
Muscle Amino Acid and Adenine Nucleotide Metabolism during Exercise and in Liver Cirrhosis: Speculations on How to Reduce the Harmful Effects of Ammonia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
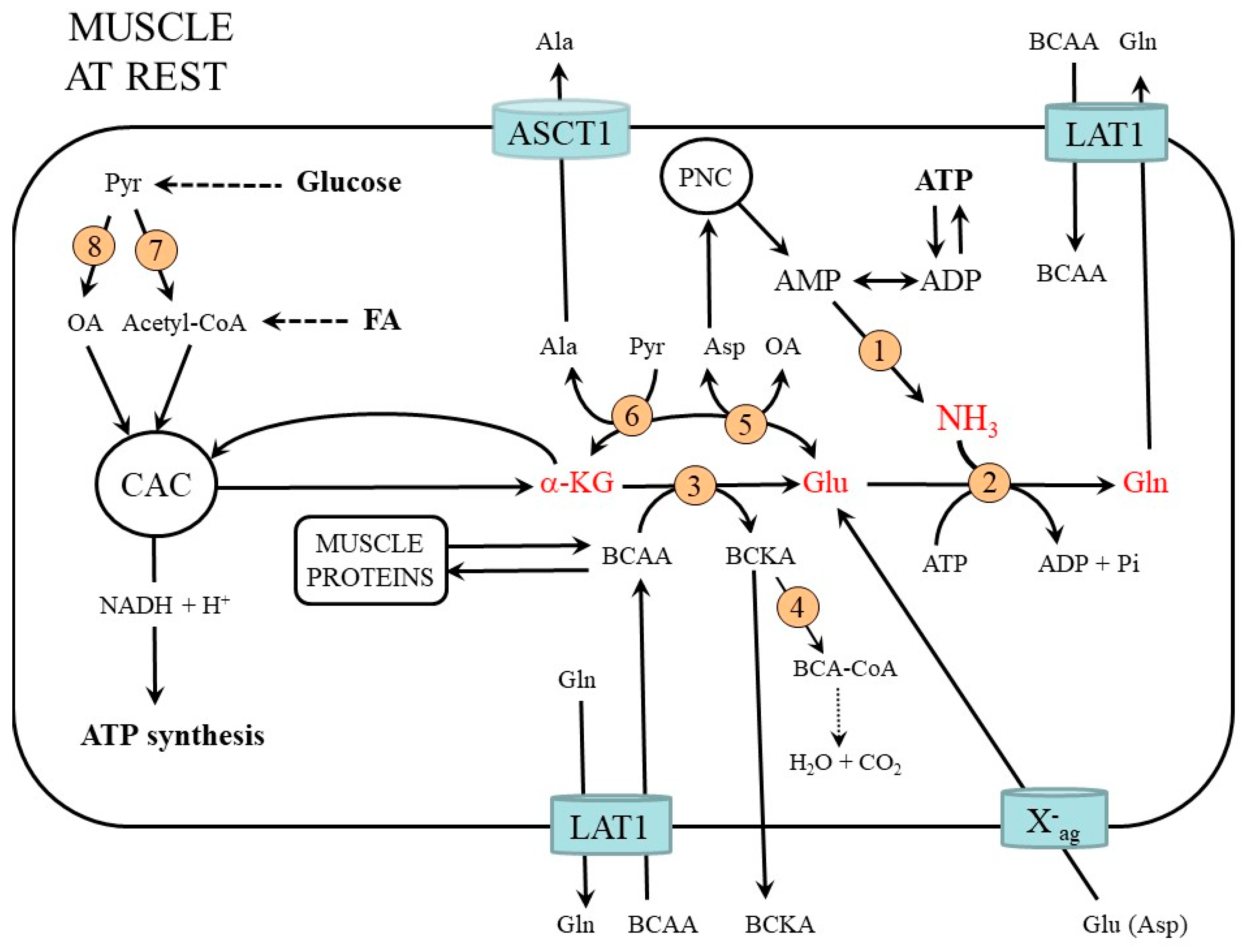
2. Muscle Ammonia and Amino Acid Metabolism at Rest
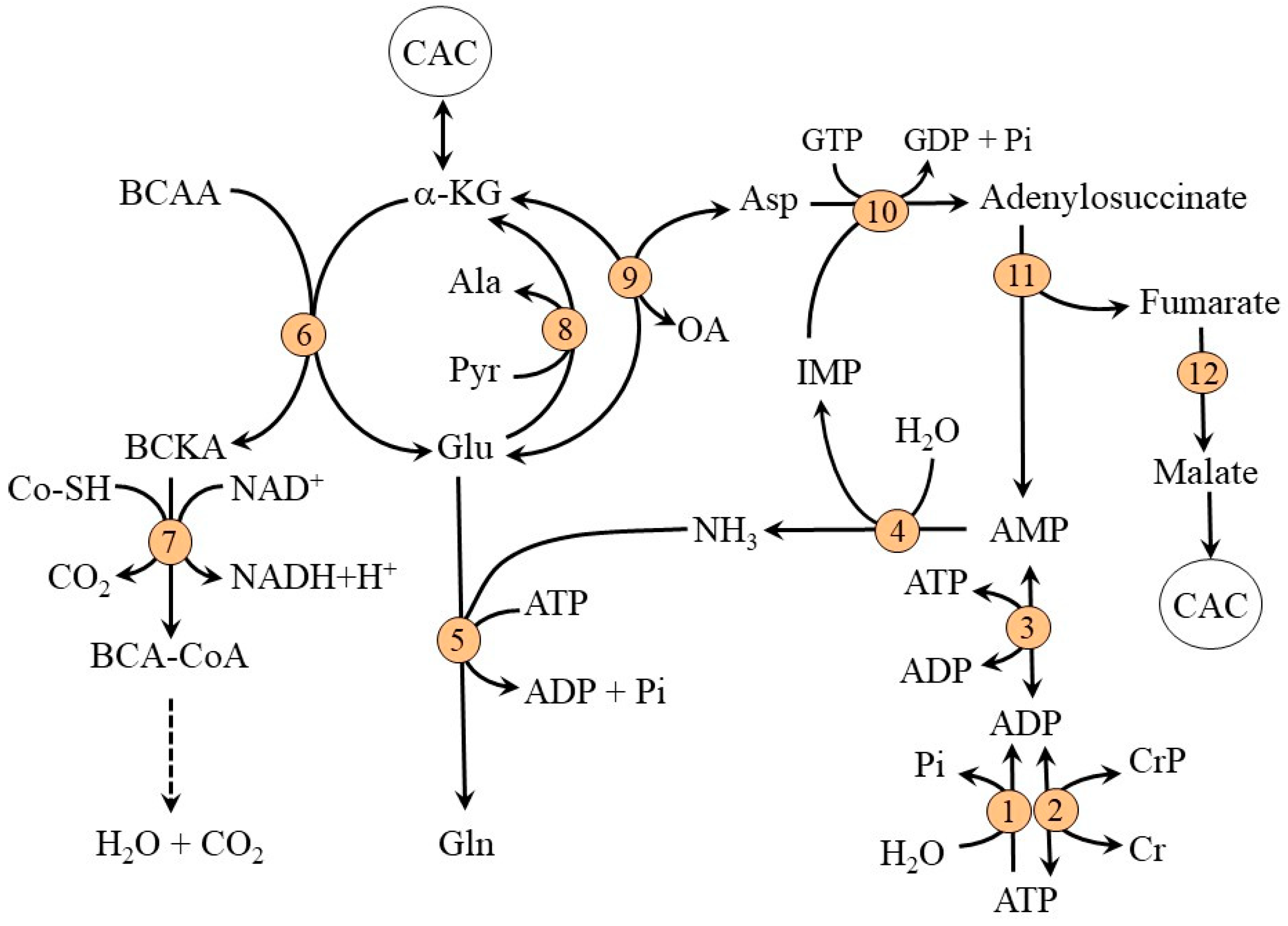
2.1. Ammonia Synthesis in Muscles
2.2. Ammonia Detoxification to Glutamine in Muscles
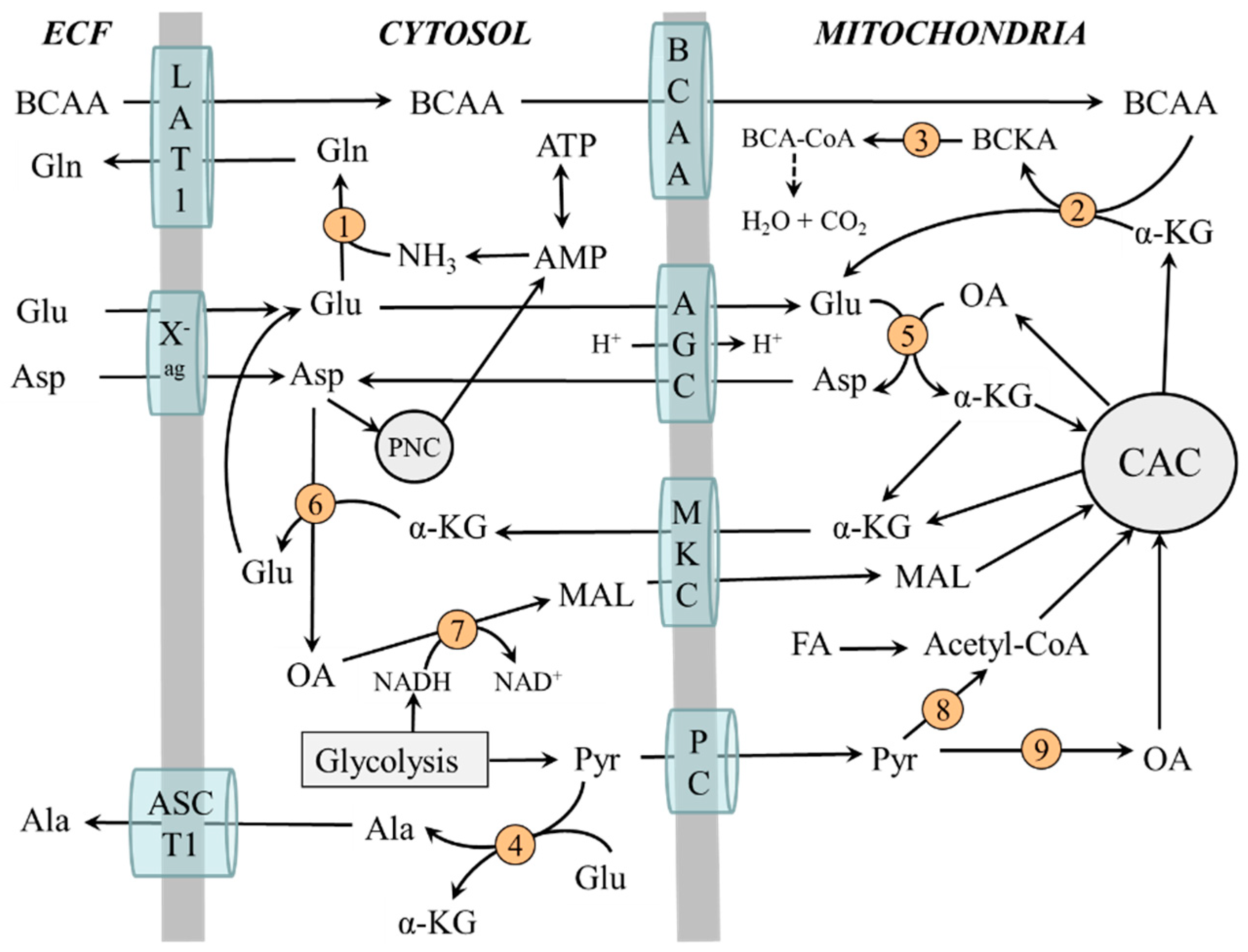
2.3. Compartmentation of Ammonia and Amino Acid Metabolism in Muscles
2.4. The Role of Glycolysis and Citric Acid Cycle (CAC)
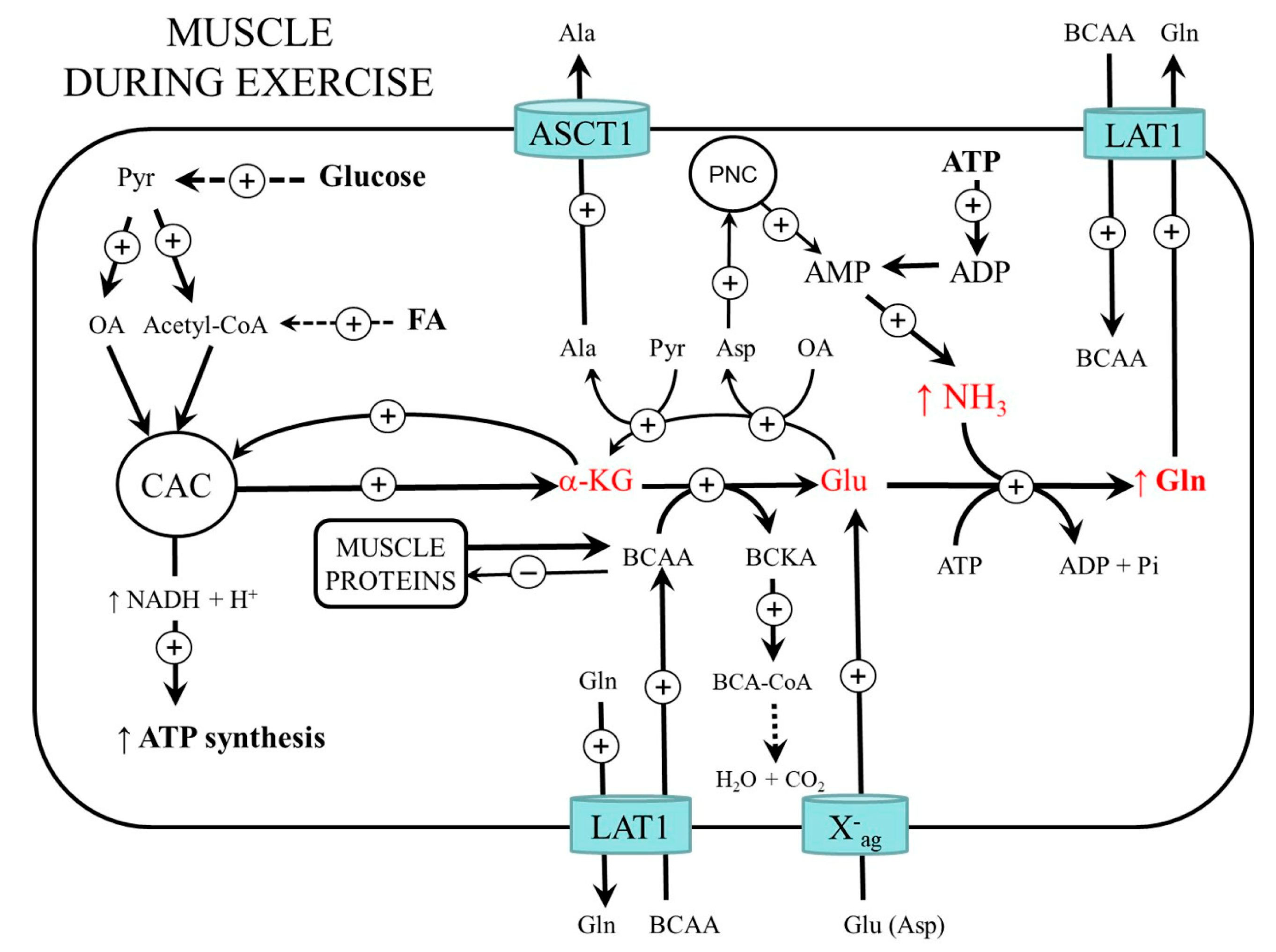
3. Muscle Ammonia and Amino Acid Metabolism during Exercise
3.1. BCAA Metabolism
3.2. Glycolysis and CAC Activity
3.3. Protein Metabolism during and after Muscle Work

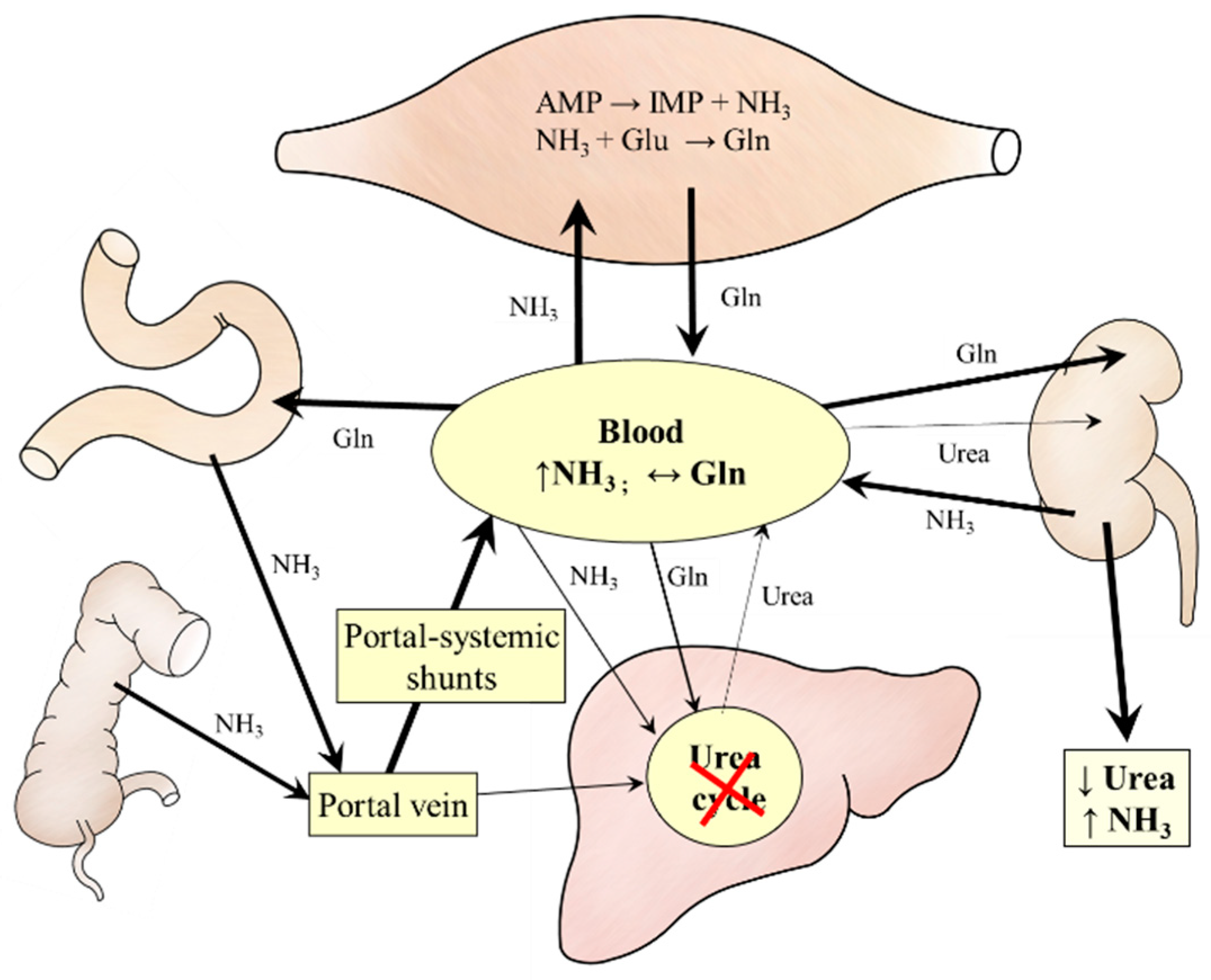
4. Ammonia Metabolism in Liver Cirrhosis
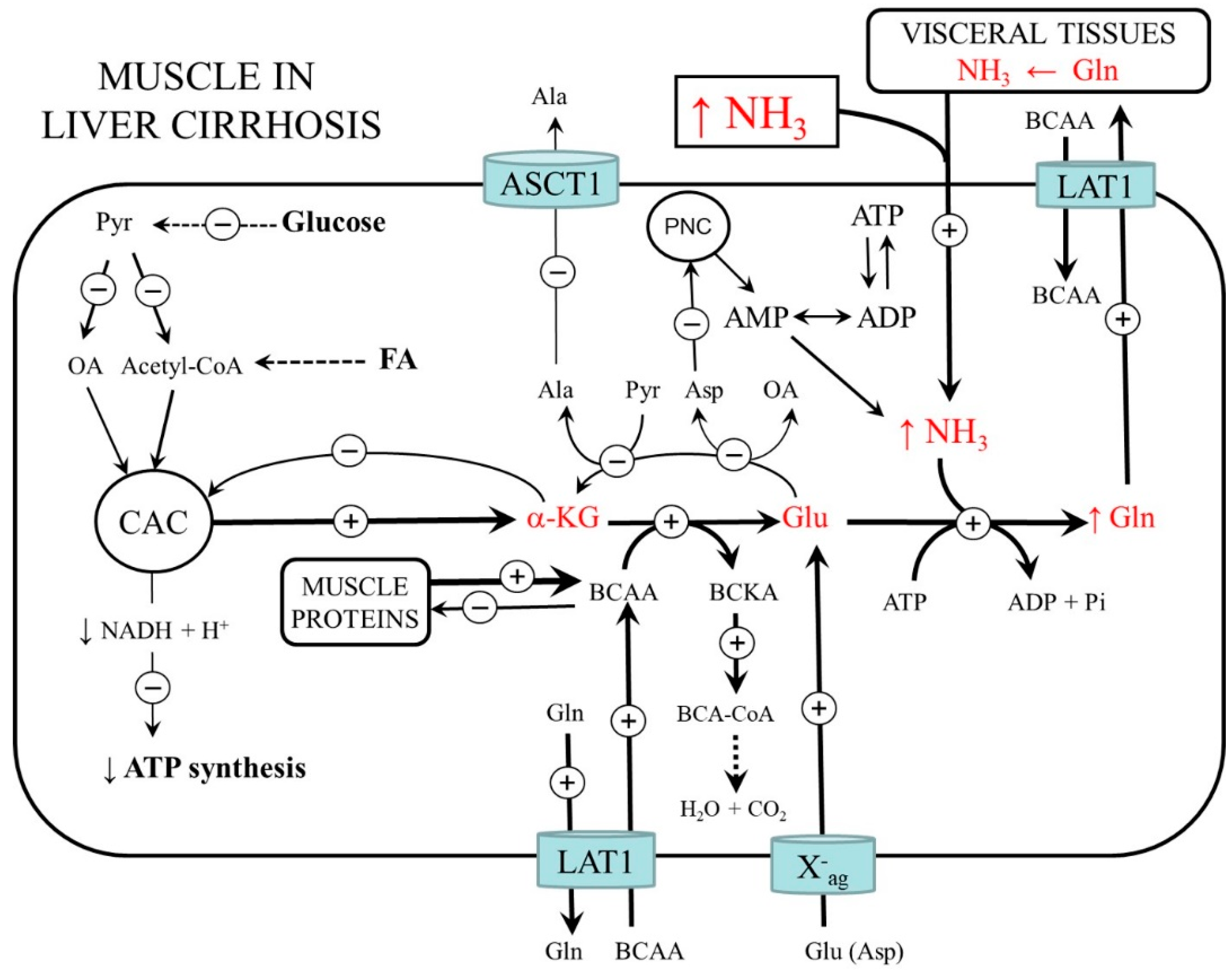
4.1. BCAA Metabolism
4.2. Protein Metabolism
5. Possibilities to Reduce the Harmful Effects of Ammonia on Muscles
5.1. Glutamate
5.2. α-KG
5.3. BCAA
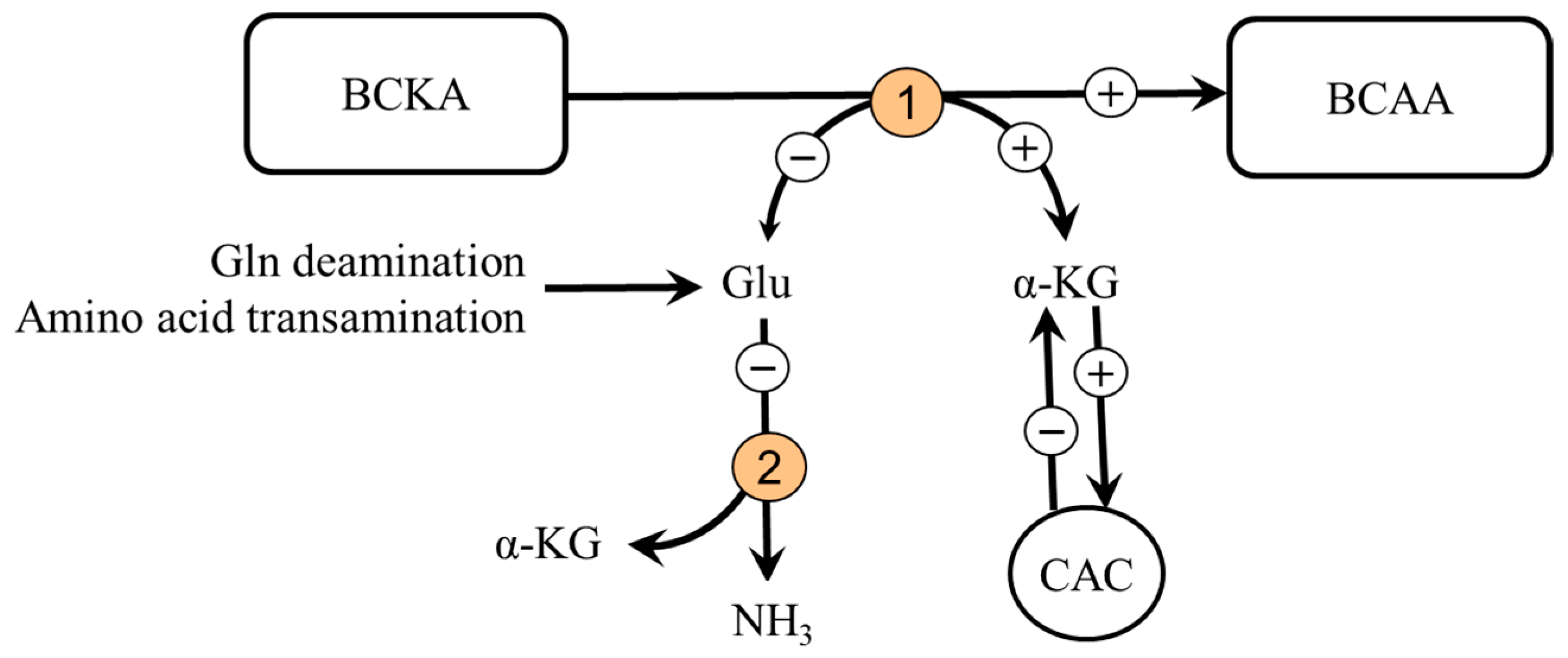
5.4. Branched-Chain Keto Acids (BCKA)
5.5. Aspartic Acid
5.6. Glutamine
5.7. Ammonia Removal
6. Summary
6.1. Similarities and Differences in Ammonia Metabolism in Muscles during Exercise and in Liver Cirrhosis
6.2. Considerations on How to Reduce the Harmful Effects of Ammonia on Muscles
- Glutamate and α-KG—administration could promote ammonia detoxification to glutamine, reduce α-KG drain from CAC, and increase the supply of reduced nucleotides for respiratory chains in mitochondria. However, studies examining the effects of glutamate on muscles in humans with hyperammonemia are rare [68]. Glutamate administration may be detrimental in liver cirrhosis due to the increased synthesis of glutamine that is catabolized to ammonia in visceral tissues.
- BCKA—administration could correct BCAA deficiency in the blood, improve muscle protein balance, decrease the drain of α-KG from the CAC (cataplerosis), and decrease ammonia production in glutamate dehydrogenase reaction. The BCKA administration is not associated with an increase in ammonia levels observed after BCAA administration [95,96,97]. Unfortunately, studies examining specifically effects of BCKA supplementation are not existing.
7. Conclusions
- The similarities in the influence of increased levels of ammonia due to strenuous exercise and liver cirrhosis on BCAA, glutamate, α-KG, aspartate, and adenine nucleotide metabolism in muscles indicate that ammonia can significantly contribute to muscle wasting regardless of the cause of its increased levels.
- Similar strategies can be designed to reduce the adverse effects of ammonia on the muscle, increase muscle performance in athletes, and reduce muscle loss in patients with hyperammonemia.
- To avoid harmful effects of ammonia on muscles, ammonia and plasma amino acid concentrations should be monitored in individuals with diseases in which ammonia levels are often elevated.
- Systematic investigation is needed to understand better the relationships between ammonia metabolism and the metabolism of other amino acids in the pathogenesis of muscle wasting due to increased ammonia levels.
Funding
Conflicts of Interest
References
- Walker, V. Ammonia metabolism and hyperammonemic disorders. Adv. Clin. Chem. 2014, 67, 73–150. [Google Scholar]
- Walker, V. Severe hyperammonaemia in adults not explained by liver disease. Ann. Clin. Biochem. 2012, 49, 214–228. [Google Scholar] [CrossRef]
- Yao, Z.P.; Li, Y.; Liu, Y.; Wang, H.L. Relationship between the incidence of non-hepatic hyperammonemia and the prognosis of patients in the intensive care unit. World J. Gastroenterol. 2020, 26, 7222–7231. [Google Scholar] [CrossRef]
- Balcerac, A.; Bihan, K.; Lebrun-Vignes, B.; Thabut, D.; Salem, J.E.; Weiss, N. Drug-associated hyperammonaemia: A Bayesian analysis of the WHO Pharmacovigilance Database. Ann. Intensive Care 2022, 12, 55. [Google Scholar] [CrossRef]
- McDaniel, J.; Davuluri, G.; Hill, E.A.; Moyer, M.; Runkana, A.; Prayson, R.; van Lunteren, E.; Dasarathy, S. Hyperammonemia results in reduced muscle function independent of muscle mass. Am. J. Physiol. 2016, 310, G163–G170. [Google Scholar] [CrossRef]
- Leweling, H.; Breitkreutz, R.; Behne, F.; Staedt, U.; Striebel, J.P.; Holm, E. Hyperammonemia-induced depletion of glutamate and branched-chain amino acids in muscle and plasma. J. Hepatol. 1996, 25, 756–762. [Google Scholar] [CrossRef]
- Kumar, A.; Davuluri, G.; Silva, R.N.E.; Engelen, M.P.K.J.; Ten Have, G.A.M.; Prayson, R.; Deutz, N.E.P.; Dasarathy, S. Ammonia lowering reverses sarcopenia of cirrhosis by restoring skeletal muscle proteostasis. Hepatology 2017, 65, 2045–2058. [Google Scholar] [CrossRef]
- Holecek, M.; Sprongl, L.; Tichý, M. Effect of hyperammonemia on leucine and protein metabolism in rats. Metabolism 2000, 49, 1330–1334. [Google Scholar] [CrossRef]
- Holecek, M.; Kandar, R.; Sispera, L.; Kovarik, M. Acute hyperammonemia activates branched-chain amino acid catabolism and decreases their extracellular concentrations: Different sensitivity of red and white muscle. Amino Acids 2011, 40, 575–584. [Google Scholar] [CrossRef]
- Holeček, M.; Vodeničarovová, M. Muscle wasting and branched-chain amino acid, alpha-ketoglutarate, and ATP depletion in a rat model of liver cirrhosis. Int. J. Exp. Pathol. 2018, 99, 274–281. [Google Scholar] [CrossRef]
- Lowenstein, J.M. Ammonia production in muscle and other tissues: The purine nucleotide cycle. Physiol. Rev. 1972, 52, 382–414. [Google Scholar] [CrossRef]
- Mavrothalassitis, G.; Tzimagiorgis, G.; Mitsialis, A.; Zannis, V.; Plaitakis, A.; Papamatheakis, J.; Moschonas, N. Isolation and characterization of cDNA clones encoding human liver glutamate dehydrogenase: Evidence for a small gene family. Proc. Natl. Acad. Sci. USA 1988, 85, 3494–3498. [Google Scholar] [CrossRef]
- Wiechetek, M.; Breves, G.; Höller, H. Effects of increased blood ammonia concentrations on the concentrations of some metabolites in rat tissues. Q. J. Exp. Physiol. 1981, 66, 423–429. [Google Scholar] [CrossRef]
- Van Hall, G.; van der Vusse, G.J.; Söderlund, K.; Wagenmakers, A.J. Deamination of amino acids as a source for ammonia production in human skeletal muscle during prolonged exercise. J. Physiol. 1995, 489, 251–261. [Google Scholar] [CrossRef]
- Adeva, M.M.; Souto, G.; Blanco, N.; Donapetry, C. Ammonium metabolism in humans. Metabolism 2012, 61, 1495–1511. [Google Scholar] [CrossRef]
- Harper, A.E.; Miller, R.H.; Block, K.P. Branched-chain amino acid metabolism. Annu. Rev. Nutr. 1984, 4, 409–454. [Google Scholar] [CrossRef]
- Shimomura, Y.; Fujii, H.; Suzuki, M.; Murakami, T.; Fujitsuka, N.; Nakai, N. Branched-chain alpha-keto acid dehydrogenase complex in rat skeletal muscle: Regulation of the activity and gene expression by nutrition and physical exercise. J. Nutr. 1995, 125, 1762S–1765S. [Google Scholar]
- Yoneshiro, T.; Wang, Q.; Tajima, K.; Matsushita, M.; Maki, H.; Igarashi, K.; Dai, Z.; White, P.J.; McGarrah, R.W.; Ilkayeva, O.R.; et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature 2019, 572, 614–619. [Google Scholar] [CrossRef]
- Cooper, A.J.; Jeitner, T.M. Central role of glutamate metabolism in the maintenance of nitrogen homeostasis in normal and hyperammonemic brain. Biomolecules 2016, 6, 16. [Google Scholar] [CrossRef]
- Abrahams, S.L.; Younathan, E.S. Modulation of the kinetic properties of phosphofructokinase by ammonium ions. J. Biol. Chem. 1971, 246, 2464–2467. [Google Scholar] [CrossRef]
- Holeček, M. The role of skeletal muscle in the pathogenesis of altered concentrations of branched-chain amino acids (valine, leucine, and isoleucine) in liver cirrhosis, diabetes, and other diseases. Physiol. Res. 2021, 70, 293–305. [Google Scholar] [CrossRef]
- Wagenmakers, A.J. Amino acid metabolism, muscular fatigue and muscle wasting. Speculations on adaptations at high altitude. Int. J. Sports Med. 1992, 13, S110–S113. [Google Scholar] [CrossRef]
- Wagenmakers, A.J.; Coakley, J.H.; Edwards, R.H. Metabolism of branched-chain amino acids and ammonia during exercise: Clues from McArdle’s disease. Int. J. Sports Med. 1990, 11, S101–S113. [Google Scholar] [CrossRef]
- Tornheim, K.; Lowenstein, J.M. The purine nucleotide cycle. The production of ammonia from aspartate by extracts of rat skeletal muscle. J. Biol. Chem. 1972, 247, 162–169. [Google Scholar] [CrossRef]
- Graham, T.E.; MacLean, D.A. Ammonia and amino acid metabolism in human skeletal muscle during exercise. Can. J. Physiol. Pharmacol. 1992, 70, 132–141. [Google Scholar] [CrossRef]
- Meyer, R.A.; Terjung, R.L. AMP deamination and IMP reamination in working skeletal muscle. Am. J. Physiol. 1980, 239, C32–C38. [Google Scholar] [CrossRef]
- Katz, A.; Broberg, S.; Sahlin, K.; Wahren, J. Muscle ammonia and amino acid metabolism during dynamic exercise in man. Clin. Physiol. 1986, 6, 365–379. [Google Scholar] [CrossRef]
- Aragón, J.J.; Lowenstein, J.M. The purine-nucleotide cycle. Comparison of the levels of citric acid cycle intermediates with the operation of the purine nucleotide cycle in rat skeletal muscle during exercise and recovery from exercise. Eur. J. Biochem. 1980, 110, 371–377. [Google Scholar] [CrossRef]
- Banister, E.W.; Cameron, B.J. Exercise-induced hyperammonemia: Peripheral and central effects. Int. J. Sports Med. 1990, S2, S129–S142. [Google Scholar] [CrossRef]
- Gibala, M.J.; MacLean, D.A.; Graham, T.E.; Saltin, B. Anaplerotic processes in human skeletal muscle during brief dynamic exercise. J. Physiol. 1997, 502, 703–713. [Google Scholar] [CrossRef]
- Dos Santos, R.V.; Caperuto, E.C.; de Mello, M.T.; Batista, M.L.; Rosa, L.F. Effect of exercise on glutamine synthesis and transport in skeletal muscle from rats. Clin. Exp. Pharmacol. Physiol. 2009, 36, 770–775. [Google Scholar] [CrossRef]
- Henriksson, J. Effect of exercise on amino acid concentrations in skeletal muscle and plasma. J. Exp. Biol. 1991, 160, 149–165. [Google Scholar] [CrossRef]
- Rowbottom, D.G.; Keast, D.; Morton, A.R. The emerging role of glutamine as an indicator of exercise stress and overtraining. Sports Med. 1996, 21, 80–97. [Google Scholar] [CrossRef]
- Bergström, J.; Fürst, P.; Hultman, E. Free amino acids in muscle tissue and plasma during exercise in man. Clin. Physiol. 1985, 5, 155–160. [Google Scholar] [CrossRef]
- Poortmans, J.R.; Siest, G.; Galteau, M.M.; Houot, O. Distribution of plasma amino acids in humans during submaximal prolonged exercise. Eur. J. Appl. Physiol. Occup. Physiol. 1974, 32, 143–147. [Google Scholar] [CrossRef]
- Okamura, K.; Matsubara, F.; Yoshioka, Y.; Kikuchi, N.; Kikuchi, Y.; Kohri, H. Exercise-induced changes in branched chain amino acid/aromatic amino acid ratio in the rat brain and plasma. Jpn. J. Pharmacol. 1987, 45, 243–248. [Google Scholar] [CrossRef]
- Kasperek, G.J.; Dohm, G.L.; Snider, R.D. Activation of branched-chain keto acid dehydrogenase by exercise. Am. J. Physiol. 1985, 248, R166–R171. [Google Scholar] [CrossRef]
- Wolfe, R.R.; Goodenough, R.D.; Wolfe, M.H.; Royle, G.T.; Nadel, E.R. Isotopic analysis of leucine and urea metabolism in exercising humans. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1982, 52, 458–466. [Google Scholar] [CrossRef]
- Knapik, J.; Meredith, C.; Jones, B.; Fielding, R.; Young, V.; Evans, W. Leucine metabolism during fasting and exercise. J. Appl. Physiol. 1991, 70, 43–47. [Google Scholar] [CrossRef]
- Wagenmakers, A.J.; Brookes, J.H.; Coakley, J.H.; Reilly, T.; Edwards, R.H. Exercise-induced activation of the branched-chain 2-oxo acid dehydrogenase in human muscle. Eur. J. Appl. Physiol. Occup. Physiol. 1989, 59, 159–167. [Google Scholar] [CrossRef]
- Ramaiah, A. Regulation of glycolysis in skeletal muscle. Life Sci. 1976, 19, 455–465. [Google Scholar] [CrossRef]
- Lancha, A.H.; Recco, M.B.; Curi, R. Pyruvate carboxylase activity in the heart and skeletal muscles of the rat. Evidence for a stimulating effect of exercise. Biochem. Mol. Biol. Int. 1994, 32, 483–489. [Google Scholar]
- Gibala, M.J.; MacLean, D.A.; Graham, T.E.; Saltin, B. Tricarboxylic acid cycle intermediate pool size and estimated cycle flux in human muscle during exercise. Am. J. Physiol. 1998, 275, E235–E242. [Google Scholar] [CrossRef] [PubMed]
- Galassetti, P.; Gibbons, F.K.; Hamilton, K.S.; Lacy, D.B.; Cherrington, A.D.; Wasserman, D.H. Enhanced muscle glucose uptake facilitates nitrogen efflux from exercised muscle. J. Appl. Physiol. 1998, 84, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Sahlin, K.; Katz, A.; Broberg, S. Tricarboxylic acid cycle intermediates in human muscle during prolonged exercise. Am. J. Physiol. 1990, 259, C834–C841. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, J.L.; Marwood, S.; Bruce, M.; Constantin-Teodosiu, D.; Greenhaff, P.L. Tricarboxylic acid cycle intermediate pool size: Functional importance for oxidative metabolism in exercising human skeletal muscle. Sports Med. 2007, 37, 1071–1088. [Google Scholar] [CrossRef] [PubMed]
- Dohm, G.L.; Puente, F.R.; Smith, C.P.; Edge, A. Changes in tissue protein levels as a result of endurance exercise. Life Sci. 1978, 23, 845–849. [Google Scholar] [CrossRef]
- Refsum, H.E.; Gjessing, L.R.; Strømme, S.B. Changes in plasma amino acid distribution and urine amino acids excretion during prolonged heavy exercise. Scand. J. Clin. Lab. Investig. 1979, 39, 407–413. [Google Scholar] [CrossRef]
- Dohm, G.L.; Kasperek, G.J.; Tapscott, E.B.; Barakat, H.A. Protein metabolism during endurance exercise. Fed. Proc. 1985, 44, 348–352. [Google Scholar]
- Young, V.R.; Torún, B. Physical activity: Impact on protein and amino acid metabolism and implications for nutritional requirements. Prog. Clin. Biol. Res. 1981, 77, 57–85. [Google Scholar]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.H.; Moon, K.M.; Min, K.W. Exercise-induced myokines can explain the importance of physical activity in the elderly: An overview. Healthcare 2020, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Holeček, M.; Mráz, J.; Tilšer, I. Plasma amino acids in four models of experimental liver injury in rats. Amino Acids 1996, 10, 229–241. [Google Scholar] [CrossRef]
- Meyer, H.P.; Chamuleau, R.A.; Legemate, D.A.; Mol, J.A.; Rothuizen, J. Effects of a branched-chain amino acid-enriched diet on chronic hepatic encephalopathy in dogs. Metab. Brain Dis. 1999, 14, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Hayashi, S.; Higashi, T.; Obata, T.; Sakata, T.; Takei, N.; Shiota, T.; Nagashima, H. Characteristics change in serum amino acid levels in different types of hepatic encephalopathy. Gastroenterol. Jpn. 1982, 17, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Morrison, W.L.; Bouchier, I.A.; Gibson, J.N.; Rennie, M.J. Skeletal muscle and whole-body protein turnover in cirrhosis. Clin. Sci. 1990, 78, 613–619. [Google Scholar] [CrossRef]
- Jacobsen, E.B.; Hamberg, O.; Quistorff, B.; Ott, P. Reduced mitochondrial adenosine triphosphate synthesis in skeletal muscle in patients with Child-Pugh class B and C cirrhosis. Hepatology 2001, 34, 7–12. [Google Scholar] [CrossRef]
- Möller, P.; Bergström, J.; Fürst, P.; Hellström, K. Muscle biopsy studies in patients with moderate liver cirrhosis with special reference to energy-rich phosphagens and electrolytes. Scand. J. Gastroenterol. 1984, 19, 267–272. [Google Scholar] [CrossRef]
- Davuluri, G.; Allawy, A.; Thapaliya, S.; Rennison, J.H.; Singh, D.; Kumar, A.; Sandlers, Y.; Van Wagoner, D.R.; Flask, C.A.; Hoppel, C.; et al. Hyperammonaemia-induced skeletal muscle mitochondrial dysfunction results in cataplerosis and oxidative stress. J. Physiol. 2016, 594, 7341–7360. [Google Scholar] [CrossRef]
- Holeček, M. Evidence of a vicious cycle in glutamine synthesis and breakdown in pathogenesis of hepatic encephalopathy-therapeutic perspectives. Metab. Brain Dis. 2014, 29, 9–17. [Google Scholar] [CrossRef]
- Holecek, M.; Tilser, I.; Skopec, F.; Sprongl, L. Leucine metabolism in rats with cirrhosis. J. Hepatol. 1996, 24, 209–216. [Google Scholar] [CrossRef]
- McCullough, A.J.; Mullen, K.D.; Kalhan, S.C. Body cell mass and leucine metabolism in cirrhosis. Gastroenterology 1992, 102 Pt 1, 1325–1333. [Google Scholar] [CrossRef]
- Peng, S.; Plank, L.D.; McCall, J.L.; Gillanders, L.K.; McIlroy, K.; Gane, E.J. Body composition, muscle function, and energy expenditure in patients with liver cirrhosis: A comprehensive study. Am. J. Clin. Nutr. 2007, 85, 1257–1266. [Google Scholar] [CrossRef]
- Holecek, M.; Sispera, L. Glutamine deficiency in extracellular fluid exerts adverse effects on protein and amino acid metabolism in skeletal muscle of healthy, laparotomized, and septic rats. Amino Acids 2014, 46, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Mourtzakis, M.; Graham, T.E. Glutamate ingestion and its effects at rest and during exercise in humans. J. Appl. Physiol. 2002, 93, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.E.; Sgro, V.; Friars, D.; Gibala, M.J. Glutamate ingestion: The plasma and muscle free amino acid pools of resting humans. Am. J. Physiol. 2000, 278, E83–E89. [Google Scholar] [CrossRef]
- Stegink, L.D.; Filer, L.J.; Baker, G.L. Effect of aspartame plus monosodium L-glutamate ingestion on plasma and erythrocyte amino acid levels in normal adult subjects fed a high protein meal. Am. J. Clin. Nutr. 1982, 36, 1145–1152. [Google Scholar] [CrossRef]
- Thomassen, A.; Bøtker, H.E.; Nielsen, T.T.; Thygesen, K.; Henningsen, P. Effects of glutamate on exercise tolerance and circulating substrate levels in stable angina pectoris. Am. J. Cardiol. 1990, 65, 173–178. [Google Scholar] [CrossRef]
- Alexander, J.W.; Porter, C.E. The treatment of a patient in hepatic coma with intravenous sodium glutamate and ACTH. Gastroenterology 1954, 26, 926–929. [Google Scholar] [CrossRef]
- Schwartz, I.R.; Lehman, E.; Hammond, J.; Seibel, J.M.; Goldson, F. The failure of monosodium glutamate in the treatment of hepatic coma. Gastroenterology 1956, 30, 869–881. [Google Scholar] [CrossRef]
- Webster, L.T., Jr.; Davidson, C.S. The effect of sodium glutamate on hepatic coma. J. Clin. Investig. 1956, 35, 191–199. [Google Scholar] [CrossRef] [PubMed]
- McDermott, W.V., Jr.; Wareham, J.; Riddell, A.G. Treatment of hepatic coma with L-glutamic acid. N. Engl. J. Med. 1955, 253, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Janeczko, M.J.; Stoll, B.; Chang, X.; Guan, X.; Burrin, D.G. Extensive gut metabolism limits the intestinal absorption of excessive supplemental dietary glutamate loads in infant pigs. J. Nutr. 2007, 137, 2384–2390. [Google Scholar] [CrossRef] [PubMed]
- Windmueller, H.G.; Spaeth, A.E. Intestinal metabolism of glutamine and glutamate from the lumen as compared to glutamine from blood. Arch. Biochem. Biophys. 1975, 171, 662–672. [Google Scholar] [CrossRef]
- Yao, K.; Yin, Y.; Li, X.; Xi, P.; Wang, J.; Lei, J.; Hou, Y.; Wu, G. Alpha-ketoglutarate inhibits glutamine degradation and enhances protein synthesis in intestinal porcine epithelial cells. Amino Acids 2012, 42, 2491–2500. [Google Scholar] [CrossRef]
- Tischler, M.E.; Desautels, M.; Goldberg, A.L. Does leucine, leucyl-tRNA, or some metabolite of leucine regulate protein synthesis and degradation in skeletal and cardiac muscle? J. Biol. Chem. 1982, 257, 1613–1621. [Google Scholar] [CrossRef]
- Holecek, M.; Muthny, T.; Kovarik, M.; Sispera, L. Effect of beta-hydroxy-beta-methylbutyrate (HMB) on protein metabolism in whole body and in selected tissues. Food Chem. Toxicol. 2009, 47, 255–259. [Google Scholar] [CrossRef]
- Kovarik, M.; Muthny, T.; Sispera, L.; Holecek, M. Effects of β-hydroxy-β-methylbutyrate treatment in different types of skeletal muscle of intact and septic rats. J. Physiol. Biochem. 2010, 66, 311–319. [Google Scholar] [CrossRef]
- Blomstrand, E.; Hassmén, P.; Ekblom, B.; Newsholme, E.A. Administration of branched-chain amino acids during sustained exercise—Effects on performance and on plasma concentration of some amino acids. Eur. J. Appl. Physiol. Occup. Physiol. 1991, 63, 83–88. [Google Scholar] [CrossRef]
- Holeček, M.; Vodeničarovová, M. Effects of branched-chain amino acids on muscles under hyperammonemic conditions. J. Physiol. Biochem. 2018, 74, 523–530. [Google Scholar] [CrossRef]
- Parry-Billings, M.; Budgett, R.; Koutedakis, Y.; Blomstrand, E.; Brooks, S.; Williams, C.; Calder, P.C.; Pilling, S.; Baigrie, R.; Newsholme, E.A. Plasma amino acid concentrations in the overtraining syndrome: Possible effects on the immune system. Med. Sci. Sports Exerc. 1992, 24, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M. Interrelationship between physical activity and branched-chain amino acids. J. Nutr. 2005, 135, 1591S–1595S. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Shirreffs, S.M.; Maughan, R.J. The effect of acute branched-chain amino acid supplementation on prolonged exercise capacity in a warm environment. Eur. J. Appl. Physiol. 2004, 93, 306–314. [Google Scholar] [CrossRef] [PubMed]
- MacLean, D.A.; Graham, T.E. Branched-chain amino acid supplementation augments plasma ammonia responses during exercise in humans. J. Appl. Physiol. 1993, 74, 2711–2717. [Google Scholar] [CrossRef]
- Falavigna, G.; Alves de Araújo, J., Jr.; Rogero, M.M.; Pires, I.S.; Pedrosa, R.G.; Martins, E., Jr.; Alves de Castro, I.; Tirapegui, J. Effects of diets supplemented with branched-chain amino acids on the performance and fatigue mechanisms of rats submitted to prolonged physical exercise. Nutrients 2012, 4, 1767–1780. [Google Scholar] [CrossRef]
- Als-Nielsen, B.; Koretz, R.L.; Kjaergard, L.L.; Gluud, C. Branched-chain amino acids for hepatic encephalopathy. Cochrane Database Syst. Rev. 2003, 2, CD001939. [Google Scholar]
- Gluud, L.L.; Dam, G.; Les, I.; Córdoba, J.; Marchesini, G.; Borre, M.; Aagaard, N.K.; Vilstrup, H. Branched-chain amino acids for people with hepatic encephalopathy. Cochrane Database Syst. Rev. 2015, 9, CD001939. [Google Scholar]
- Calders, P.; Pannier, J.L.; Matthys, D.M.; Lacroix, E.M. Pre-exercise branched-chain amino acid administration increases endurance performance in rats. Med. Sci. Sports Exerc. 1997, 29, 1182–1186. [Google Scholar] [CrossRef]
- Holecek, M.; Siman, P.; Vodenicarovova, M.; Kandar, R. Alterations in protein and amino acid metabolism in rats fed a branched-chain amino acid- or leucine-enriched diet during postprandial and postabsorptive states. Nutr. Metab. 2016, 13, 12. [Google Scholar] [CrossRef]
- Yagi, M.; Matthews, D.E.; Walser, M. Nitrogen sparing by 2-ketoisocaproate in parenterally fed rats. Am. J. Physiol. 1990, 259, E633–E638. [Google Scholar] [CrossRef]
- Mitch, W.E.; Walser, M.; Sapir, D.G. Nitrogen sparing induced by leucine compared with that induced by its keto analogue, alpha-ketoisocaproate, in fasting obese man. J. Clin. Investig. 1981, 67, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.M.; Walser, M.; Drachman, D.B. Branched-chain ketoacids reduce muscle protein degradation in Duchenne muscular dystrophy. Muscle Nerve 1982, 5, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Sapir, D.G.; Stewart, P.M.; Walser, M.; Moreadith, C.; Moyer, E.D.; Imbembo, A.L.; Rosenshein, N.B.; Munoz, S. Effects of alpha-ketoisocaproate and of leucine on nitrogen metabolism in postoperative patients. Lancet 1983, 1, 1010–1014. [Google Scholar] [CrossRef]
- Imura, K.; Shiota, T.; Swain, L.M.; Walser, M. Utilization for protein synthesis of 2-ketoisocaproate relative to utilization of leucine, as estimated from exhalation of labelled CO2. Clin. Sci. 1988, 75, 301–307. [Google Scholar] [CrossRef]
- Jungers, P.; Chauveau, P. Amino acids and keto acids in the treatment of chronic renal failure. Blood Purif. 1988, 6, 299–314. [Google Scholar] [CrossRef]
- Walser, M.; Mitch, W.E.; Abras, E. Supplements containing amino acids and keto acids in the treatment of chronic uremia. Kidney Int. 1983, 16, S285–S289. [Google Scholar]
- Teplan, V.; Schück, O.; Horácková, M.; Skibová, J.; Holecek, M. Effect of a keto acid-amino acid supplement on the metabolism and renal elimination of branched-chain amino acids in patients with chronic renal insufficiency on a low protein diet. Wien. Klin. Wochenschr. 2000, 112, 876–881. [Google Scholar]
- Muñoz, S.; Walser, M. Effect of experimental liver disease on the utilization for protein synthesis of orally administered alpha-ketoisocaproate. Hepatology 1986, 6, 472–476. [Google Scholar] [CrossRef]
- De Almeida, R.D.; Prado, E.S.; Llosa, C.D.; Magalhães-Neto, A.; Cameron, L.C. Acute supplementation with keto analogues and amino acids in rats during resistance exercise. Br. J. Nutr. 2010, 104, 1438–1442. [Google Scholar] [CrossRef]
- Prado, E.S.; de Rezende Neto, J.M.; de Almeida, R.D.; Dória de Melo, M.G.; Cameron, L.C. Keto analogue and amino acid supplementation affects the ammonaemia response during exercise under ketogenic conditions. Br. J. Nutr. 2011, 105, 1729–1733. [Google Scholar] [CrossRef]
- Camerino, S.R.; Lima, R.C.; França, T.C.; Herculano Ede, A.; Rodrigues, D.S.; Gouveia, M.G.; Cameron, L.C.; Prado, E.S. Keto analogue and amino acid supplementation and its effects on ammonemia and performance under thermoneutral conditions. Food Funct. 2016, 7, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.T.; Gonçalves, S.C.; Pedrosa, M.L.; Silva, M.E.; Bassini, A.; Coelho, W.S.; de Magalhães-Neto, A.M.; Prado, E.S.; Cameron, L.C. Keto analogues and amino acid supplementation and its effects on ammonaemia during extenuating endurance exercise in ketogenic diet-fed rats. Br. J. Nutr. 2018, 120, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Herlong, H.F.; Maddrey, W.C.; Walser, M. The use of ornithine salts of branched-chain ketoacids in portal-systemic encephalopathy. Ann. Intern. Med. 1980, 93, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Maddrey, W.C.; Weber, F.L., Jr.; Coulter, A.W.; Chura, C.M.; Chapanis, N.P.; Walser, M. Effects of keto analogues of essential amino acids in portal-systemic encephalopathy. Gastroenterology 1976, 71, 190–195. [Google Scholar] [CrossRef]
- Batshaw, M.L.; Brusilow, S.; Walser, M. Long-term management of a case of carbamyl phosphate synthetase deficiency using ketanalogues and hydroxyanalogues of essential amino acids. Pediatrics 1976, 58, 227–235. [Google Scholar]
- Thoene, J.; Batshaw, M.; Spector, E.; Kulovich, S.; Brusilow, S.; Walser, M.; Nyhan, W. Neonatal citrllinemia: Treatment with keto-analogues of essential amino acids. J. Pediatr. 1977, 90, 218–224. [Google Scholar] [CrossRef]
- McReynolds, J.W.; Mantagos, S.; Brusilow, S.; Rosenberg, L.E. Treatment of complete ornithine transcarbamylase deficiency with nitrogen-free analogues of essential amino acids. J. Pediatr. 1978, 93, 421–427. [Google Scholar] [CrossRef]
- Ji, L.L.; Miller, R.H.; Nagle, F.J.; Lardy, H.A.; Stratman, F.W. Amino acid metabolism during exercise in trained rats: The potential role of carnitine in the metabolic fate of branched-chain amino acids. Metabolism 1987, 36, 748–752. [Google Scholar] [CrossRef]
- Adán, C.; Ardévol, A.; Rafecas, I.; Remesar, X.; Alemany, M.; Fernández-López, J.A. Amino acid nitrogen handling by hind leg muscle of the rat during exercise. Arch. Physiol. Biochem. 1997, 105, 478–486. [Google Scholar] [CrossRef]
- Trudeau, F. Aspartate as an ergogenic supplement. Sports Med. 2008, 38, 9–16. [Google Scholar] [CrossRef]
- Lancha, A.H., Jr.; Recco, M.B.; Abdalla, D.S.; Curi, R. Effect of aspartate, asparagine, and carnitine supplementation in the diet on metabolism of skeletal muscle during a moderate exercise. Physiol. Behav. 1995, 57, 367–371. [Google Scholar] [CrossRef]
- Marquezi, M.L.; Roschel, H.A.; dos Santa Costa, A.; Sawada, L.A.; Lancha, A.H., Jr. Effect of aspartate and asparagine supplementation on fatigue determinants in intense exercise. Int. J. Sport Nutr. Exerc. Metab. 2003, 13, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. Ammonia removal by metabolic scavengers for the prevention and treatment of hepatic encephalopathy in cirrhosis. Drugs R D 2021, 21, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Hammarqvist, F.; Wernerman, J.; Ali, R.; von der Decken, A.; Vinnars, E. Addition of glutamine to total parenteral nutrition after elective abdominal surgery spares free glutamine in muscle, counteracts the fall in muscle protein synthesis, and improves nitrogen balance. Ann. Surg. 1989, 209, 455–461. [Google Scholar] [CrossRef]
- Hardy, G.; Hardy, I.J. Can glutamine enable the critically ill to cope better with infection? JPEN J. Parenter. Enter. Nutr. 2008, 32, 489–491. [Google Scholar] [CrossRef]
- Legault, Z.; Bagnall, N.; Kimmerly, D.S. The influence of oral L-glutamine supplementation on muscle strength recovery and soreness following unilateral knee extension eccentric exercise. Int. J. Sport Nutr. Exerc. Metab. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Castell, L.M. Can glutamine modify the apparent immunodepression observed after prolonged, exhaustive exercise? Nutrition 2002, 18, 371–375. [Google Scholar] [CrossRef]
- Córdova-Martínez, A.; Caballero-García, A.; Bello, H.J.; Pérez-Valdecantos, D.; Roche, E. Effect of glutamine supplementation on muscular damage biomarkers in professional basketball players. Nutrients 2021, 13, 2073. [Google Scholar] [CrossRef]
- Phongsamran, P.V.; Kimm, J.W.; Cupo Abbottm, J.; Rosenblatt, A. Pharmacotherapy for hepatic encephalopathy. Drugs 2010, 70, 1131–1148. [Google Scholar] [CrossRef]
- Shih, V.E. Alternative-pathway therapy for hyperammonemia. N. Engl. J. Med. 2007, 356, 2321–2322. [Google Scholar] [CrossRef]
- Van Straten, G.; van Dalen, D.; Mesu, S.J.; Rothuizen, J.; Teske, E.; Spee, B.; Favier, R.P.; van Geijlswijk, I.M. Efficacy of orally administered sodium benzoate and sodium phenylbutyrate in dogs with congenital portosystemic shunts. J. Vet. Intern. Med. 2019, 33, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, H.D.; Zacharias, A.P.; Gluud, L.L.; Morgan, M.Y. Pharmacotherapies that specifically target ammonia for the prevention and treatment of hepatic encephalopathy in adults with cirrhosis. Cochrane Database Syst. Rev. 2019, 6, CD012334. [Google Scholar] [CrossRef] [PubMed]
- Holecek, M.; Vodenicarovova, M.; Siman, P. Acute effects of phenylbutyrate on glutamine, branched-chain amino acid and protein metabolism in skeletal muscles of rats. Int. J. Exp. Pathol. 2017, 98, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Holecek, M.; Vodenicarovova, M. Phenylbutyrate exerts adverse effects on liver regeneration and amino acid concentrations in partially hepatectomized rats. Int. J. Exp. Pathol. 2016, 97, 278–284. [Google Scholar] [CrossRef]
- Dam, G.; Ott, P.; Aagaard, N.K.; Vilstrup, H. Branched-chain amino acids and muscle ammonia detoxification in cirrhosis. Metab. Brain Dis. 2013, 28, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Dam, G.; Keiding, S.; Munk, O.L.; Ott, P.; Buhl, M.; Vilstrup, H.; Bak, L.K.; Waagepetersen, H.S.; Schousboe, A.; Møller, N.; et al. Branched-chain amino acids increase arterial blood ammonia in spite of enhanced intrinsic muscle ammonia metabolism in patients with cirrhosis and healthy subjects. Am. J. Physiol. 2011, 301, G269–G277. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holeček, M. Muscle Amino Acid and Adenine Nucleotide Metabolism during Exercise and in Liver Cirrhosis: Speculations on How to Reduce the Harmful Effects of Ammonia. Metabolites 2022, 12, 971. https://doi.org/10.3390/metabo12100971
Holeček M. Muscle Amino Acid and Adenine Nucleotide Metabolism during Exercise and in Liver Cirrhosis: Speculations on How to Reduce the Harmful Effects of Ammonia. Metabolites. 2022; 12(10):971. https://doi.org/10.3390/metabo12100971
Chicago/Turabian StyleHoleček, Milan. 2022. "Muscle Amino Acid and Adenine Nucleotide Metabolism during Exercise and in Liver Cirrhosis: Speculations on How to Reduce the Harmful Effects of Ammonia" Metabolites 12, no. 10: 971. https://doi.org/10.3390/metabo12100971
APA StyleHoleček, M. (2022). Muscle Amino Acid and Adenine Nucleotide Metabolism during Exercise and in Liver Cirrhosis: Speculations on How to Reduce the Harmful Effects of Ammonia. Metabolites, 12(10), 971. https://doi.org/10.3390/metabo12100971