
Isotope-Assisted Metabolic Flux Analysis: A Powerful Technique to Gain New Insights into the Human Metabolome in Health and Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Isotope-Assisted Metabolic Flux Analysis (iMFA)
- iMFA provides quantitative information on the entire metabolic network, including metabolic flux values, confidence intervals, and statistical analysis.
- The iMFA model can easily be modified with new pathways, compartments, or influxes, since the iMFA software automatically reformulates the underlying metabolic network when a reaction is added or removed [32]. This feature is especially valuable when the preformulated metabolic network does not satisfactorily fit the data and can be advantageous over other flux analysis methods that require analytical formula derivations for each flux [6,33].
- A poor fit between the iMFA model and labeling data suggests either measurement errors or incorrect model assumptions. Modifying the metabolic model to achieve an acceptable fit can uncover previously unknown metabolic features and may ultimately lead to new insights into the metabolic system, such as unconventional metabolite channeling [34] or novel major carbon sources [35].
- iMFA rigorously accounts for network complexities [20,23], including: reaction reversibilities, which are common in the pentose phosphate pathway (PPP) and the tricarboxylic acid (TCA) cycle; pathway cyclicity (a TCA cycle feature); high network connectivity (common in central carbon metabolism); and isotope natural abundance.
- iMFA is particularly useful for discerning fluxes in complex mammalian cells that have multiple inputs, which can complicate interpretation of labeling patterns. For example, in glucose labeling experiments, the labeling of TCA metabolites is diluted by anaplerotic compounds. By integrating isotope labeling data with extracellular flux values, iMFA can address whether decreased labeling is due to decreased entry of labeled nutrients, increased incorporation of unlabeled compounds, or both.
- iMFA is scalable and can incorporate large metabolic data sets. In addition, the network-based approach of iMFA ensures that labeling patterns are analyzed in the context of the whole network, rather than as standalone elements. These attributes allow for the iMFA approach to fill holes in the model and buffer measurement error effects in the data [6,32].
- Although iMFA and ISA are closely related and can often be performed using the same software, ISA is typically used to estimate fractional contributions of different metabolites to de novo fatty acid biosynthesis, whereas iMFA estimates absolute metabolic fluxes throughout the metabolic network [20,36,37].
- While in silico genome-scale model flux analysis is a powerful fluxomic tool, it relies on numerous assumptions for flux prediction. iMFA estimates metabolic fluxes from actual isotope labeling experiment data and offers superior flux resolution [38].
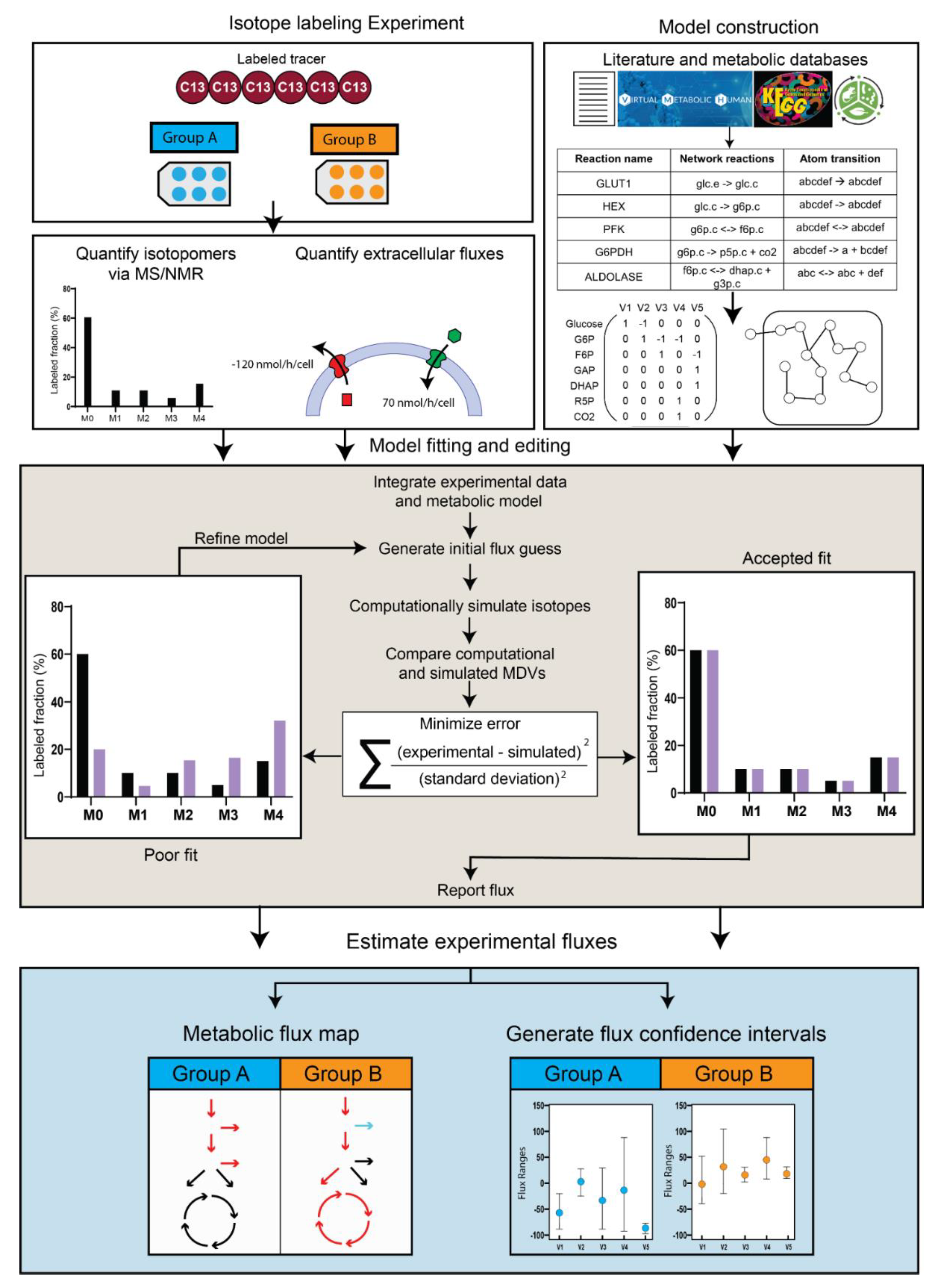
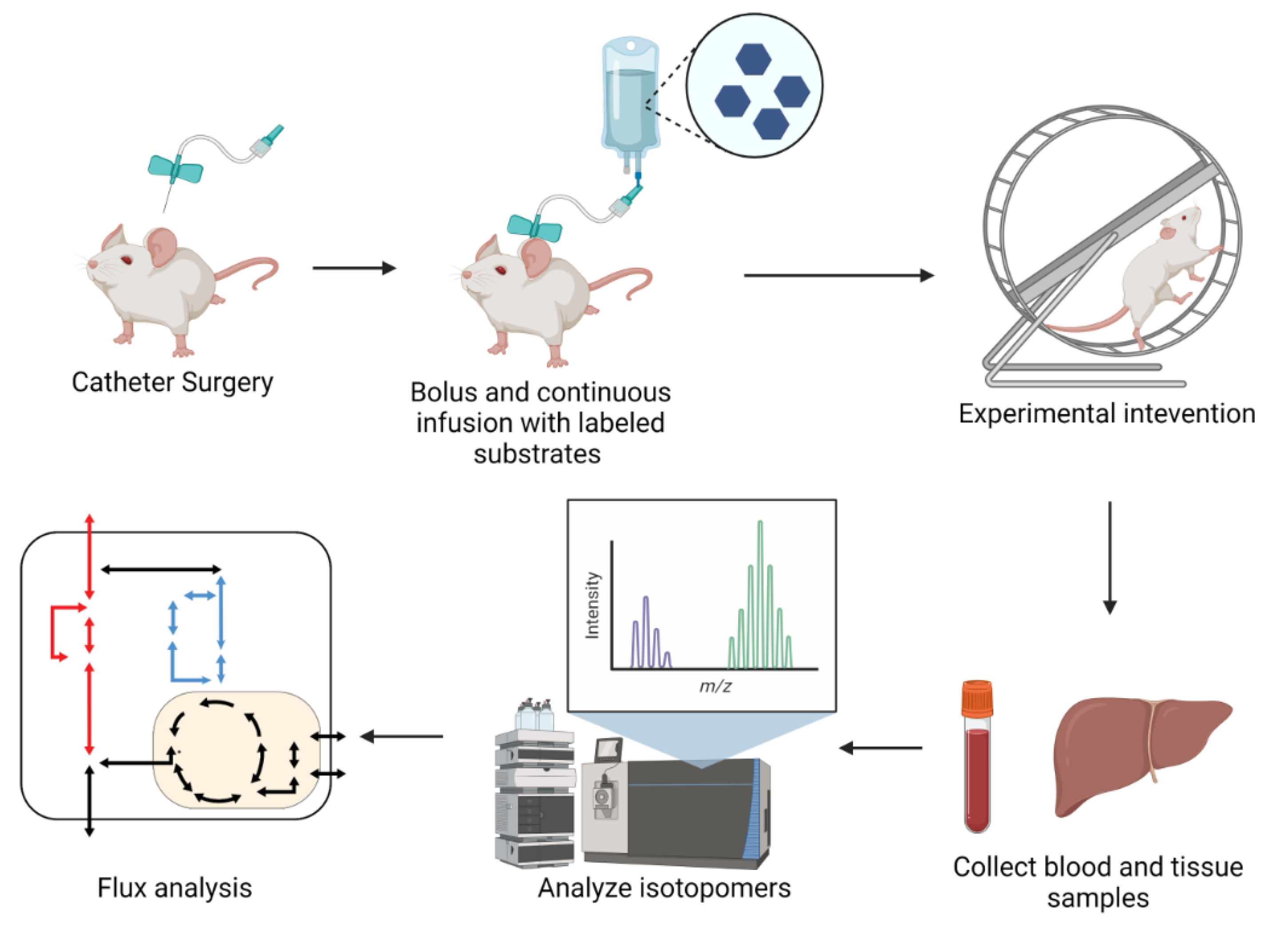
iMFA Workflow
3. iMFA Considerations for Mammalian Cells
3.1. Tracer Selection
3.2. Steady State Considerations
3.3. Quenching
3.4. Measuring MDVs: MS vs. NMR
3.5. Natural Isotope Abundance
3.6. Compartmentalization
3.7. Dilution Reactions
3.8. Tissue-Specific Model
3.9. In Vivo iMFA Considerations
4. iMFA Applications in Human Physiology and Disease
4.1. Stem Cell Differentiation and Proliferation
4.2. Cellular Activation
4.3. Cancer
4.4. Infection
4.5. Compensating for Genetic Loss of Function
4.6. Drug Effects
4.7. Extracellular Vesicles
4.8. In Vivo Studies
5. New Frontiers in iMFA
5.1. Dynamic MFA
5.2. Genome-Scale MFA
5.3. Co-Culture and Cross-Talk
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.N.; Schuck, P.F.; Wenk, G.L.; Ferreira, G.C. Metabolic Disturbances in Diseases with Neurological Involvement. Aging Dis. 2014, 5, 238–255. [Google Scholar] [CrossRef] [PubMed]
- Lamri, A.; Pigeyre, M.; Garver, W.S.; Meyre, D. The Extending Spectrum of NPC1-Related Human Disorders: From Niemann-Pick C1 Disease to Obesity. Endocr. Rev. 2018, 39, 192–220. [Google Scholar] [CrossRef] [PubMed]
- Supandi, F.; van Beek, J.H.G.M. Computational Prediction of Changes in Brain Metabolic Fluxes during Parkinson’s Disease from MRNA Expression. PLoS ONE 2018, 13, e0203687. [Google Scholar] [CrossRef] [PubMed]
- Agana, M.; Frueh, J.; Kamboj, M.; Patel, D.R.; Kanungo, S. Common Metabolic Disorder (Inborn Errors of Metabolism) Concerns in Primary Care Practice. Ann. Transl. Med. 2018, 6, 469. [Google Scholar] [CrossRef]
- Crown, S.B.; Kelleher, J.K.; Rouf, R.; Muoio, D.M.; Antoniewicz, M.R. Comprehensive Metabolic Modeling of Multiple 13C-Isotopomer Data Sets to Study Metabolism in Perfused Working Hearts. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H881–H891. [Google Scholar] [CrossRef]
- McGarrity, S.; Halldórsson, H.; Palsson, S.; Johansson, P.I.; Rolfsson, Ó. Understanding the Causes and Implications of Endothelial Metabolic Variation in Cardiovascular Disease through Genome-Scale Metabolic Modeling. Front. Cardiovasc. Med. 2016, 3, 10. [Google Scholar] [CrossRef]
- De Berardinis, R.J.; Thompson, C.B. Cellular Metabolism and Disease: What Do Metabolic Outliers Teach Us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef]
- De Queiroz, R.M.; Oliveira, I.A.; Piva, B.; Bouchuid Catão, F.; da Costa Rodrigues, B.; da Costa Pascoal, A.; Diaz, B.L.; Todeschini, A.R.; Caarls, M.B.; Dias, W.B. Hexosamine Biosynthetic Pathway and Glycosylation Regulate Cell Migration in Melanoma Cells. Front. Oncol. 2019, 9, 116. [Google Scholar] [CrossRef]
- Hart, G.W.; Akimoto, Y. The O-GlcNAc Modification; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009; ISBN 9780879697709. [Google Scholar]
- Basehore, S.; Bohlman, S.; Weber, C.; Swaminathan, S.; Zhang, Y.; Jang, C.; Arany, Z.; Clyne, A.M. Laminar Flow on Endothelial Cells Suppresses ENOS O-GlcNAcylation to Promote ENOS Activity. Circ. Res. 2021, 129, 1054–1066. [Google Scholar] [CrossRef]
- Clish, C.B. Metabolomics: An Emerging but Powerful Tool for Precision Medicine. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [PubMed]
- Cascante, M.; Marin, S. Metabolomics and Fluxomics Approaches. Essays Biochem. 2008, 45, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A Roadmap for Interpreting 13C Metabolite Labeling Patterns from Cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Krömer, J.O. Fluxomics—Connecting ‘omics Analysis and Phenotypes. Environ. Microbiol. 2013, 15, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Caceres, V.; Bell, L.N.; O’Rourke, B.; Paolocci, N.; Aon, M.A. From Metabolomics to Fluxomics: A Computational Procedure to Translate Metabolite Profiles into Metabolic Fluxes. Biophys. J. 2015, 108, 163–172. [Google Scholar] [CrossRef]
- Niklas, J.; Heinzle, E. Metabolic Flux Analysis in Systems Biology of Mammalian Cells. In Genomics and Systems Biology of Mammalian Cell Culture; Hu, W.S., Zeng, A.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 109–132. ISBN 978-3-642-28350-5. [Google Scholar]
- Sauer, U. Metabolic Networks in Motion: 13C-Based Flux Analysis. Mol. Syst. Biol. 2006, 2, 62. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. A Guide to 13C Metabolic Flux Analysis for the Cancer Biologist. Exp. Mol. Med. 2018, 50, 19. [Google Scholar] [CrossRef]
- Rahim, M.; Ragavan, M.; Deja, S.; Merritt, M.E.; Burgess, S.C.; Young, J.D. INCA 2.0: A Tool for Integrated, Dynamic Modeling of NMR- and MS-Based Isotopomer Measurements and Rigorous Metabolic Flux Analysis. Metab. Eng. 2022, 69, 275–285. [Google Scholar] [CrossRef]
- Dai, Z.; Locasale, J.W. Understanding Metabolism with Flux Analysis: From Theory to Application. Metab. Eng. 2017, 43, 94–102. [Google Scholar] [CrossRef]
- Nargund, S.; Sriram, G. Mathematical Modeling of Isotope Labeling Experiments for Metabolic Flux Analysis. Methods Mol. Biol. 2014, 1083, 109–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wondisford, F.E.; Song, C.; Zhang, T.; Su, X. Metabolic Flux Analysis—Linking Isotope Labeling and Metabolic Fluxes. Metabolites 2020, 10, 447. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. Methods and Advances in Metabolic Flux Analysis: A Mini-Review. J. Ind. Microbiol. Biotechnol. 2015, 42, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W.; Möllney, M.; Petersen, S.; De Graaf, A.A. A Universal Framework for 13C Metabolic Flux Analysis. Metab. Eng. 2001, 3, 265–283. [Google Scholar] [CrossRef]
- Lugar, D.J.; Sriram, G. Isotope-Assisted Metabolic Flux Analysis as an Equality-Constrained Nonlinear Program for Improved Scalability and Robustness. PLoS Comput. Biol. 2022, 18, e1009831. [Google Scholar] [CrossRef]
- Fischer, E.; Sauer, U. Metabolic Flux Profiling of Escherichia Coli Mutants in Central Carbon Metabolism Using GC-MS. Eur. J. Biochem. 2003, 270, 880–891. [Google Scholar] [CrossRef]
- Hörl, M.; Schnidder, J.; Sauer, U.; Zamboni, N. Non-Stationary 13C-Metabolic Flux Ratio Analysis. Biotechnol. Bioeng. 2013, 110, 3164–3176. [Google Scholar] [CrossRef]
- Yuan, J.; Bennett, B.D.; Rabinowitz, J.D. Kinetic Flux Profiling for Quantitation of Cellular Metabolic Fluxes. Nat. Protoc. 2008, 3, 1328–1340. [Google Scholar] [CrossRef]
- Niedenführ, S.; Wiechert, W.; Nöh, K. How to Measure Metabolic Fluxes: A Taxonomic Guide for 13C Fluxomics. Curr. Opin. Biotechnol. 2015, 34, 82–90. [Google Scholar]
- Hasenour, C.M.; Wall, M.L.; Ridley, D.E.; Hughey, C.C.; James, F.D.; Wasserman, D.H.; Young, J.D. Mass Spectrometry-Based Microassay of 2H and 13C Plasma Glucose Labeling to Quantify Liver Metabolic Fluxes In Vivo. Am. J. Physiol.-Endocrinol. Metab. 2015, 309, E191–E203. [Google Scholar] [CrossRef]
- Rantanen, A.; Rousu, J.; Jouhten, P.; Zamboni, N.; Maaheimo, H.; Ukkonen, E. An Analytic and Systematic Framework for Estimating Metabolic Flux Ratios from 13C Tracer Experiments. BMC Bioinform. 2008, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive Carboxylation Supports Redox Homeostasis during Anchorage-Independent Growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Moiz, B.; Garcia, J.; Basehore, S.; Sun, A.; Li, A.; Padmanabhan, S.; Albus, K.; Jang, C.; Sriram, G.; Clyne, A.M. 13C Metabolic Flux Analysis Indicates Endothelial Cells Attenuate Metabolic Perturbations by Modulating TCA Activity. Metabolites 2021, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Badur, M.G.; Spiering, S.; Divakaruni, A.; Meurs, N.E.; Yu, M.S.; Colas, A.R.; Murphy, A.N.; Mercola, M.; Metallo, C.M. Lipid Availability Influences the Metabolic Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, B.; Lin, L.; Zhang, H.; Luan, T. 13C Isotope-Based Metabolic Flux Analysis Revealing Cellular Landscape of Glucose Metabolism in Human Liver Cells Exposed to Perfluorooctanoic Acid. Sci. Total Environ. 2021, 770, 145329. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Pakrasi, H.B.; Maranas, C.D. Elucidation of Photoautotrophic Carbon Flux Topology in Synechocystis PCC 6803 Using Genome-Scale Carbon Mapping Models. Metab. Eng. 2018, 47, 190–199. [Google Scholar] [CrossRef]
- Bergman, T.; Carlquist, M.; Jörnvall, H. Amino Acid Analysis by High Performance Liquid Chromatography of Phenylthiocarbamyl Derivatives. In Advanced Methods in Protein Microsequence Analysis; Wittmann-Liebold, B., Salnikow, J., Erdmann, V.A., Eds.; Springer: Berlin/Heidelberg, Germany, 1986; pp. 45–55. ISBN 978-3-642-71534-1. [Google Scholar]
- Sherwood, R.A. Amino Acid Measurement by High-Performance Liquid Chromatography Using Electrochemical Detection. J. Neurosci. Methods 1990, 34, 17–22. [Google Scholar] [CrossRef]
- Ling, Z.; Xu, P.; Zhong, Z.; Wang, F.; Shu, N.; Zhang, J.; Tang, X.; Liu, L.; Liu, X. Sensitive Determination of Glucose in Dulbecco’s Modified Eagle Medium by High-Performance Liquid Chromatography with 1-Phenyl-3-Methyl-5-Pyrazolone Derivatization: Application to Gluconeogenesis Studies. Biomed. Chromatogr. 2016, 30, 601–605. [Google Scholar] [CrossRef]
- Robitaille, L.; Hoffer, L.J. Measurement of Branched Chain Amino Acids in Blood Plasma by High Performance Liquid Chromatography. Can. J. Physiol. Pharmacol. 1988, 66, 613–617. [Google Scholar] [CrossRef]
- Zhu, W.; Stevens, A.P.; Dettmer, K.; Gottfried, E.; Hoves, S.; Kreutz, M.; Holler, E.; Canelas, A.B.; Kema, I.; Oefner, P.J. Quantitative Profiling of Tryptophan Metabolites in Serum, Urine, and Cell Culture Supernatants by Liquid Chromatography–Tandem Mass Spectrometry. Anal. Bioanal. Chem. 2011, 401, 3249–3261. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Lam, C.-W.; Curreem, S.O.T.; Lee, K.-C.; Lau, C.C.Y.; Chow, W.-N.; Ngan, A.H.Y.; To, K.K.W.; Chan, J.F.W.; Hung, I.F.N.; et al. Identification of Specific Metabolites in Culture Supernatant of Mycobacterium Tuberculosis Using Metabolomics: Exploration of Potential Biomarkers. Emerg. Microbes Infect. 2015, 4, e6. [Google Scholar] [CrossRef]
- Quek, L.-E.; Nielsen, L.K. Customization of 13C-MFA Strategy According to Cell Culture System. Methods Mol. Biol. 2014, 1191, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive Glutamine Metabolism by IDH1 Mediates Lipogenesis under Hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.W.; Parker, S.J.; Metallo, C.M. Deuterium Tracing to Interrogate Compartment-Specific NAD(P)H Metabolism in Cultured Mammalian Cells. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2020; Volume 2088, pp. 51–71. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for Linking Genomes to Life and the Environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Daníelsdóttir, A.D.; Krecke, M.; Merten, D.; Haraldsdóttir, H.S.; et al. The Virtual Metabolic Human Database: Integrating Human and Gut Microbiome Metabolism with Nutrition and Disease. Nucleic Acids Res. 2018, 47, D614–D624. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc Database of Metabolic Pathways and Enzymes. Nucleic Acids Res. 2017, 46, D633–D639. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc Database of Metabolic Pathways and Enzymes—A 2019 Update. Nucleic Acids Res. 2020, 48, D453–D455. [Google Scholar] [CrossRef]
- Jin, H.; Mitchell, J.M.; Moseley, H.N.B. Atom Identifiers Generated by a Neighborhood-Specific Graph Coloring Method Enable Compound Harmonization across Metabolic Databases. Metabolites 2020, 10, 368. [Google Scholar] [CrossRef]
- Jaworski, W.; Szymkuć, S.; Mikulak-Klucznik, B.; Piecuch, K.; Klucznik, T.; Kaźmierowski, M.; Rydzewski, J.; Gambin, A.; Grzybowski, B.A. Automatic Mapping of Atoms across Both Simple and Complex Chemical Reactions. Nat. Commun. 2019, 10, 1434. [Google Scholar] [CrossRef]
- Young, J.D. INCA: A Computational Platform for Isotopically Non-Stationary Metabolic Flux Analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef]
- Yoo, H.; Antoniewicz, M.R.; Stephanopoulos, G.; Kelleher, J.K. Quantifying Reductive Carboxylation Flux of Glutamine to Lipid in a Brown Adipocyte Cell Line. J. Biol. Chem. 2008, 283, 20621–20627. [Google Scholar] [CrossRef] [PubMed]
- Kajihata, S.; Furusawa, C.; Matsuda, F.; Shimizu, H. OpenMebius: An Open Source Software for Isotopically Nonstationary 13C-Based Metabolic Flux Analysis. Biomed. Res. Int. 2014, 2014, 627014. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, M.; Nöh, K.; Dalman, T.; Niedenführ, S.; Stute, B.; Wiechert, W. 13CFLUX2—High-Performance Software Suite for (13)C-Metabolic Flux Analysis. Bioinformatics 2013, 29, 143–145. [Google Scholar] [CrossRef]
- Mairinger, T.; Wegscheider, W.; Peña, D.A.; Steiger, M.G.; Koellensperger, G.; Zanghellini, J.; Hann, S. Comprehensive Assessment of Measurement Uncertainty in 13C-Based Metabolic Flux Experiments. Anal. Bioanal. Chem. 2018, 410, 3337–3348. [Google Scholar] [CrossRef]
- Kumar, A.; Mitchener, J.; King, Z.A.; Metallo, C.M. Escher-Trace: A Web Application for Pathway-Based Visualization of Stable Isotope Tracing Data. BMC Bioinformatics 2020, 21, 297. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Determination of Confidence Intervals of Metabolic Fluxes Estimated from Stable Isotope Measurements. Metab. Eng. 2006, 8, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.; Mazat, J.-P. FluxVisualizer, a Software to Visualize Fluxes through Metabolic Networks. Processes 2018, 6, 39. [Google Scholar] [CrossRef]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine Transporter Regulation of Pentose Phosphate Pathway Dependency and Disulfide Stress Exposes a Targetable Metabolic Vulnerability in Cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef]
- Nargund, S.; Sriram, G. Designer Labels for Plant Metabolism: Statistical Design of Isotope Labeling Experiments for Improved Quantification of Flux in Complex Plant Metabolic Networks. Mol. Biosyst. 2013, 9, 99–112. [Google Scholar] [CrossRef]
- Carinhas, N.; Koshkin, A.; Pais, D.A.M.; Alves, P.M.; Teixeira, A.P. 13C-Metabolic Flux Analysis of Human Adenovirus Infection: Implications for Viral Vector Production. Biotechnol. Bioeng. 2017, 114, 195–207. [Google Scholar] [CrossRef]
- Metallo, C.M.; Walther, J.L.; Stephanopoulos, G. Evaluation of 13C Isotopic Tracers for Metabolic Flux Analysis in Mammalian Cells. J. Biotechnol. 2009, 144, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Walther, J.L.; Metallo, C.M.; Zhang, J.; Stephanopoulos, G. Optimization of 13C Isotopic Tracers for Metabolic Flux Analysis in Mammalian Cells. Metab. Eng. 2012, 14, 162–171. [Google Scholar] [CrossRef] [PubMed]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 Depletion Inhibits Glycolysis and Induces Oxidative Phosphorylation in Hepatocellular Carcinoma and Sensitizes to Metformin. Nat. Commun. 2018, 9, 446. [Google Scholar] [CrossRef] [PubMed]
- Crown, S.B.; Long, C.P.; Antoniewicz, M.R. Optimal Tracers for Parallel Labeling Experiments and 13C Metabolic Flux Analysis: A New Precision and Synergy Scoring System. Metab. Eng. 2016, 38, 10–18. [Google Scholar] [CrossRef]
- Zheng, Y.; Sriram, G. Steady-State and Instationary Modeling of Proteinogenic and Free Amino Acid Isotopomers for Flux Quantification. In Plant Metabolic Flux Analysis: Methods and Protocols; Dieuaide-Noubhani, M., Alonso, A.P., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 155–179. ISBN 978-1-62703-688-7. [Google Scholar]
- Nicolae, A.; Wahrheit, J.; Bahnemann, J.; Zeng, A.P.; Heinzle, E. Non-Stationary 13C Metabolic Flux Analysis of Chinese Hamster Ovary Cells in Batch Culture Using Extracellular Labeling Highlights Metabolic Reversibility and Compartmentation. BMC Syst. Biol. 2014, 8, 50. [Google Scholar] [CrossRef]
- Jazmin, L.J.; Young, J.D.; Smallbone, K.; Stanford, N.J. Isotopically Nonstationary 13C Metabolic Flux Analysis. Methods Mol. Biol. 2013, 985, 367–390. [Google Scholar] [CrossRef]
- Wiechert, W.; Niedenführ, S.; Nöh, K. A Primer to 13C Metabolic Flux Analysis. Fundam. Bioeng. 2015, 97–142. [Google Scholar]
- Cheah, Y.E.; Young, J.D. Isotopically Nonstationary Metabolic Flux Analysis (INST-MFA): Putting Theory into Practice. Curr. Opin. Biotechnol. 2018, 54, 80–87. [Google Scholar] [CrossRef]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite Measurement: Pitfalls to Avoid and Practices to Follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef]
- Emwas, A.H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Nagana Gowda, G.A.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. Nmr Spectroscopy for Metabolomics Research. Metabolites 2019, 9, 123. [Google Scholar]
- Dunn, W.B.; Ellis, D.I. Metabolomics: Current Analytical Platforms and Methodologies. TrAC—Trends Anal. Chem. 2005, 24, 285–294. [Google Scholar] [CrossRef]
- Gowda, G.A.N.; Djukovic, D. Overview of Mass Spectrometry-Based Metabolomics: Opportunities and Challenges. Methods Mol. Biol. 2014, 1198, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Deja, S.; Fu, X.; Fletcher, J.A.; Kucejova, B.; Browning, J.D.; Young, J.D.; Burgess, S.C. Simultaneous Tracers and a Unified Model of Positional and Mass Isotopomers for Quantification of Metabolic Flux in Liver. Metab. Eng. 2020, 59, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Aretz, I.; Meierhofer, D. Advantages and Pitfalls of Mass Spectrometry Based Metabolome Profiling in Systems Biology. Int. J. Mol. Sci. 2016, 17, 632. [Google Scholar] [CrossRef]
- Long, C.P.; Antoniewicz, M.R. High-Resolution 13C Metabolic Fl Ux Analysis. Nat. Protoc. 2019, 14, 2856–2877. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. Tandem Mass Spectrometry for Measuring Stable-Isotope Labeling. Curr. Opin. Biotechnol. 2013, 24, 48–53. [Google Scholar] [CrossRef]
- Shi, X.; Xi, B.; Jasbi, P.; Turner, C.; Jin, Y.; Gu, H. Comprehensive Isotopic Targeted Mass Spectrometry: Reliable Metabolic Flux Analysis with Broad Coverage. Anal. Chem. 2020, 92, 11728–11738. [Google Scholar] [CrossRef]
- Choi, J.; Antoniewicz, M.R. Tandem Mass Spectrometry for 13C Metabolic Flux Analysis: Methods and Algorithms Based on EMU Framework. Front. Microbiol. 2019, 10, 31. [Google Scholar] [CrossRef]
- Sriram, G.; Rahib, L.; He, J.S.; Campos, A.E.; Parr, L.S.; Liao, J.C.; Dipple, K.M. Global Metabolic Effects of Glycerol Kinase Overexpression in Rat Hepatoma Cells. Mol. Genet. Metab. 2008, 93, 145–159. [Google Scholar] [CrossRef]
- Van Winden, W.A.; Wittmann, C.; Heinzle, E.; Heijnen, J.J. Correcting Mass Isotopomer Distributions for Naturally Occurring Isotopes. Biotechnol. Bioeng. 2002, 80, 477–479. [Google Scholar] [CrossRef]
- Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K.; Elia, I.; Buescher, J.M.; Orth, M.F.; Davidson, S.M.; et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016, 17, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Lagziel, S.; Lee, W.D.; Shlomi, T. Studying Metabolic Flux Adaptations in Cancer through Integrated Experimental-Computational Approaches. BMC Biol. 2019, 17, 51. [Google Scholar] [CrossRef]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 23, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shah, S.; Fan, J.; Park, J.O.; Wellen, K.E.; Rabinowitz, J.D. Malic Enzyme Tracers Reveal Hypoxia-Induced Switch in Adipocyte NADPH Pathway Usage. Nat. Chem. Biol. 2016, 12, 345–352. [Google Scholar] [CrossRef]
- Klie, S.; Krueger, S.; Krall, L.; Giavalisco, P.; Flügge, U.I.; Willmitzer, L.; Steinhauser, D. Analysis of the Compartmentalized Metabolome—A Validation of the Non-Aqueous Fractionation Technique. Front. Plant Sci. 2011, 2, 55. [Google Scholar] [CrossRef]
- Pan, D.; Lindau, C.; Lagies, S.; Wiedemann, N.; Kammerer, B. Metabolic Profiling of Isolated Mitochondria and Cytoplasm Reveals Compartment-Specific Metabolic Responses. Metabolomics 2018, 14, 59. [Google Scholar] [CrossRef]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 2016, 166, 1324–1337.e11. [Google Scholar] [CrossRef]
- Gravel, S.-P.; Andrzejewski, S.; Avizonis, D.; St-Pierre, J. Stable Isotope Tracer Analysis in Isolated Mitochondria from Mammalian Systems. Metabolites 2014, 4, 166–183. [Google Scholar] [CrossRef]
- Nonnenmacher, Y.; Palorini, R.; Hiller, K. Determining Compartment-Specific Metabolic Fluxes. Methods Mol. Biol. 2019, 1862, 137–149. [Google Scholar] [CrossRef]
- Lee, W.D.; Mukha, D.; Aizenshtein, E.; Shlomi, T. Spatial-Fluxomics Provides a Subcellular-Compartmentalized View of Reductive Glutamine Metabolism in Cancer Cells. Nat. Commun. 2019, 10, 1351. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, M.H.; He, L.; Tang, Y.J. Channeling in Native Microbial Pathways: Implications and Challenges for Metabolic Engineering. Biotechnol. Adv. 2017, 35, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.C.R.; Sweetlove, L.J.; George Ratcliffe, R. Capturing Metabolite Channeling in Metabolic Flux Phenotypes. Plant Physiol. 2011, 157, 981. [Google Scholar] [CrossRef] [PubMed]
- Orman, M.A.; Mattick, J.; Androulakis, I.P.; Berthiaume, F.; Ierapetritou, M.G. Stoichiometry Based Steady-State Hepatic Flux Analysis: Computational and Experimental Aspects. Metabolites 2012, 2, 268–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Cheng, J.; Xu, F.; Chen, Y.E.; Du, J.B.; Yuan, M.; Zhu, F.; Xu, X.C.; Yuan, S. Red Blood Cell Extrudes Nucleus and Mitochondria against Oxidative Stress. IUBMB Life 2011, 63, 560–565. [Google Scholar] [CrossRef]
- Wiback, S.J.; Palsson, B.O. Extreme Pathway Analysis of Human Red Blood Cell Metabolism. Biophys. J. 2002, 83, 808–818. [Google Scholar] [CrossRef]
- Beylot, M.; Soloviev, M.V.; David, F.; Landau, B.R.; Brunengraber, H. Tracing Hepatic Gluconeogenesis Relative to Citric Acid Cycle Activity In Vitro and In Vivo: Comparisons in the Use of [3-13C]Lactate, [2-13C]Acetate, and α-Keto[3-13C]Isocaproate. J. Biol. Chem. 1995, 270, 1509–1514. [Google Scholar] [CrossRef]
- Jin, E.S.; Jones, J.G.; Burgess, S.C.; Merritt, M.E.; Dean Sherry, A.; Malloy, C.R. Comparison of [3,4-13C2]Glucose to [6,6-2H2]Glucose as a Tracer for Glucose Turnover by Nuclear Magnetic Resonance. Magn. Reason. Med. 2005, 53, 1479–1483. [Google Scholar] [CrossRef][Green Version]
- Chung, S.T.; Chacko, S.K.; Sunehag, A.L.; Haymond, M.W. Measurements of Gluconeogenesis and Glycogenolysis: A Methodological Review. Diabetes 2015, 64, 3996–4010. [Google Scholar] [CrossRef]
- Junghans, P.; Görs, S.; Lang, I.S.; Steinhoff, J.; Hammon, H.M.; Metges, C.C. A Simplified Mass Isotopomer Approach to Estimate Gluconeogenesis Rate In Vivo Using Deuterium Oxide. Rapid Commun. Mass Spectrom. 2010, 24, 1287–1295. [Google Scholar] [CrossRef]
- Sherry, D.; Malloy, C. Metabolic Networks in the Liver by 2H and 13C NMR. In Metabolomics by In Vivo NMR; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005; pp. 159–174. ISBN 9780470011508. [Google Scholar]
- Hasenour, C.M.; Rahim, M.; Young, J.D. In Vivo Estimates of Liver Metabolic Flux Assessed by 13C-Propionate and 13C-Lactate Are Impacted by Tracer Recycling and Equilibrium Assumptions. Cell Rep. 2020, 32, 107986. [Google Scholar] [CrossRef] [PubMed]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957. [Google Scholar] [CrossRef]
- Teslaa, T.; Teitell, M.A. Pluripotent Stem Cell Energy Metabolism: An Update. EMBO J. 2015, 34, 138. [Google Scholar] [CrossRef]
- Sá, J.V.; Kleiderman, S.; Brito, C.; Sonnewald, U.; Leist, M.; Teixeira, A.P.; Alves, P.M. Quantification of Metabolic Rearrangements During Neural Stem Cells Differentiation into Astrocytes by Metabolic Flux Analysis. Neurochem. Res. 2017, 42, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Carido, M.; Sebastião, M.J.; Gomes-Alves, P.; Elliott, D.A.; Domian, I.J.; Teixeira, A.P.; et al. 3D Aggregate Culture Improves Metabolic Maturation of Human Pluripotent Stem Cell Derived Cardiomyocytes. Biotechnol. Bioeng. 2017, 115, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Odenwelder, D.C.; Lu, X.; Harcum, S.W. Induced Pluripotent Stem Cells Can Utilize Lactate as a Metabolic Substrate to Support Proliferation. Biotechnol. Prog. 2021, 37, e3090. [Google Scholar] [CrossRef]
- Fensterheim, B.A.; Young, J.D.; Luan, L.; Kleinbard, R.R.; Stothers, C.L.; Patil, N.K.; Mcatee-pereira, A.G.; Guo, Y.; Trenary, I.; Hernandez, A.; et al. The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism. J. Immunol. 2018, 200, 3777–3789. [Google Scholar] [CrossRef]
- Held, N.M.; Kuipers, E.N.; van Weeghel, M.; van Klinken, J.B.; Denis, S.W.; Lombès, M.; Wanders, R.J.; Vaz, F.M.; Rensen, P.C.N.; Verhoeven, A.J.; et al. Pyruvate Dehydrogenase Complex Plays a Central Role in Brown Adipocyte Energy Expenditure and Fuel Utilization during Short-Term Beta-Adrenergic Activation. Sci. Rep. 2018, 8, 9562. [Google Scholar] [CrossRef]
- Wang, Z.; Ning, T.; Song, A.; Rutter, J.; Wang, Q.A.; Jiang, L. Chronic Cold Exposure Enhances Glucose Oxidation in Brown Adipose Tissue. EMBO Rep. 2020, 21, e50085. [Google Scholar] [CrossRef]
- Thomas, A.; Rahmanian, S.; Bordbar, A.; Palsson, B.Ø.; Jamshidi, N. Network Reconstruction of Platelet Metabolism Identifies Metabolic Signature for Aspirin Resistance. Sci. Rep. 2014, 4, 3925. [Google Scholar] [CrossRef]
- Sake, C.L.; Metcalf, A.J.; Di Paola, J.; Neeves, K.B.; Boyle, N.R. Isotopically Nonstationary 13C Metabolic Flux Analysis in Resting and Activated Human Platelets. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.M.J.; Ji, X.; Zimmerman, L.J.; Li, M.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Zou, Y.; Qian, J.; Slebos, R.J.C.; et al. The Airway Epithelium Undergoes Metabolic Reprogramming in Individuals at High Risk for Lung Cancer. JCI Insight 2016, 1, e88814. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, A.N.; Odenwelder, D.C.; Harcum, S.W. High Extracellular Lactate Causes Reductive Carboxylation in Breast Tissue Cell Lines Grown under Normoxic Conditions. PLoS ONE 2019, 14, e0213419. [Google Scholar] [CrossRef]
- Kumar, A.; Cordes, T.; Thalacker-Mercer, A.E.; Pajor, A.M.; Murphy, A.N.; Metallo, C.M. NaCT/SLC13A5 Facilitates Citrate Import and Metabolism under Nutrient-Limited Conditions. Cell Rep. 2021, 36, 109701. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.M.; Lopatkin, A.J.; Lobritz, M.A.; Collins, J.J. Bacterial Metabolism and Antibiotic Efficacy. Cell Metab. 2019, 30, 251. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.S.; Lamprecht, D.A.; Asmal, R.; Adamson, J.H.; Borah, K.; Beste, D.J.V.; Lee, B.S.; Pethe, K.; Rousseau, S.; Krieger, I.; et al. Bedaquiline Reprograms Central Metabolism to Reveal Glycolytic Vulnerability in Mycobacterium Tuberculosis. Nat. Commun. 2020, 11, 6092. [Google Scholar] [CrossRef]
- Sanchez, E.L.; Lagunoff, M. Viral Activation of Cellular Metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef]
- Lesner, N.P.; Gokhale, A.S.; Kota, K.; DeBerardinis, R.J.; Mishra, P. α-Ketobutyrate Links Alterations in Cystine Metabolism to Glucose Oxidation in MtDNA Mutant Cells. Metab. Eng. 2020, 60, 157–167. [Google Scholar] [CrossRef]
- Jiang, L.; Boufersaoui, A.; Yang, C.; Ko, B.; Rakheja, D.; Guevara, G.; Hu, Z.; DeBerardinis, R.J. Quantitative Metabolic Flux Analysis Reveals an Unconventional Pathway of Fatty Acid Synthesis in Cancer Cells Deficient for the Mitochondrial Citrate Transport Protein. Metab. Eng. 2017, 43, 198–207. [Google Scholar] [CrossRef]
- Noguchi, S.; Ishikawa, H.; Wakita, K.; Matsuda, F.; Shimizu, H. Direct and Quantitative Analysis of Altered Metabolic Flux Distributions and Cellular ATP Production Pathway in Fumarate Hydratase-Diminished Cells. Sci. Rep. 2020, 10, 13065. [Google Scholar] [CrossRef]
- Parker, S.J.; Svensson, R.U.; Divakaruni, A.S.; Lefebvre, A.E.; Murphy, A.N.; Shaw, R.J.; Metallo, C.M. LKB1 Promotes Metabolic Flexibility in Response to Energy Stress. Metab. Eng. 2017, 43, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 Inactivation Dictates Therapeutic Response of Non-Small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.J. ATP Synthase: From Sequence to Ring Size to the P/O Ratio. Proc. Natl. Acad. Sci. USA 2010, 107, 16755–16756. [Google Scholar] [CrossRef]
- Araki, C.; Okahashi, N.; Maeda, K.; Shimizu, H.; Matsuda, F. Mass Spectrometry-Based Method to Study Inhibitor-Induced Metabolic Redirection in the Central Metabolism of Cancer Cells. Mass Spectrom. 2018, 7, A0067. [Google Scholar] [CrossRef] [PubMed]
- Palviainen, M.; Saari, H.; Kärkkäinen, O.; Pekkinen, J.; Auriola, S.; Yliperttula, M.; Puhka, M.; Hanhineva, K.; Siljander, P.R.M. Metabolic Signature of Extracellular Vesicles Depends on the Cell Culture Conditions. J. Extracell. Vesicles 2019, 8, 1596669. [Google Scholar] [CrossRef] [PubMed]
- Born, L.J.; Harmon, J.W.; Jay, S.M. Therapeutic Potential of Extracellular Vesicle-Associated Long Noncoding RNA. Bioeng. Transl. Med. 2020, 5, e10172. [Google Scholar] [CrossRef]
- Achreja, A.; Meurs, N.; Nagrath, D. Quantifying Metabolic Transfer Mediated by Extracellular Vesicles Using Exo-MFA: An Integrated Empirical and Computational Platform. In Metabolic Flux Analysis in Eukaryotic Cells: Methods and Protocols; Springer: New York, NY, USA, 2020; pp. 205–221. ISBN 978-1-0716-0158-7 978-1-0716-0159-4. [Google Scholar]
- Achreja, A.; Zhao, H.; Yang, L.; Yun, T.H.; Marini, J.; Nagrath, D. Exo-MFA—A 13C Metabolic Flux Analysis Framework to Dissect Tumor Microenvironment-Secreted Exosome Contributions towards Cancer Cell Metabolism. Metab. Eng. 2017, 43, 156–172. [Google Scholar] [CrossRef]
- Hughey, C.C.; James, F.D.; Bracy, D.P.; Donahue, E.P.; Young, J.D.; Viollet, B.; Foretz, M.; Wasserman, D.H. Loss of Hepatic AMP-Activated Protein Kinase Impedes the Rate of Glycogenolysis but Not Gluconeogenic Fluxes in Exercising Mice. J. Biol. Chem. 2017, 292, 20125–20140. [Google Scholar] [CrossRef]
- Cappel, D.A.; Kucejova, B.; Fletcher, J.A.; Fu, X.; Berglund, E.D.; Liu, T.; Joel, K.; Hammer, S.; Mishra, P.; Browning, J.D.; et al. Pyruvate-Carboxylase-Mediated Anaplerosis Promotes Antioxidant Capacity by Sustaining TCA Cycle and Redox Metabolism in Liver. Cell Metab. 2019, 29, 1291–1305.e8. [Google Scholar] [CrossRef]
- Hasenour, C.M.; Kennedy, A.J.; Bednarski, T.; Trenary, I.A.; Eudy, B.J.; DaSilva, R.P.; Boyd, K.L.; Young, J.D. Vitamin E Does Not Prevent Western Diet-Induced NASH Progression and Increases Metabolic Flux Dysregulation in Mice. J. Lipid Res. 2020, 61, 707–721. [Google Scholar] [CrossRef]
- Campit, S.; Chandrasekaran, S. Inferring Metabolic Flux from Time-Course Metabolomics. Methods Mol. Biol. 2020, 2088, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, C.; Illanes, A.; Becerra, S.; Cairó, J.J.; Gòdia, F. Considerations on the Lactate Consumption by CHO Cells in the Presence of Galactose. J. Biotechnol. 2006, 125, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Niklas, J.; Schräder, E.; Sandig, V.; Noll, T.; Heinzle, E. Quantitative Characterization of Metabolism and Metabolic Shifts during Growth of the New Human Cell Line AGE1.HN Using Time Resolved Metabolic Flux Analysis. Bioprocess Biosyst. Eng. 2011, 34, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Martínez, V.S.; Buchsteiner, M.; Gray, P.; Nielsen, L.K.; Quek, L.E. Dynamic Metabolic Flux Analysis Using B-Splines to Study the Effects of Temperature Shift on CHO Cell Metabolism. Metab. Eng. Commun. 2015, 2, 46–57. [Google Scholar] [CrossRef]
- Quek, L.E.; Krycer, J.R.; Ohno, S.; Yugi, K.; Fazakerley, D.J.; Scalzo, R.; Elkington, S.D.; Dai, Z.; Hirayama, A.; Ikeda, S.; et al. Dynamic 13C Flux Analysis Captures the Reorganization of Adipocyte Glucose Metabolism in Response to Insulin. iScience 2020, 23, 100855. [Google Scholar] [CrossRef]
- Basler, G.; Fernie, A.R.; Nikoloski, Z. Advances in Metabolic Flux Analysis toward Genome-Scale Profiling of Higher Organisms. Biosci. Rep. 2018, 38, BSR20170224. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Maranas, C.D. 13C Metabolic Flux Analysis at a Genome-Scale. Metab. Eng. 2015, 32, 12–22. [Google Scholar] [CrossRef]
- Hendry, J.I.; Gopalakrishnan, S.; Ungerer, J.; Pakrasi, H.B.; Tang, Y.J.; Maranas, C.D. Genome-Scale Fluxome of Synechococcus Elongatus UTEX 2973 Using Transient 13C-Labeling Data. Plant Physiol. 2019, 179, 761–769. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Z.; Meng, Y.; Chen, Z.; Han, J.; Wei, Y.; Shen, T.; Yi, Y.; Xie, X. Parallel Isotope Differential Modeling for Instationary 13C Fluxomics at the Genome Scale. Biotechnol. Biofuels 2020, 13, 103. [Google Scholar] [CrossRef]
- Backman, T.W.H.; Ando, D.; Singh, J.; Keasling, J.D.; Martín, H.G. Constraining Genome-Scale Models to Represent the Bow Tie Structure of Metabolism for 13C Metabolic Flux Analysis. Metabolites 2018, 8, 3. [Google Scholar] [CrossRef]
- Ando, D.; Garcia, M.H. Two-Scale 13C Metabolic Flux Analysis for Metabolic Engineering. Methods Mol. Biol. 2018, 1671, 170–179. [Google Scholar] [CrossRef][Green Version]
- García Martín, H.; Kumar, V.S.; Weaver, D.; Ghosh, A.; Chubukov, V.; Mukhopadhyay, A.; Arkin, A.; Keasling, J.D. A Method to Constrain Genome-Scale Models with 13C Labeling Data. PLoS Comput. Biol. 2015, 11, e1004363. [Google Scholar] [CrossRef] [PubMed]
- Foguet, C.; Jayaraman, A.; Marin, S.; Selivanov, V.A.; Moreno, P.; Messeguer, R.; de Atauri, P.; Cascante, M. P13CMFA: Parsimonious 13C Metabolic Flux Analysis. PLoS Comput. Biol. 2019, 15, e1007310. [Google Scholar] [CrossRef]
- Canfield, S.G.; Stebbins, M.J.; Morales, B.S.; Asai, S.W.; Vatine, G.D.; Svendsen, C.N.; Palecek, S.P.; Shusta, E.V. An Isogenic Blood–Brain Barrier Model Comprising Brain Endothelial Cells, Astrocytes, and Neurons Derived from Human Induced Pluripotent Stem Cells. J. Neurochem. 2017, 140, 874–888. [Google Scholar] [CrossRef]
- Gebreselassie, N.A.; Antoniewicz, M.R. 13C-Metabolic Flux Analysis of Co-Cultures: A Novel Approach. Metab. Eng. 2015, 31, 132–139. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moiz, B.; Li, A.; Padmanabhan, S.; Sriram, G.; Clyne, A.M. Isotope-Assisted Metabolic Flux Analysis: A Powerful Technique to Gain New Insights into the Human Metabolome in Health and Disease. Metabolites 2022, 12, 1066. https://doi.org/10.3390/metabo12111066
Moiz B, Li A, Padmanabhan S, Sriram G, Clyne AM. Isotope-Assisted Metabolic Flux Analysis: A Powerful Technique to Gain New Insights into the Human Metabolome in Health and Disease. Metabolites. 2022; 12(11):1066. https://doi.org/10.3390/metabo12111066
Chicago/Turabian StyleMoiz, Bilal, Andrew Li, Surya Padmanabhan, Ganesh Sriram, and Alisa Morss Clyne. 2022. "Isotope-Assisted Metabolic Flux Analysis: A Powerful Technique to Gain New Insights into the Human Metabolome in Health and Disease" Metabolites 12, no. 11: 1066. https://doi.org/10.3390/metabo12111066
APA StyleMoiz, B., Li, A., Padmanabhan, S., Sriram, G., & Clyne, A. M. (2022). Isotope-Assisted Metabolic Flux Analysis: A Powerful Technique to Gain New Insights into the Human Metabolome in Health and Disease. Metabolites, 12(11), 1066. https://doi.org/10.3390/metabo12111066