholar] [CrossRef]
- Tsachaki, M.; Odermatt, A. Subcellular localization and membrane topology of 17β-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2019, 489, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Drury, J.E. Human aldo-keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch. Biochem. Biophys. 2007, 464, 241–250. [Google Scholar] [CrossRef]
- Steckelbroeck, S.; Jin, Y.; Gopishetty, S.; Oyesanmi, B.; Penning, T.M. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity—Implications for steroid hormone metabolism and action. J. Biol. Chem. 2004, 279, 10784–10795. [Google Scholar] [CrossRef]
- Byrns, M.C.; Steckelbroeck, S.; Penning, T.M. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem. Pharmacol. 2008, 75, 484–493. [Google Scholar] [CrossRef]
- Penning, T.M.; Steckelbroeck, S.; Bauman, D.R.; Miller, M.W.; Jin, Y.; Peehl, D.M.; Fung, K.-M.; Lin, H.-K. Aldo-keto reductase (AKR) 1C3: Role in prostate disease and the development of specific inhibitors. Mol. Cell. Endocrinol. 2006, 248, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Puranen, T.; Poutanen, M.; Ghosh, D.; Vihko, P.; Vihko, R. Characterization of Structural and Functional Properties of Human 17β-Hydroxysteroid Dehydrogenase Type 1 Using Recombinant Enzymes and Site-Directed Mutagenesis. Mol. Endocrinol. 1997, 11, 77–86. [Google Scholar] [CrossRef][Green Version]
- Heinosalo, T.; Saarinen, N.; Poutanen, M. Role of hydroxysteroid (17beta) dehydrogenase type 1 in reproductive tissues and hormone-dependent diseases. Mol. Cell. Endocrinol. 2019, 489, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, S.; Mannaerts, G.P.; Baes, M.; Van Veldhoven, P.P. Peroxisomal multifunctional protein-2: The enzyme, the patients and the knockout mouse model. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2006, 1761, 973–994. [Google Scholar] [CrossRef] [PubMed]
- Marijanovic, Z.; Laubner, D.; Möller, G.; Gege, C.; Husen, B.; Adamski, J.; Breitling, R. Closing the Gap: Identification of Human 3-Ketosteroid Reductase, the Last Unknown Enzyme of Mammalian Cholesterol Biosynthesis. Mol. Endocrinol. 2003, 17, 1715–1725. [Google Scholar] [CrossRef]
- Vinklarova, L.; Schmidt, M.; Benek, O.; Kuca, K.; Gunn-Moore, F.; Musilek, K. Friend or enemy? Review of 17β-HSD10 and its role in human health or disease. J. Neurochem. 2020, 155, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Lukacik, P.; Keller, B.; Bunkoczi, G.; Kavanagh, K.; Lee, W.H.; Adamski, J.; Oppermann, U. Structural and biochemical characterization of human orphan DHRS10 reveals a novel cytosolic enzyme with steroid dehydrogenase activity. Biochem. J. 2007, 402, 419–427. [Google Scholar] [CrossRef]
- Schuster, D.; Kowalik, D.; Kirchmair, J.; Laggner, C.; Markt, P.; Aebischer-Gumy, C.; Ströhle, F.; Möller, G.; Wolber, G.; Wilckens, T.; et al. Identification of chemically diverse, novel inhibitors of 17β-hydroxysteroid dehydrogenase type 3 and 5 by pharmacophore-based virtual screening. J. Steroid Biochem. Mol. Biol. 2011, 125, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Braun, F.; Bertoletti, N.; Möller, G.; Adamski, J.; Frotscher, M.; Guragossian, N.; Gírio, P.A.M.; Le Borgne, M.; Ettouati, L.; Falson, P.; et al. Structure-based design and profiling of novel 17β-HSD14 inhibitors. Eur. J. Med. Chem. 2018, 155, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Sager, C.P.; Weber, S.; Negri, M.; Banachowicz, P.; Möller, G.; Adamski, J.; Hartmann, R.W.; Marchais-Oberwinkler, S. Homology modeling meets site-directed mutagenesis: An ideal combination to elucidate the topology of 17β-HSD2. J. Steroid Biochem. Mol. Biol. 2021, 206, 105790. [Google Scholar] [CrossRef]
- Möller, G.; Deluca, D.; Gege, C.; Rosinus, A.; Kowalik, D.; Peters, O.; Droescher, P.; Elger, W.; Adamski, J.; Hillisch, A. Structure-based design, synthesis and in vitro characterization of potent 17β-hydroxysteroid dehydrogenase type 1 inhibitors based on 2-substitutions of estrone and D-homo-estrone. Bioorg. Med. Chem. Lett. 2009, 19, 6740–6744. [Google Scholar] [CrossRef]
- Jackson, V.J.; Yosaatmadja, Y.; Flanagan, J.U.; Squire, C.J. Structure of AKR1C3 with 3-phenoxybenzoic acid bound. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 409–413. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Nicholls, A. Conformer Generation with OMEGA: Learning from the Data Set and the Analysis of Failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substitution Pattern | AKR1C3 Inhibition | Docking and SAR Analyses | ||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R6‘ | R2 | R3 | R4 | Inhibition at 10 µM [%] | IC50 [µM] | Key Interactions | Docking Score |
| Isoliquiritigenin (16) | H | H | H | OH | 31.1 ± 3.2 | nd | 65.81 | |
| Butein (17) | H | H | OH | OH | 37.2 ± 1.4 | nd | 63.11 | |
| 18 | OCH3 | H | H | OCH3 | 47.3 ± 6.4 | 11.91 ± 2.03 | 74.55 | |
| 19 | OH | H | H | OCH3 | 78.1 ± 2.7 | 2.36 ± 0.54 | Tyr319 | 67.71 |
| 20 | OH | H | CH3 | H | 89.2 ± 6.3 | 1.94 ± 0.32 | Tyr319 | 69.80 |
| 21 | OH | H | H | CH3 | 87.9 ± 4.9 | 5.18 ± 1.64 | Tyr319 | 69.09 |
| 22 | OH | H | H | Cl | 70.8 ± 3.8 | nd | Tyr319 | 66.90 |
| 23 | OH | Cl | H | Cl | 90.9 ± 4.6 | 1.08 ± 0.27 | Tyr319, Tyr216 | 58.91 |
| 24 | OCH3 | H | H | OH | 24.6 ± 4.8 | nd | Tyr216 | 62.77 |
| 25 | OCH3 | Cl | H | Cl | 33.3 ± 6.0 | nd | out of binding pocket | 48.26 |
| 26 | OH | H | H | F | 68.5 ± 6.4 | nd | Tyr319 | 62.76 |
| 27 | OCH3 | H | H | F | 20.3 ± 8.4 | nd | out of binding pocket | 45.19 |
| 28 | OCH3 | H | H | Cl | 26.1 ± 2.0 | nd | out of binding pocket | 48.32 |
| 29 | OH | H | H | OH | 40.6 ± 7.7 | nd | 64.83 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Möller, G.; Temml, V.; Cala Peralta, A.; Gruet, O.; Richomme, P.; Séraphin, D.; Viault, G.; Kraus, L.; Huber-Cantonati, P.; Schopfhauser, E.; et al. Analogues of Natural Chalcones as Efficient Inhibitors of AKR1C3. Metabolites 2022, 12, 99. https://doi.org/10.3390/metabo12020099
Möller G, Temml V, Cala Peralta A, Gruet O, Richomme P, Séraphin D, Viault G, Kraus L, Huber-Cantonati P, Schopfhauser E, et al. Analogues of Natural Chalcones as Efficient Inhibitors of AKR1C3. Metabolites. 2022; 12(2):99. https://doi.org/10.3390/metabo12020099
Chicago/Turabian StyleMöller, Gabriele, Veronika Temml, Antonio Cala Peralta, Océane Gruet, Pascal Richomme, Denis Séraphin, Guillaume Viault, Luisa Kraus, Petra Huber-Cantonati, Elisabeth Schopfhauser, and et al. 2022. "Analogues of Natural Chalcones as Efficient Inhibitors of AKR1C3" Metabolites 12, no. 2: 99. https://doi.org/10.3390/metabo12020099
APA StyleMöller, G., Temml, V., Cala Peralta, A., Gruet, O., Richomme, P., Séraphin, D., Viault, G., Kraus, L., Huber-Cantonati, P., Schopfhauser, E., Pachmayr, J., Tokarz, J., Schuster, D., Helesbeux, J.-J., & Dyar, K. A. (2022). Analogues of Natural Chalcones as Efficient Inhibitors of AKR1C3. Metabolites, 12(2), 99. https://doi.org/10.3390/metabo12020099