
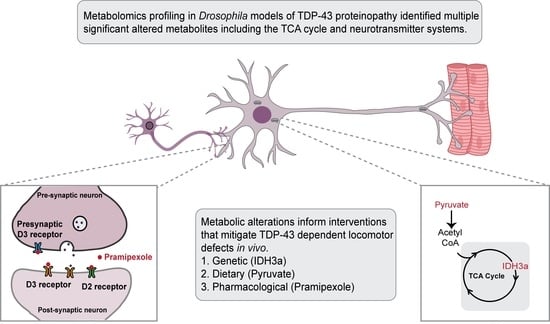
TDP-43 Proteinopathy Causes Broad Metabolic Alterations including TCA Cycle Intermediates and Dopamine Levels in Drosophila Models of ALS

 , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. TDP-43 Proteinopathy in Motor Neurons Alters Several Metabolic Pathways In Vivo, in Drosophila Models of ALS
2.2. TDP-43 Proteinopathy Causes Alterations in the TCA Cycle and the Glutaminolysis Pathway
2.3. Overexpression of Isocitrate Dehydrogenase 3 (IDH3) in MNs Rescues TDP-43 Induced Locomotor Deficits
2.4. Pyruvate Supplementation Mitigates TDP-43 Induced Locomotor Deficits in Drosophila Models of ALS
2.5. TDP-43 Proteinopathy Causes Alterations in Neurotransmitter Levels
2.6. Dopamine Agonist Feeding Mitigates TDP-43 Induced Locomotor Deficits in Drosophila Models of ALS
3. Discussion
4. Materials and Methods
4.1. Drosophila Genetics
4.2. Fly Food Supplementation
4.3. Metabolomics
4.4. Locomotor Assays
4.5. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef]
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 2015, 7, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Pradat, P.F.; Ludolph, A.C.; Loeffler, J.P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Tefera, T.W.; Borges, K. Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci. 2016, 10, 611. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 2011, 44, 20–24. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Estes, P.S.; Boehringer, A.; Zwick, R.; Tang, J.E.; Grigsby, B.; Zarnescu, D.C. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 2011, 20, 2308–2321. [Google Scholar] [CrossRef]
- Estes, P.S.; Daniel, S.G.; McCallum, A.P.; Boehringer, A.V.; Sukhina, A.S.; Zwick, R.A.; Zarnescu, D.C. Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a Drosophila model of amyotrophic lateral sclerosis. Dis. Models Mech. 2013, 6, 721–733. [Google Scholar] [CrossRef]
- Lawton, K.A.; Cudkowicz, M.E.; Brown, M.V.; Alexander, D.; Caffrey, R.; Wulff, J.E.; Bowser, R.; Lawson, R.; Jaffa, M.; Milburn, M.V.; et al. Biochemical alterations associated with ALS. Amyotroph. Lateral Scler. 2012, 13, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Lawton, K.A.; Brown, M.V.; Alexander, D.; Li, Z.; Wulff, J.E.; Lawson, R.; Jaffa, M.; Milburn, M.V.; Ryals, J.A.; Bowser, R.; et al. Plasma metabolomic biomarker panel to distinguish patients with amyotrophic lateral sclerosis from disease mimics. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 362–370. [Google Scholar] [CrossRef]
- Manzo, E.; O’Conner, A.G.; Barrows, J.M.; Shreiner, D.D.; Birchak, G.J.; Zarnescu, D.C. Medium-Chain Fatty Acids, Beta-Hydroxybutyric Acid and Genetic Modulation of the Carnitine Shuttle Are Protective in a Drosophila Model of ALS Based on TDP-43. Front. Mol. Neurosci. 2018, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Manzo, E.; Lorenzini, I.; Barrameda, D.; O’Conner, A.G.; Barrows, J.M.; Starr, A.; Kovalik, T.; Rabichow, B.E.; Lehmkuhl, E.M.; Shreiner, D.D.; et al. Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. eLife 2019, 8, e45114. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef]
- Ugur, B.; Bao, H.; Stawarski, M.; Duraine, L.R.; Zuo, Z.; Lin, Y.Q.; Neely, G.G.; Macleod, G.T.; Chapman, E.R.; Bellen, H.J. The Krebs Cycle Enzyme Isocitrate Dehydrogenase 3A Couples Mitochondrial Metabolism to Synaptic Transmission. Cell Rep. 2017, 21, 3794–3806. [Google Scholar] [CrossRef]
- Kato, S.; Oda, M.; Tanabe, H. Diminution of dopaminergic neurons in the substantia nigra of sporadic amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 1993, 19, 300–304. [Google Scholar] [CrossRef]
- Borasio, G.D.; Linke, R.; Schwarz, J.; Schlamp, V.; Abel, A.; Mozley, P.D.; Tatsch, K. Dopaminergic deficit in amyotrophic lateral sclerosis assessed with [I-123] IPT single photon emission computed tomography. J. Neurol. Neurosurg. Psychiatry 1998, 65, 263–265. [Google Scholar] [CrossRef][Green Version]
- Coyne, A.N.; Siddegowda, B.B.; Estes, P.S.; Johannesmeyer, J.; Kovalik, T.; Daniel, S.G.; Pearson, A.; Bowser, R.; Zarnescu, D.C. Futsch/MAP1B mRNA Is a Translational Target of TDP-43 and Is Neuroprotective in a Drosophila Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2014, 34, 15962–15974. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Lorenzini, I.; Chou, C.C.; Torvund, M.; Rogers, R.S.; Starr, A.; Zaepfel, B.L.; Levy, J.; Johannesmeyer, J.; Schwartz, J.C.; et al. Post-transcriptional Inhibition of Hsc70-4/HSPA8 Expression Leads to Synaptic Vesicle Cycling Defects in Multiple Models of ALS. Cell Rep. 2017, 21, 110–125. [Google Scholar] [CrossRef]
- Goutman, S.A.; Boss, J.; Guo, K.; Alakwaa, F.M.; Patterson, A.; Kim, S.; Savelieff, M.G.; Hur, J.; Feldman, E.L. Untargeted metabolomics yields insight into ALS disease mechanisms. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1329–1338. [Google Scholar] [CrossRef]
- Veyrat-Durebex, C.; Corcia, P.; Piver, E.; Devos, D.; Dangoumau, A.; Gouel, F.; Vourc’h, P.; Emond, P.; Laumonnier, F.; Nadal-Desbarats, L.; et al. Disruption of TCA Cycle and Glutamate Metabolism Identified by Metabolomics in an In Vitro Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2016, 53, 6910–6924. [Google Scholar] [CrossRef] [PubMed]
- Ng Kee Kwong, K.C.; Gregory, J.M.; Pal, S.; Chandran, S.; Mehta, A.R. Cerebrospinal fluid cytotoxicity in amyotrophic lateral sclerosis: A systematic review of in vitro studies. Brain Commun. 2020, 2, fcaa121. [Google Scholar] [CrossRef]
- Andreadou, E.; Kapaki, E.; Kokotis, P.; Paraskevas, G.P.; Katsaros, N.; Libitaki, G.; Zis, V.; Sfagos, C.; Vassilopoulos, D. Plasma glutamate and glycine levels in patients with amyotrophic lateral sclerosis: The effect of riluzole treatment. Clin. Neurol. Neurosurg. 2008, 110, 222–226. [Google Scholar] [CrossRef]
- Fiszman, M.L.; Ricart, K.C.; Latini, A.; Rodriguez, G.; Sica, R.E. In vitro neurotoxic properties and excitatory aminoacids concentration in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Relationship with the degree of certainty of disease diagnoses. Acta Neurol. Scand. 2010, 121, 120–126. [Google Scholar] [CrossRef]
- Genc, S.; Kurnaz, I.A.; Ozilgen, M. Astrocyte-neuron lactate shuttle may boost more ATP supply to the neuron under hypoxic conditions—In silico study supported by in vitro expression data. BMC Syst. Biol. 2011, 5, 162. [Google Scholar] [CrossRef]
- Tefera, T.W.; Wong, Y.; Barkl-Luke, M.E.; Ngo, S.T.; Thomas, N.K.; McDonald, T.S.; Borges, K. Triheptanoin Protects Motor Neurons and Delays the Onset of Motor Symptoms in a Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2016, 11, e0161816. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Kuo, W.T.; Huang, Y.C.; Lee, T.C.; Yu, L.C. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013, 4, e622. [Google Scholar] [CrossRef]
- Cudkowicz, M.; Bozik, M.E.; Ingersoll, E.W.; Miller, R.; Mitsumoto, H.; Shefner, J.; Moore, D.H.; Schoenfeld, D.; Mather, J.L.; Archibald, D.; et al. The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat. Med. 2011, 17, 1652–1656. [Google Scholar] [CrossRef]
- Okano, H.; Yasuda, D.; Fujimori, K.; Morimoto, S.; Takahashi, S. Ropinirole, a New ALS Drug Candidate Developed Using iPSCs. Trends Pharmacol. Sci. 2020, 41, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Joardar, A.; Menzl, J.; Podolsky, T.C.; Manzo, E.; Estes, P.S.; Ashford, S.; Zarnescu, D.C. PPAR gamma activation is neuroprotective in a Drosophila model of ALS based on TDP-43. Hum. Mol. Genet. 2014, 24, 1741–1754. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loganathan, S.; Wilson, B.A.; Carey, S.B.; Manzo, E.; Joardar, A.; Ugur, B.; Zarnescu, D.C. TDP-43 Proteinopathy Causes Broad Metabolic Alterations including TCA Cycle Intermediates and Dopamine Levels in Drosophila Models of ALS. Metabolites 2022, 12, 101. https://doi.org/10.3390/metabo12020101
Loganathan S, Wilson BA, Carey SB, Manzo E, Joardar A, Ugur B, Zarnescu DC. TDP-43 Proteinopathy Causes Broad Metabolic Alterations including TCA Cycle Intermediates and Dopamine Levels in Drosophila Models of ALS. Metabolites. 2022; 12(2):101. https://doi.org/10.3390/metabo12020101
Chicago/Turabian StyleLoganathan, Suvithanandhini, Bryce A. Wilson, Sara B. Carey, Ernesto Manzo, Archi Joardar, Berrak Ugur, and Daniela C. Zarnescu. 2022. "TDP-43 Proteinopathy Causes Broad Metabolic Alterations including TCA Cycle Intermediates and Dopamine Levels in Drosophila Models of ALS" Metabolites 12, no. 2: 101. https://doi.org/10.3390/metabo12020101
APA StyleLoganathan, S., Wilson, B. A., Carey, S. B., Manzo, E., Joardar, A., Ugur, B., & Zarnescu, D. C. (2022). TDP-43 Proteinopathy Causes Broad Metabolic Alterations including TCA Cycle Intermediates and Dopamine Levels in Drosophila Models of ALS. Metabolites, 12(2), 101. https://doi.org/10.3390/metabo12020101