
Evaluating the Accuracy of the QCEIMS Approach for Computational Prediction of Electron Ionization Mass Spectra of Purines and Pyrimidines




Abstract
:1. Introduction
2. Results
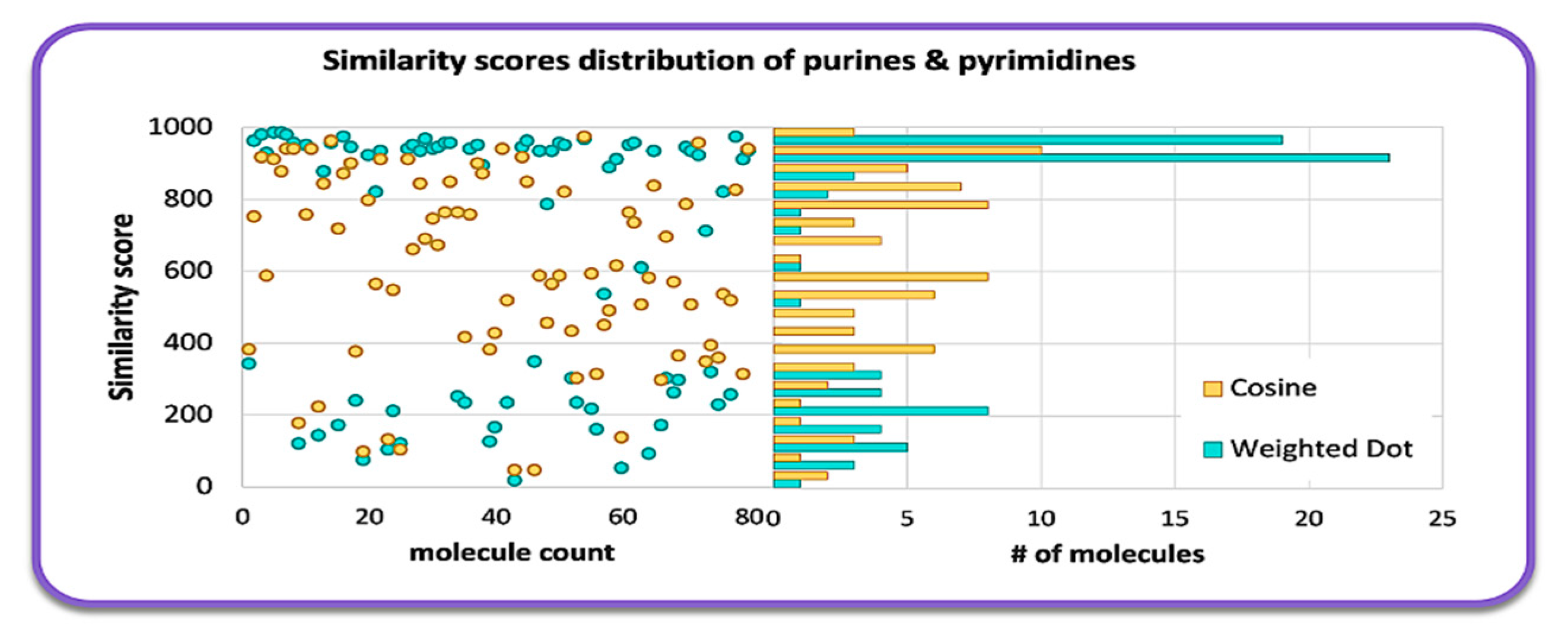
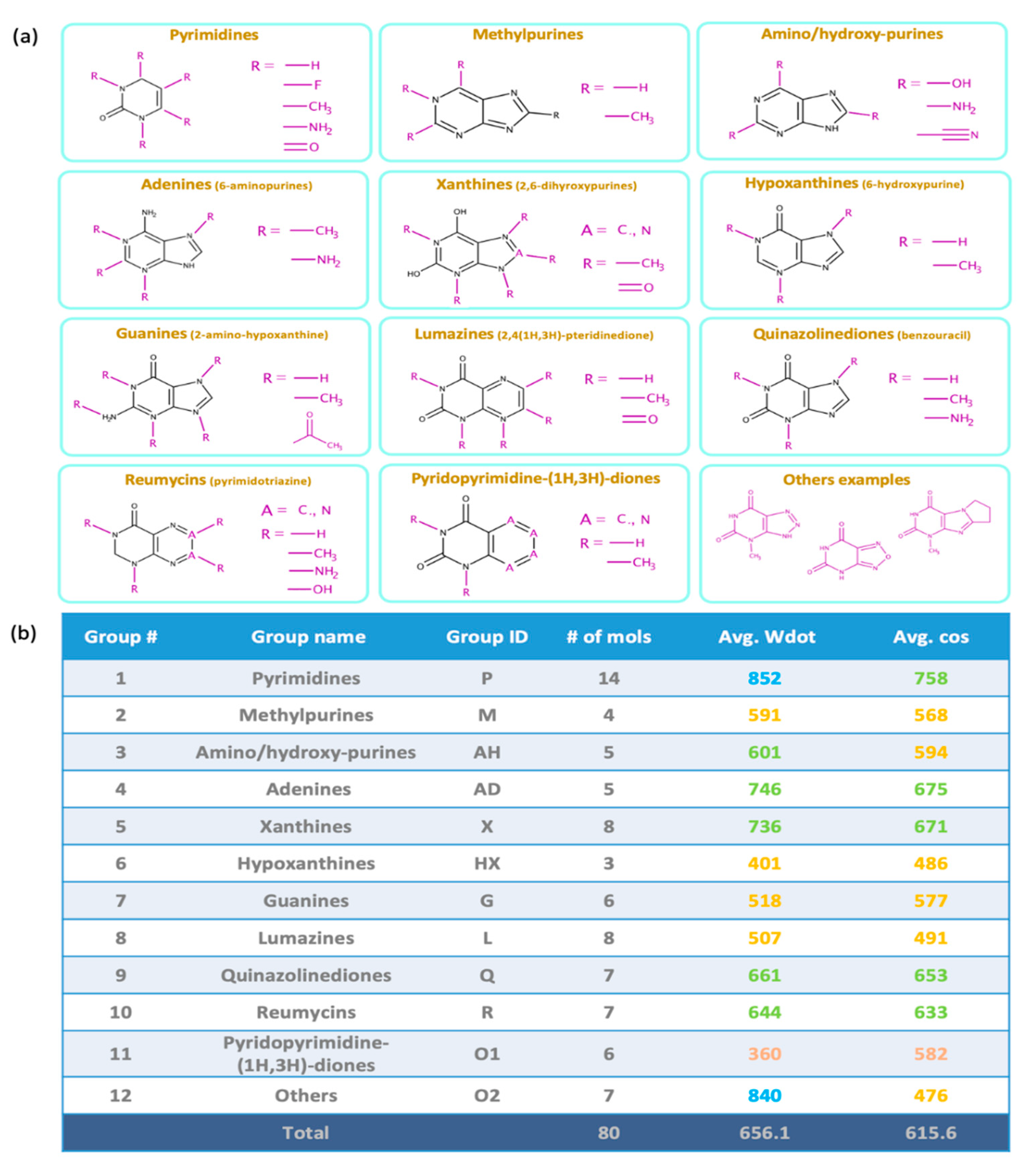
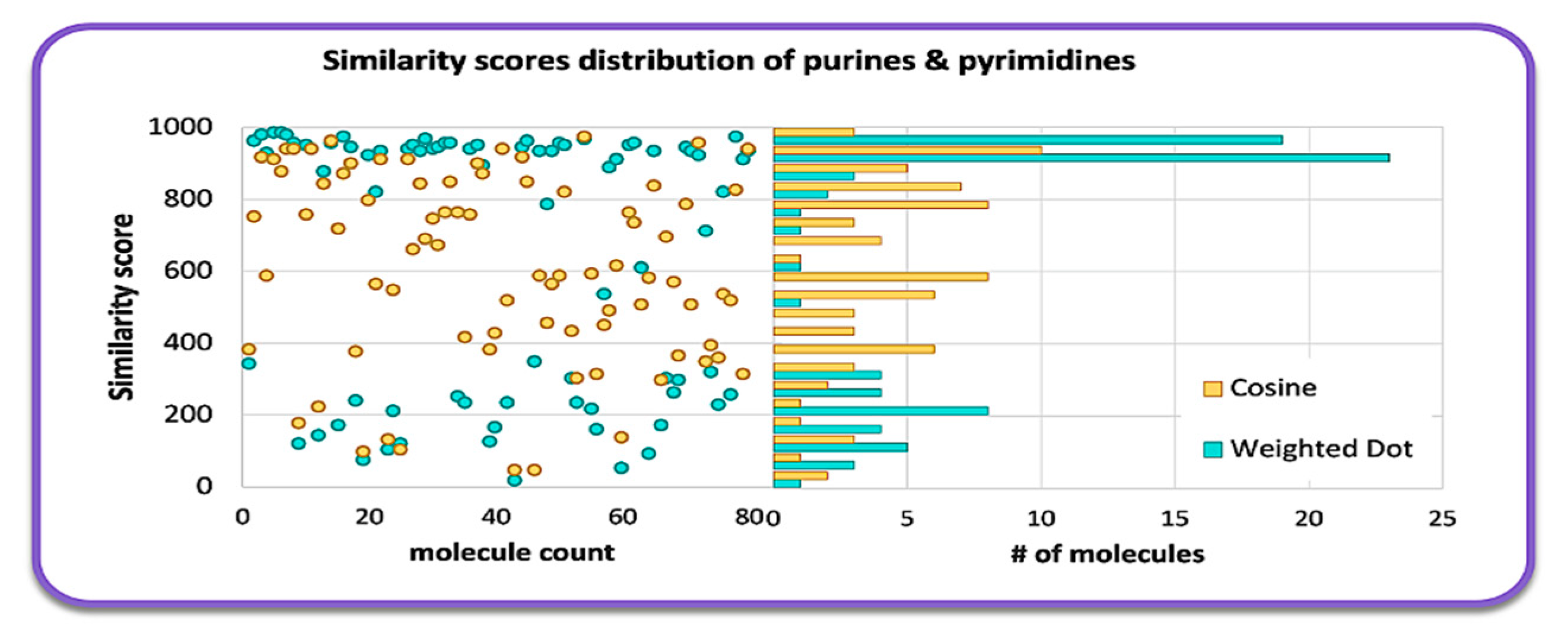
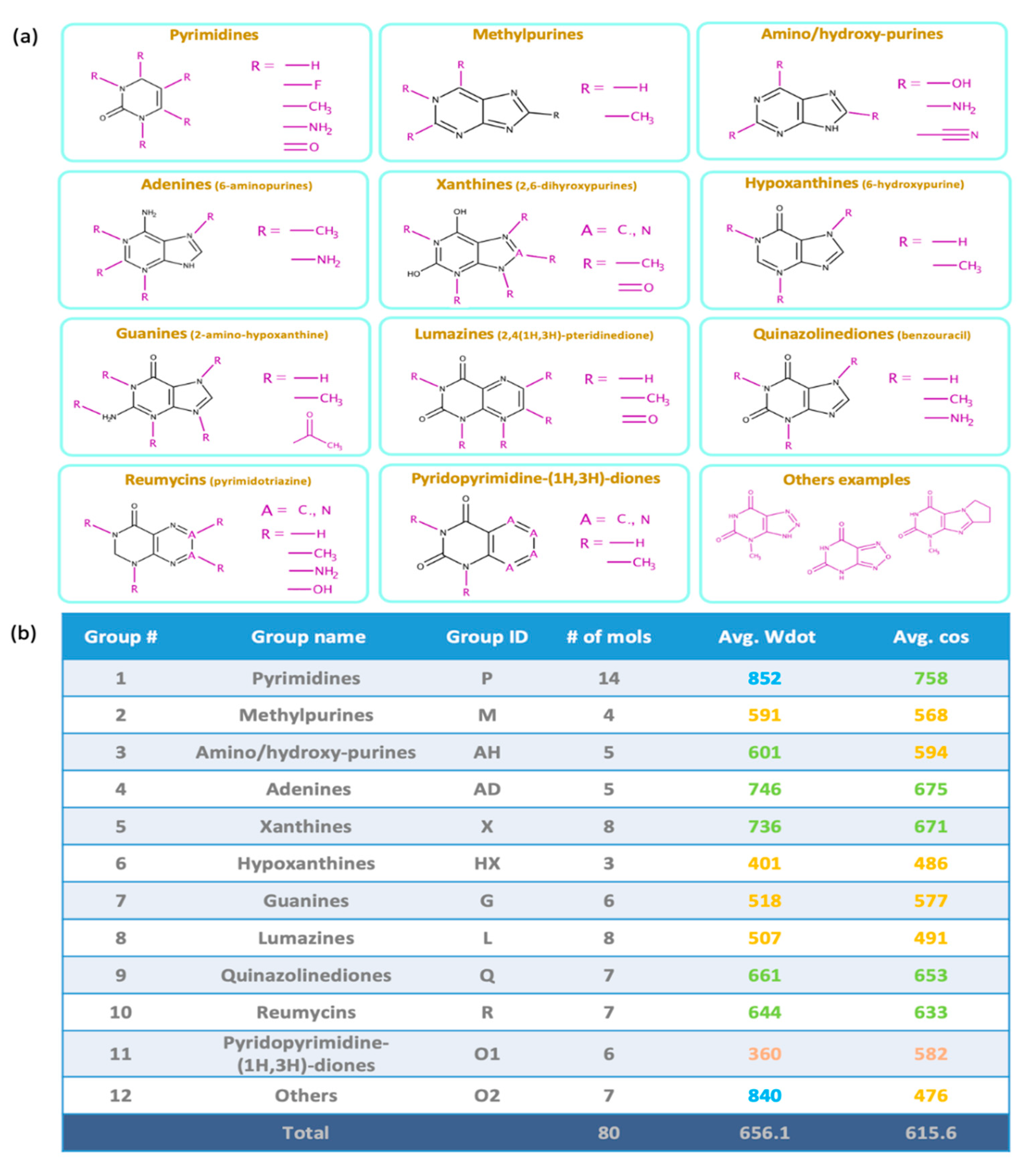
2.1. Overall Similarity Scores and Investigation of Structural Subclasses
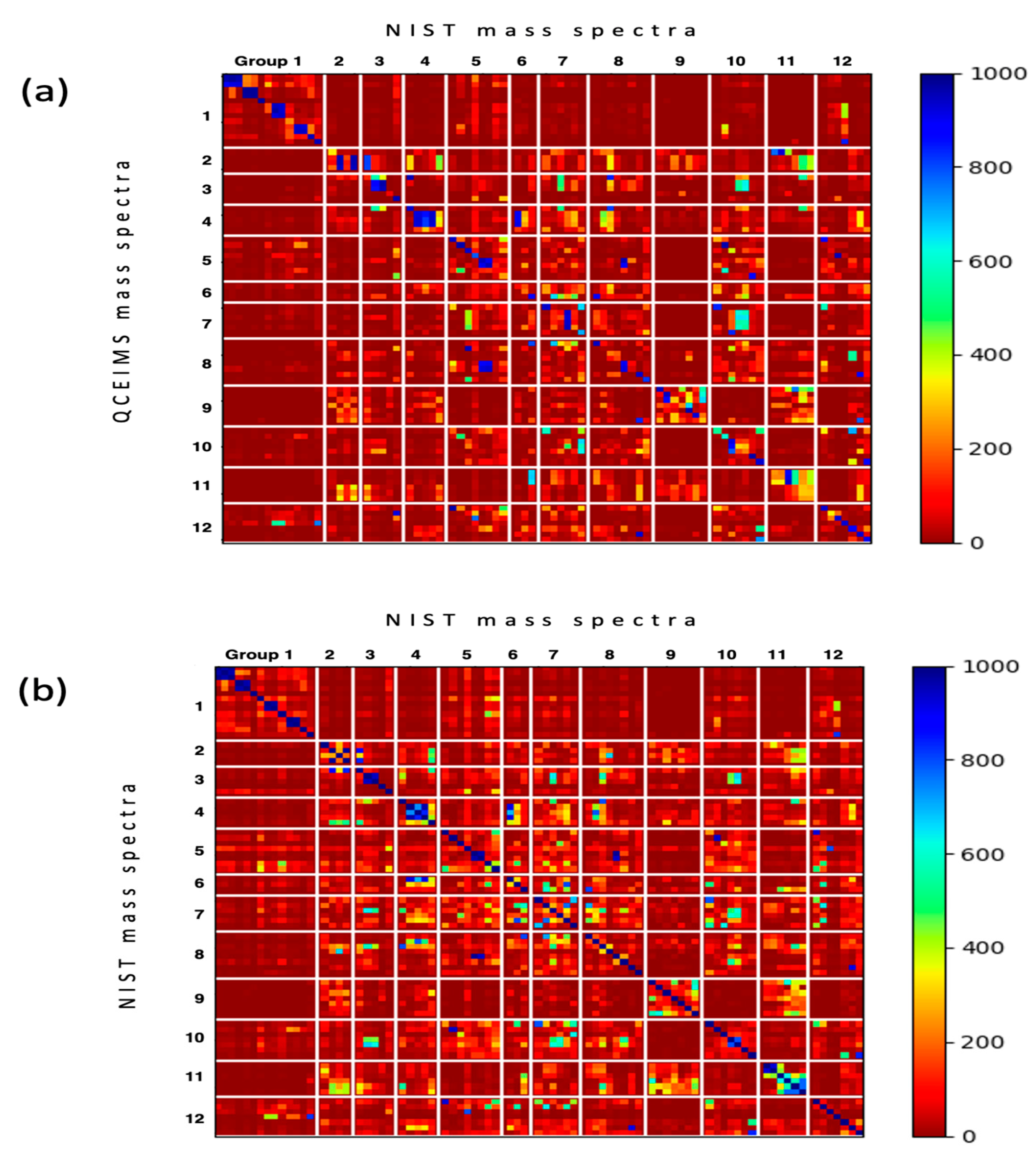
2.2. Validating QCEIMS Spectra by Matching against Large Libraries
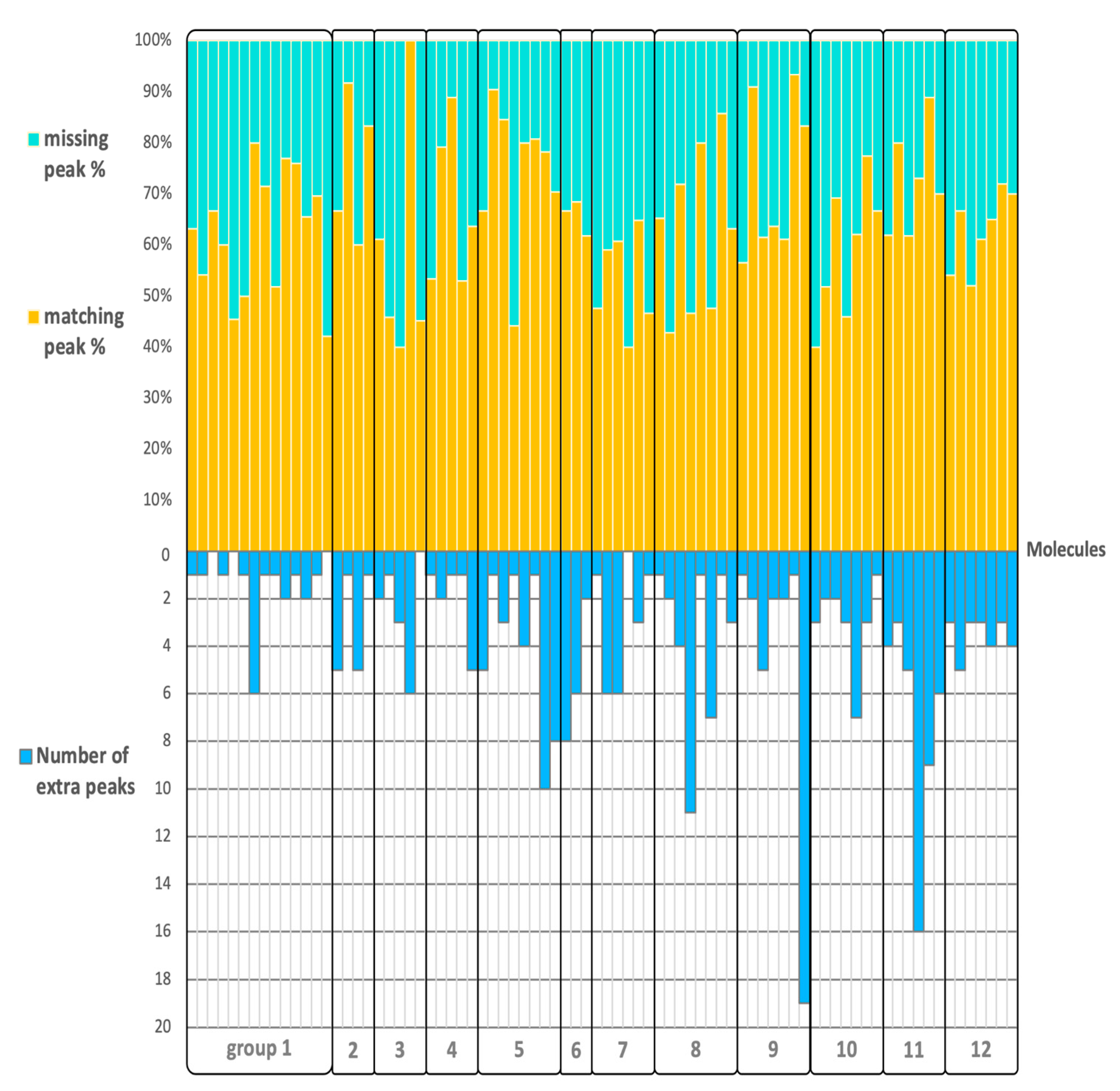
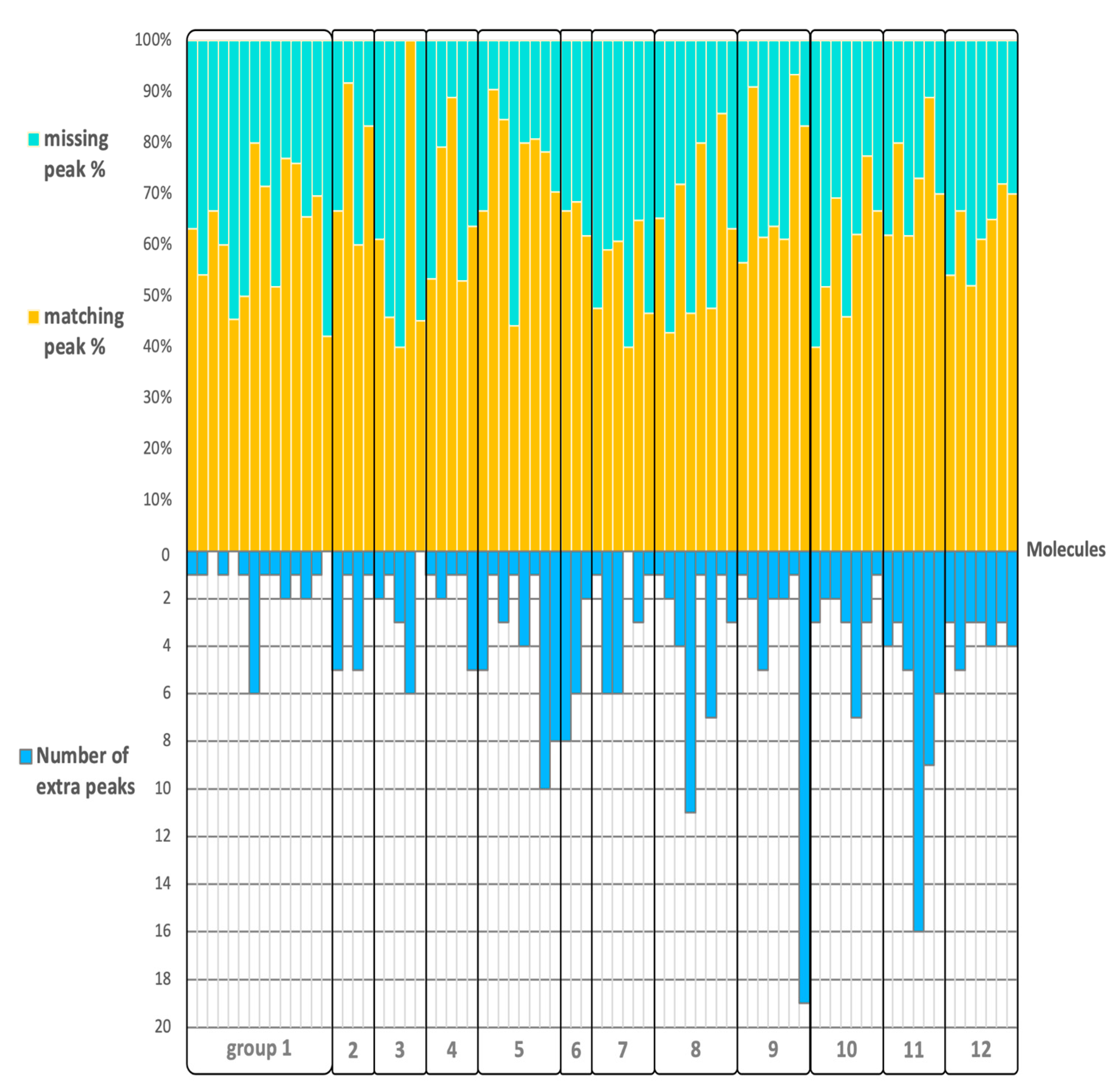
2.3. Matching, Missing, Extra Fragment Ions Versus Ion Abundances
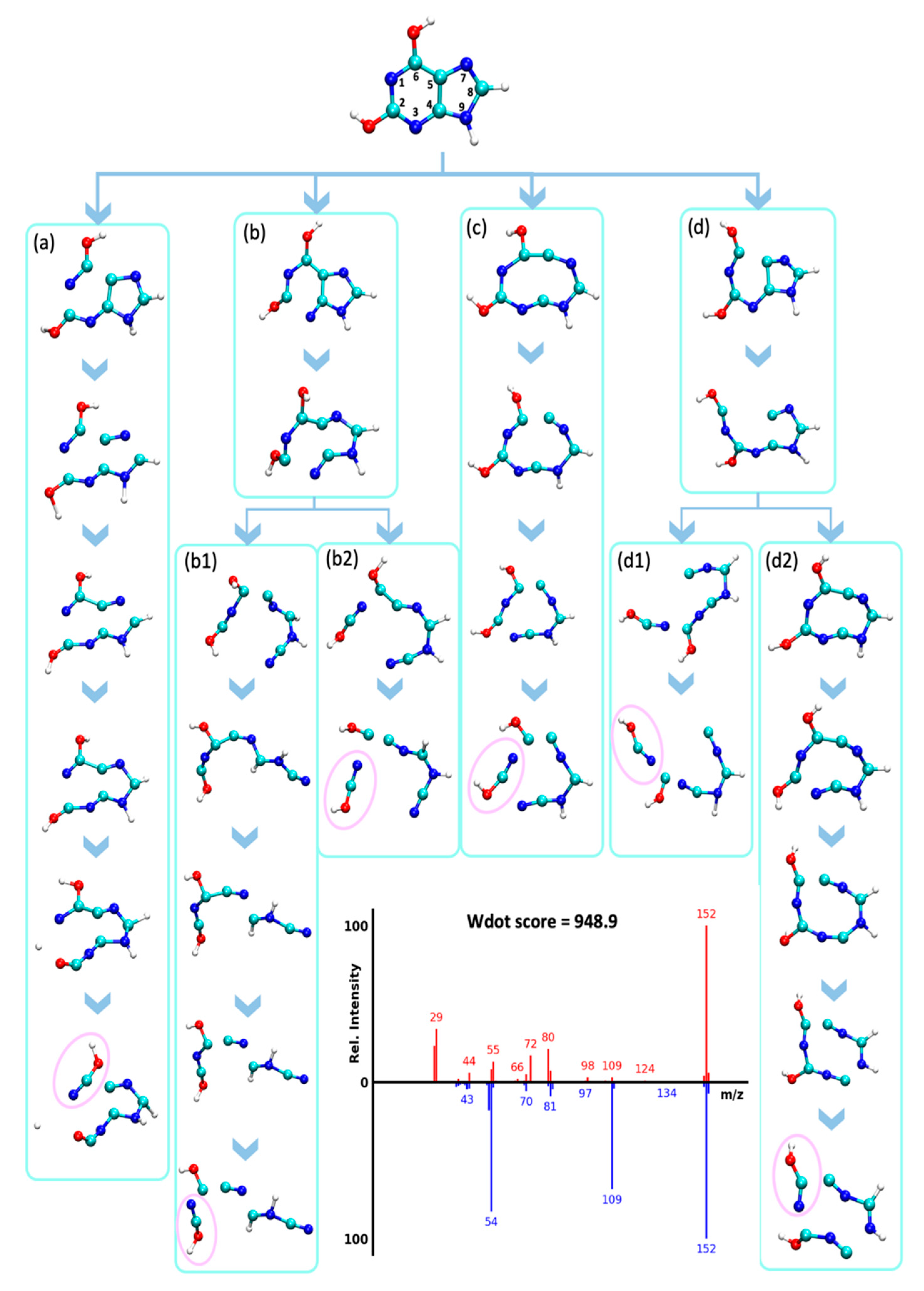
2.4. Detailed Studies of Fragmentation Pathways
2.4.1. Abundance of Molecular Ions in QCEIMS Predictions
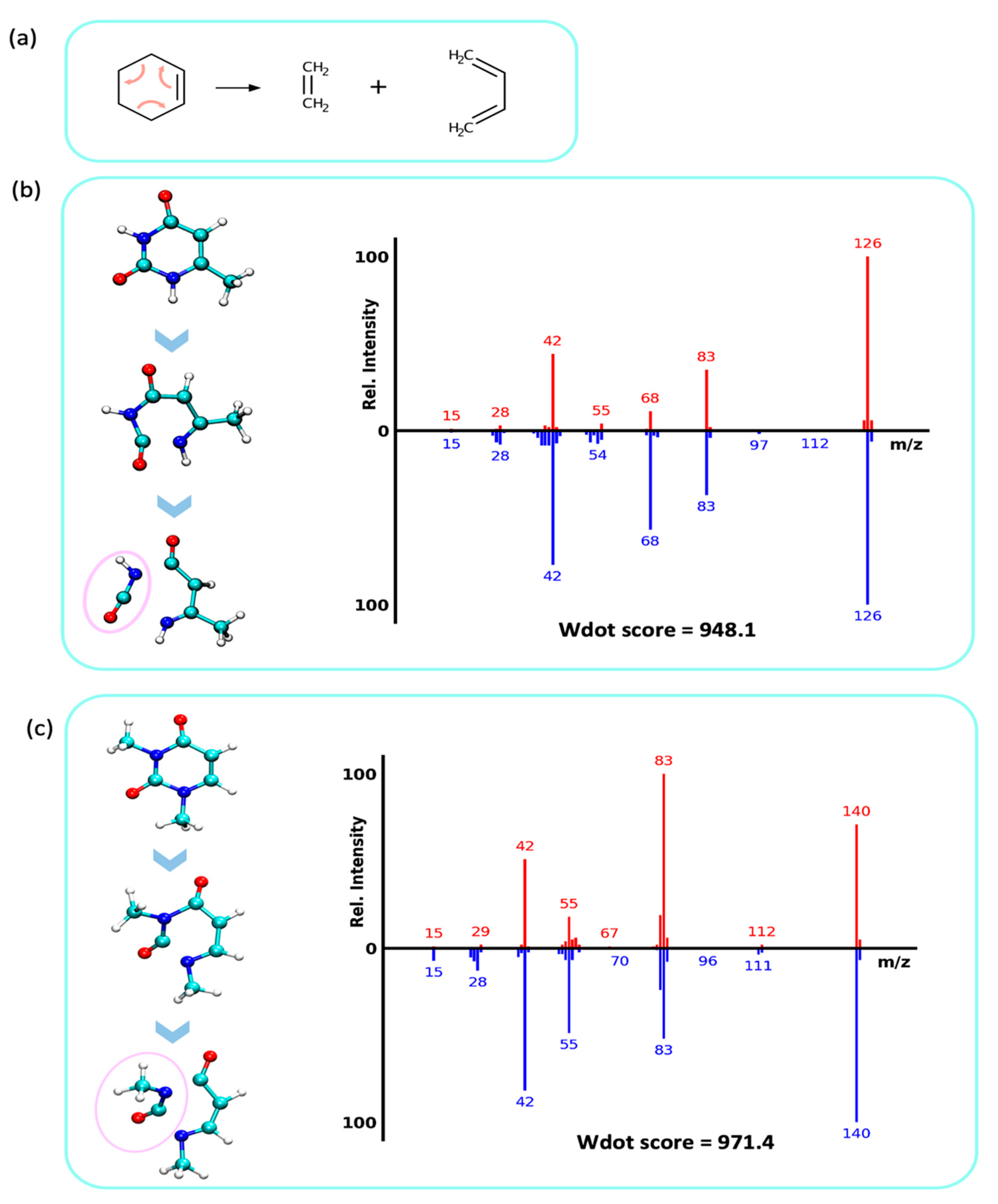
2.4.2. Retro Diels–Alder Reaction and Loss of Isocyanic- or Cyanic Acid (NCOH, HNCO, 43 u)
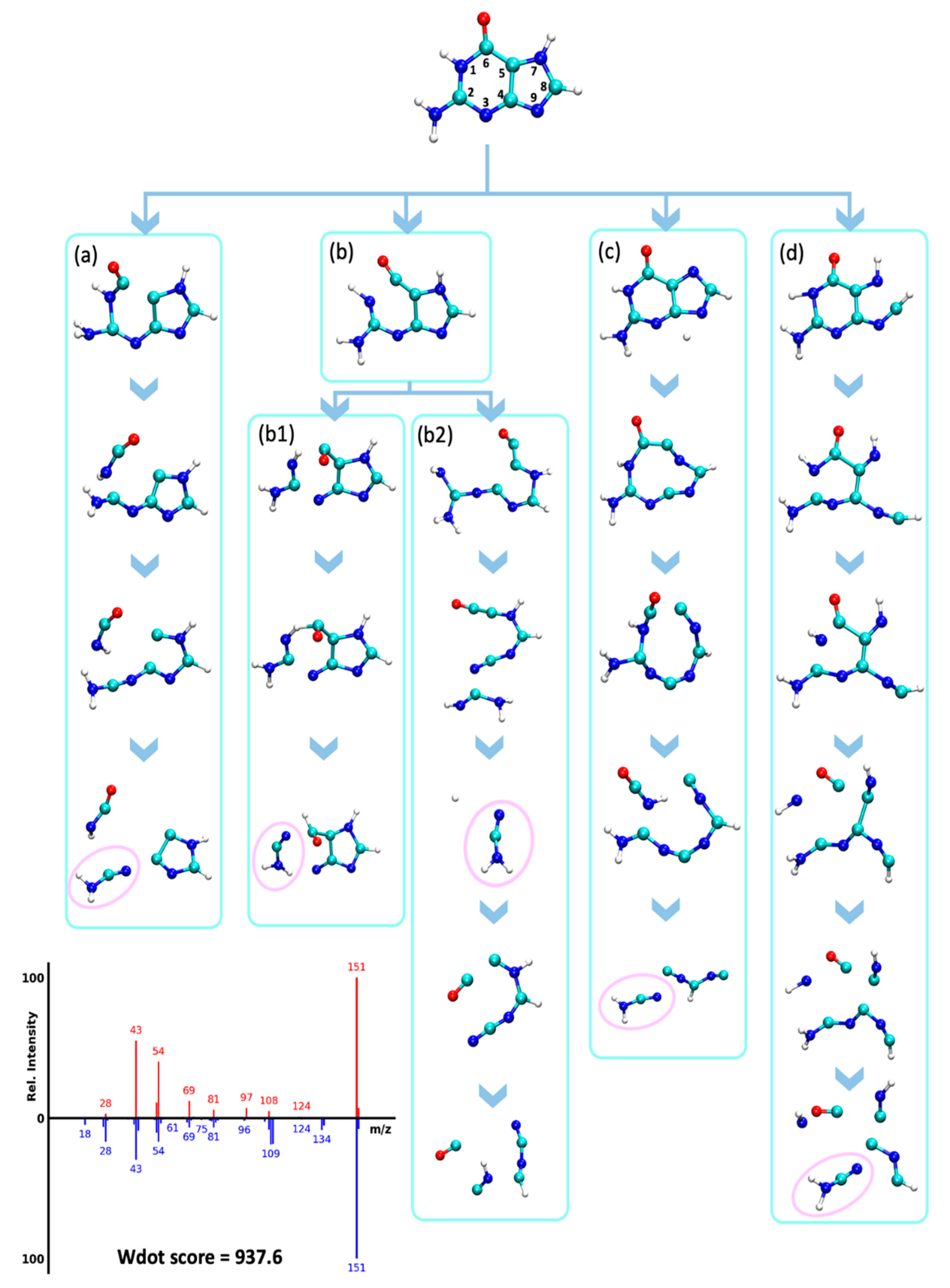
2.4.3. Loss of Hydrogen Cyanide (HCN, 27 u)
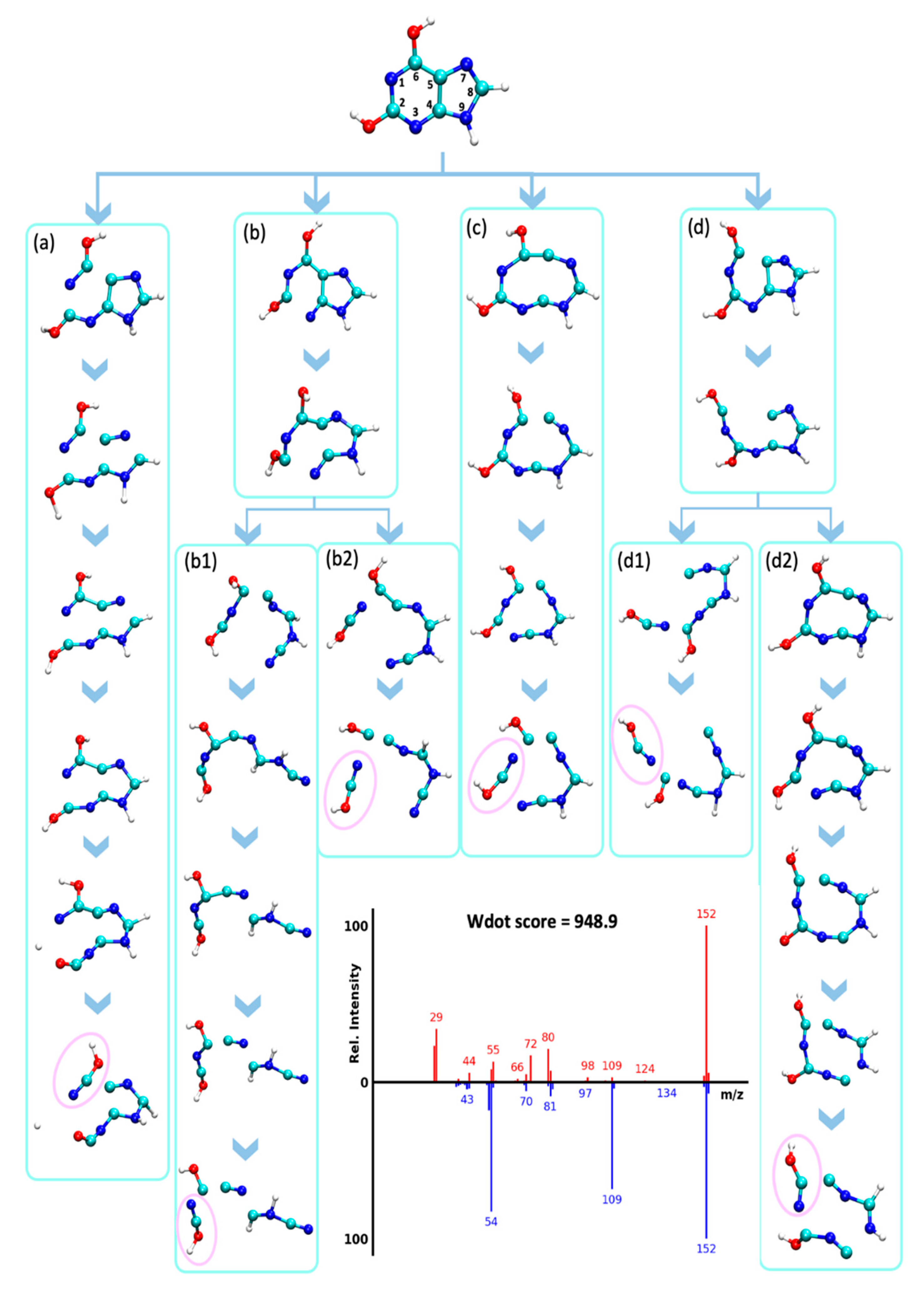
2.4.4. Retro Diels–Alder Reaction of Guanines and Loss of Cyanamide (CN2H2, 42 u)
3. Discussion
4. Methods
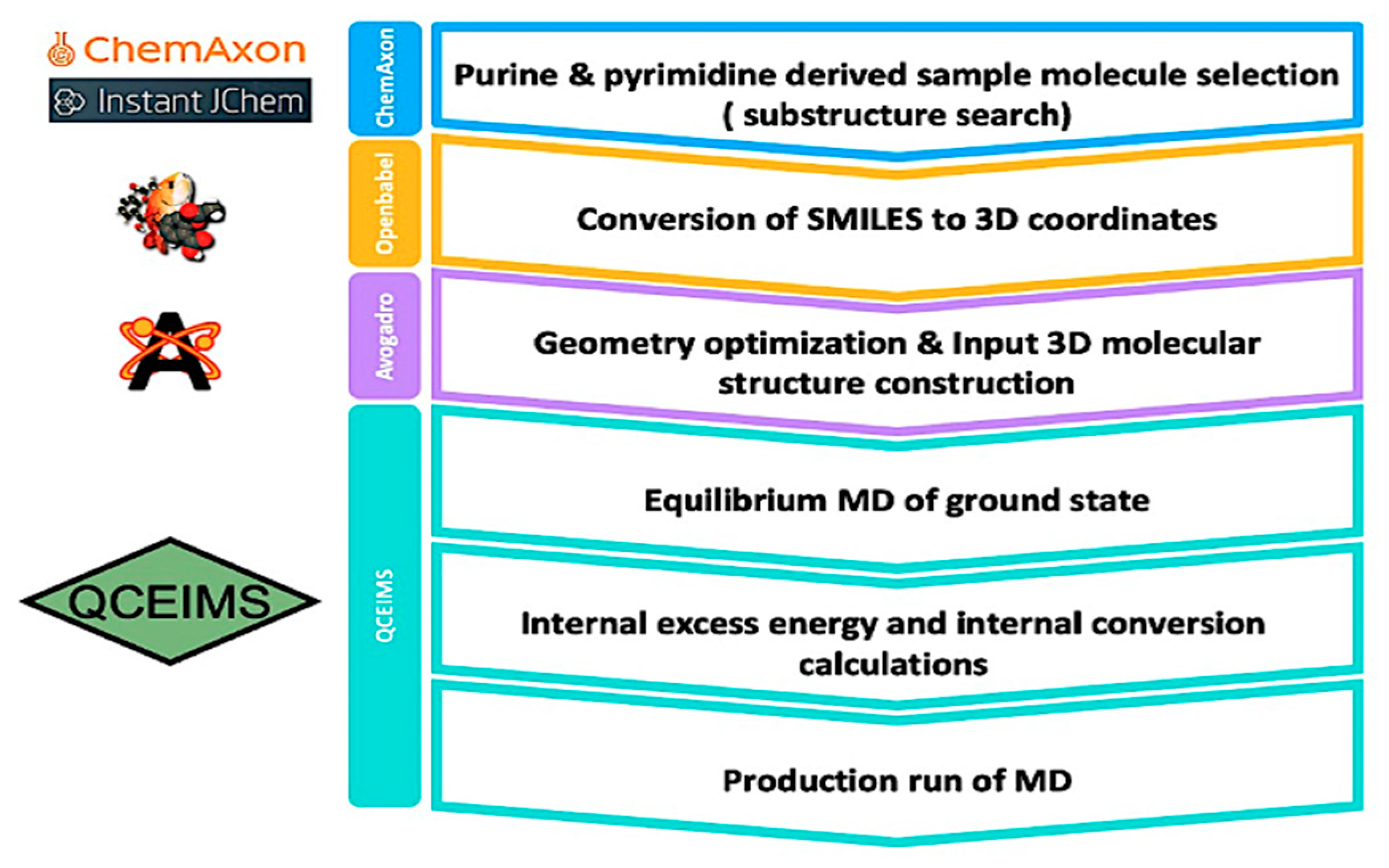
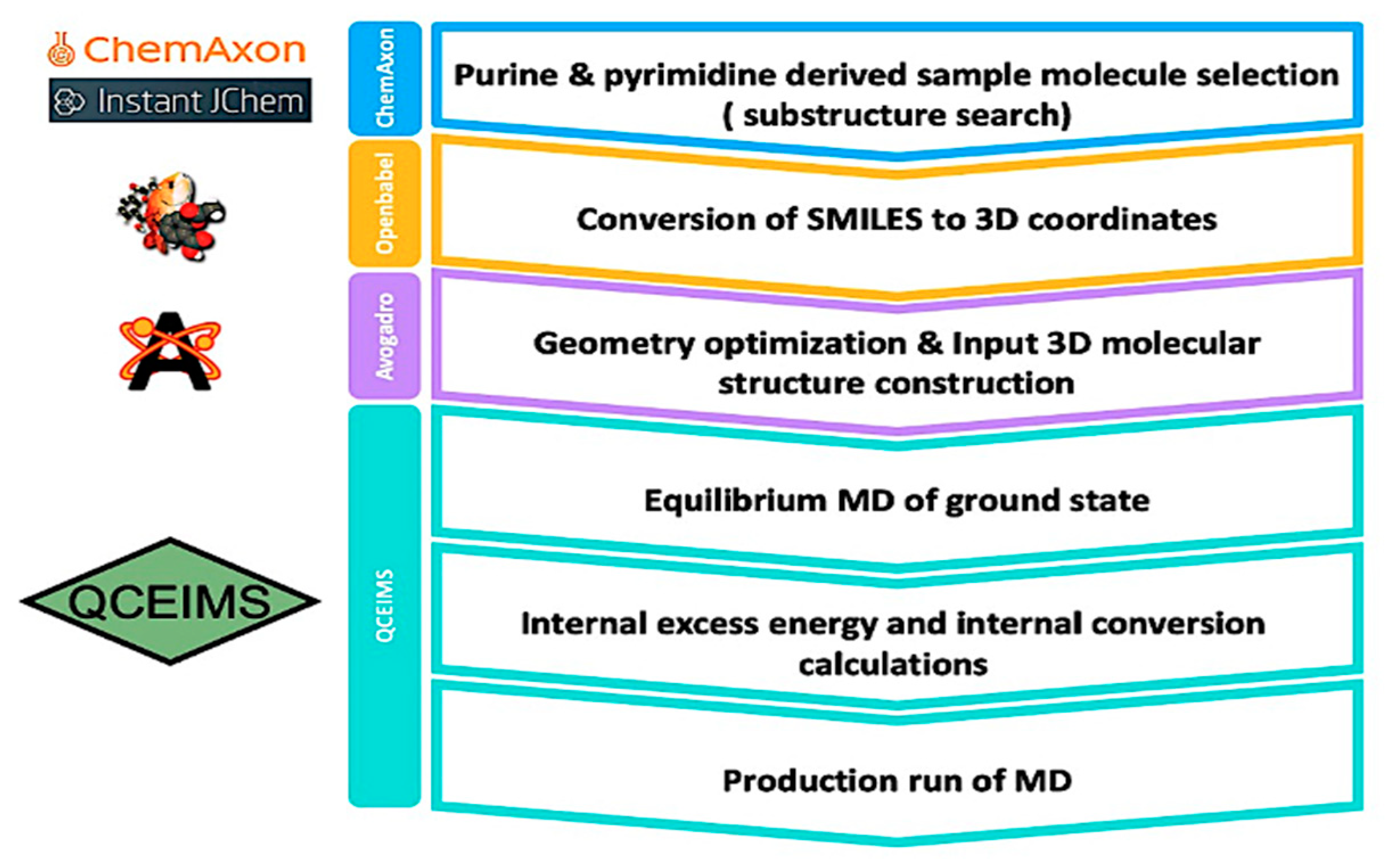
4.1. Selection of the Sample Molecules and Preparation of Their 3D Structure
4.2. QCEIMS Calculation
4.3. Analysis and Evaluation of Mass Spectra
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parker, W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar]
- Shuvalov, O.; Petukhov, A.; Daks, A.; Fedorova, O.; Vasileva, E.; Barlev, N.A. One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy. Oncotarget 2017, 8, 23955. [Google Scholar]
- Liekens, S.; Bronckaers, A.; Balzarini, J. Improvement of purine and pyrimidine antimetabolite-based anticancer treatment by selective suppression of mycoplasma-encoded catabolic enzymes. Lancet Oncol. 2009, 10, 628–635. [Google Scholar]
- Hoffmann, H.-H.; Kunz, A.; Simon, V.A.; Palese, P.; Shaw, M.L. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 5777–5782. [Google Scholar]
- Shaw, T.; Locarnini, S.A. Hepatic purine and pyrimidine metabolism: Implications for antiviral chemotherapy of viral hepatitis. Liver 1995, 15, 169–184. [Google Scholar]
- Okesli, A.; Khosla, C.; Bassik, M.C. Human pyrimidine nucleotide biosynthesis as a target for antiviral chemotherapy. Curr. Opin. Biotechnol. 2017, 48, 127–134. [Google Scholar]
- Hyde, J.E. Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr. Drug Targets 2007, 8, 31–47. [Google Scholar]
- Kim, J.N.; Blount, K.F.; Puskarz, I.; Lim, J.; Link, K.H.; Breaker, R.R. Design and antimicrobial action of purine analogues that bind Guanine riboswitches. ACS Chem. Biol. 2009, 4, 915–927. [Google Scholar]
- Abou-Ghadir, O.F.; Hayallah, A.M.; Abdel-Moty, S.G.; Hussein, M.A. Design and synthesis of some new purine-dione derivatives of potential anti-inflammatory activity. Der Pharma Chem. 2014, 6, 199–211. [Google Scholar]
- Naik, N.S.; Shastri, L.A.; Chougala, B.M.; Samundeeswari, S.; Holiyachi, M.; Joshi, S.D.; Sunagar, V. Synthesis of novel aryl and coumarin substituted pyrazolo [1, 5-a] pyrimidine derivatives as potent anti-inflammatory and anticancer agents. Chem. Data Collect. 2020, 30, 100550. [Google Scholar]
- Rodrigues, D.; Jerónimo, C.; Henrique, R.; Belo, L.; de Lourdes Bastos, M.; de Pinho, P.G.; Carvalho, M. Biomarkers in bladder cancer: A metabolomic approach using in vitro and ex vivo model systems. Int. J. Cancer 2016, 139, 256–268. [Google Scholar]
- Halliwell, B. Why and how should we measure oxidative DNA damage in nutritional studies? How far have we come? Am. J. Clin. Nutr. 2000, 72, 1082–1087. [Google Scholar]
- Margolin Eren, K.J.; Elkabets, O.; Amirav, A. A comparison of electron ionization mass spectra obtained at 70 eV, low electron energies, and with cold EI and their NIST library identification probabilities. J. Mass Spectrom. 2020, 55, e4646. [Google Scholar]
- Ausloos, P.; Clifton, C.; Lias, S.; Mikaya, A.; Stein, S.E.; Tchekhovskoi, D.V.; Sparkman, O.; Zaikin, V.; Zhu, D. The critical evaluation of a comprehensive mass spectral library. J. Am. Soc. Mass Spectrom. 1999, 10, 287–299. [Google Scholar]
- Stein, S.E.; Scott, D.R. Optimization and testing of mass spectral library search algorithms for compound identification. J. Am. Soc. Mass Spectrom. 1994, 5, 859–866. [Google Scholar]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev. 2018, 37, 513–532. [Google Scholar] [PubMed]
- Kerber, A.; Laue, R.; Meringer, M.; Varmuza, K. MOLGEN-MS: Evaluation of low resolution electron impact mass spectra with MS classification and exhaustive structure generation. Adv. Mass Spectrom. 2001, 15, 22. [Google Scholar]
- Kind, T.; Liu, K.-H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar]
- Zhou, J.; Weber, R.J.; Allwood, J.W.; Mistrik, R.; Zhu, Z.; Ji, Z.; Chen, S.; Dunn, W.B.; He, S.; Viant, M.R. HAMMER: Automated operation of mass frontier to construct in silico mass spectral fragmentation libraries. Bioinformatics 2014, 30, 581–583. [Google Scholar]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [PubMed] [Green Version]
- Allen, F.; Pon, A.; Greiner, R.; Wishart, D. Computational prediction of electron ionization mass spectra to assist in GC/MS compound identification. Analyt. Chem. 2016, 88, 7689–7697. [Google Scholar]
- Lourderaj, U.; Sun, R.; Kohale, S.C.; Barnes, G.L.; De Jong, W.A.; Windus, T.L.; Hase, W.L. The VENUS/NWChem software package. Tight coupling between chemical dynamics simulations and electronic structure theory. Comput. Phys. Commun. 2014, 185, 1074–1080. [Google Scholar]
- Grimme, S. Towards first principles calculation of electron impact mass spectra of molecules. Angew. Chem. Int. Ed. 2013, 52, 6306–6312. [Google Scholar]
- Cauët, E.; Vanhaegenborgh, Y.J.; De Proft, F.; Geerlings, P. Conceptual and Computational DFT-based In Silico Fragmentation Method for the Identification of Metabolite Mass Spectra. Chemistry-Methods 2021, 1, 101–115. [Google Scholar]
- Cautereels, J.; Van Hee, N.; Chatterjee, S.; Van Alsenoy, C.; Lemière, F.; Blockhuys, F. QCMS2 as a new method for providing insight into peptide fragmentation: The influence of the side-chain and inter–side-chain interactions. J. Mass Spectrom. 2020, 55, e4446. [Google Scholar]
- Koopman, J.; Grimme, S. From QCEIMS to QCxMS: A Tool to Routinely Calculate CID Mass Spectra Using Molecular Dynamics. J. Am. Soc. Mass Spectrom. 2021, 32, 1735–1751. [Google Scholar] [PubMed]
- Bauer, C.A.; Grimme, S. Automated quantum chemistry based molecular dynamics simulations of electron ionization induced fragmentations of the nucleobases uracil, thymine, cytosine, and guanine. Eur. J.Mass Spectrom. 2015, 21, 125–140. [Google Scholar]
- Ásgeirsson, V.; Bauer, C.A.; Grimme, S. Quantum chemical calculation of electron ionization mass spectra for general organic and inorganic molecules. Chem. Sci. 2017, 8, 4879–4895. [Google Scholar] [PubMed] [Green Version]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [PubMed] [Green Version]
- Bauer, C.A.; Grimme, S. Elucidation of electron ionization induced fragmentations of adenine by semiempirical and density functional molecular dynamics. J. Phys. Chem. A 2014, 118, 11479–11484. [Google Scholar] [PubMed]
- Koopman, J.; Grimme, S. Calculation of electron ionization mass spectra with semiempirical GFNn-xTB methods. ACS Omega 2019, 4, 15120–15133. [Google Scholar] [PubMed] [Green Version]
- Wang, S.; Kind, T.; Tantillo, D.J.; Fiehn, O. Predicting in silico electron ionization mass spectra using quantum chemistry. J. Cheminform. 2020, 12, 63. [Google Scholar] [PubMed]
- Spackman, P.R.; Bohman, B.; Karton, A.; Jayatilaka, D. Quantum chemical electron impact mass spectrum prediction for de novo structure elucidation: Assessment against experimental reference data and comparison to competitive fragmentation modeling. Int. J. Quant. Chem. 2018, 118, e25460. [Google Scholar]
- Schreckenbach, S.A.; Anderson, J.S.; Koopman, J.; Grimme, S.; Simpson, M.J.; Jobst, K.J. Predicting the Mass Spectra of Environmental Pollutants Using Computational Chemistry: A Case Study and Critical Evaluation. J. Am. Soc. Mass Spectrom. 2021, 32, 1508–1518. [Google Scholar] [PubMed]
- Nedyalkova, M.A.; Madurga, S.; Tobiszewski, M.; Simeonov, V. Calculating the partition coefficients of organic solvents in octanol/water and octanol/air. J. Chem. Inform. Model. 2019, 59, 2257–2263. [Google Scholar]
- Thiel, W. Semiempirical quantum–chemical methods. Wiley Interdiscipl. Rev. Comput. Mol. Sci. 2014, 4, 145–157. [Google Scholar]
- Warth, B.; Spangler, S.; Fang, M.; Johnson, C.H.; Forsberg, E.M.; Granados, A.; Martin, R.L.; Domingo-Almenara, X.; Huan, T.; Rinehart, D. Exposome-scale investigations guided by global metabolomics, pathway analysis, and cognitive computing. Anal. Chem. 2017, 89, 11505–11513. [Google Scholar]
- Yamamoto, H.; Sasaki, K. Metabolomics-based approach for ranking the candidate structures of unidentified peaks in capillary electrophoresis time-of-flight mass spectrometry. Electrophoresis 2017, 38, 1053–1059. [Google Scholar]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2018, 15, 53–56. [Google Scholar]
- Kenani, A.; Bernier, J.L.; Henichart, J.P. Synthesis and EIMS fragmentation analysis of [1, 3-15N2] xanthine and [1, 3-15N2] caffeine. J. Labell. Compd. Radiopharm. 1995, 36, 187–192. [Google Scholar]
- Zeeck, E.; Harder, T.; Beckmann, M. Uric acid: The sperm-release pheromone of the marine polychaete Platynereis dumerilii. J. Chem. Ecol. 1998, 24, 13–22. [Google Scholar]
- Rice, J.M.; Dudek, G.O. Mass spectra of nucleic acid derivatives. II. Guanine, adenine, and related compounds. J. Am. Chem. Soc. 1967, 89, 2719–2725. [Google Scholar]
- Bonini, P.; Kind, T.; Tsugawa, H.; Barupal, D.K.; Fiehn, O. Retip: Retention time prediction for compound annotation in untargeted metabolomics. Anal. Chem. 2020, 92, 7515–7522. [Google Scholar] [PubMed]
- Blaženović, I.; Kind, T.; Torbašinović, H.; Obrenović, S.; Mehta, S.S.; Tsugawa, H.; Wermuth, T.; Schauer, N.; Jahn, M.; Biedendieck, R. Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: Database boosting is needed to achieve 93% accuracy. J. Cheminform. 2017, 9, 32. [Google Scholar] [PubMed] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 17. [Google Scholar]
- Dewar, M.J.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar]
- Weber, W.; Thiel, W. Orthogonalization corrections for semiempirical methods. Theor. Chem. Accounts 2000, 103, 495–506. [Google Scholar]
- Neese, F. The ORCA program system. Wiley Interdiscipl. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top 1 | Top 2 | Top 3 | Top 4 | Top 5 | Top 10 | Top 100 | Out of Range | Total | |
|---|---|---|---|---|---|---|---|---|---|
| Cumulative Number of Molecules | 36 | 45 | 46 | 49 | 52 | 59 | 69 | 11 | 80 |
| Cumulative Percentage | 45% | 56% | 58% | 61% | 65% | 74% | 86% | 14% | 100% |
| Cumulative Wdot Average | 713 | 711 | 702 | 702 | 701 | 712 | 708 | 326 | 656 |
| Mol ID | Molecular Weight | Experimental Abundance (%) | QCEIMS Abundance (%) | Abundance Difference (%) | Average Difference (%) | |
|---|---|---|---|---|---|---|
| Pyrimidines | 5 | 126 | 82 | 32 | −50 | 30.3% |
| 16 | 140 | 64 | 98 | +34 | ||
| 17 | 140 | 100 | 71 | −29 | ||
| 19 | 141 | 100 | 91 | −9 | ||
| 41 | 154 | 100 | 65 | −35 | ||
| 42 | 154 | 80 | 57 | −23 | ||
| 43 | 155 | 100 | 97 | −3 | ||
| 81 | 170 | 100 | 41 | −59 | ||
| Xanthines | 39 | 153 | 94 | 100 | +6 | 39.8% |
| 75 | 168 | 27 | 100 | +73 | ||
| 100 | 180 | 100 | 57 | −43 | ||
| 106 | 181 | 25 | 67 | +42 | ||
| 132 | 195 | 14 | 49 | +35 | ||
| Hypoxanthines | 28 | 150 | 100 | 83 | −17 | 30% |
| 31 | 150 | 70 | 100 | +30 | ||
| 52 | 164 | 100 | 57 | −43 | ||
| Lumazines | 65 | 166 | 2 | 100 | +98 | 42% |
| 96 | 180 | 100 | 64 | −36 | ||
| 121 | 192 | 100 | 79 | −21 | ||
| 128 | 194 | 100 | 87 | −13 | ||
| Quinazoline- diones | 86 | 177 | 60 | 26 | −34 | 19.7% |
| 115 | 190 | 100 | 87 | −13 | ||
| 120 | 192 | 100 | 88 | −12 | ||
| Remycins | 92 | 179 | 66 | 19 | −47 | 37.5% |
| 105 | 181 | 1 | 49 | +48 | ||
| 123 | 193 | 24 | 40 | +16 | ||
| 125 | 193 | 63 | 27 | −36 | ||
| 126 | 193 | 100 | 31 | −69 | ||
| 147 | 207 | 14 | 23 | +9 | ||
| Pyridopyrimidine-(1H,3H)-diones | 47 | 163 | 100 | 82 | −18 | 20.4% |
| 50 | 163 | 88 | 100 | +12 | ||
| 116 | 191 | 100 | 84 | −16 | ||
| 117 | 191 | 100 | 54 | −46 | ||
| 118 | 191 | 100 | 90 | −10 | ||
| Others | 70 | 167 | 11 | 89 | +78 | 62.3% |
| 74 | 168 | 100 | 59 | −41 | ||
| 80 | 170 | 100 | 21 | −79 | ||
| 122 | 192 | 49 | 100 | −51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Kind, T.; Tantillo, D.J.; Wang, L.-P.; Fiehn, O. Evaluating the Accuracy of the QCEIMS Approach for Computational Prediction of Electron Ionization Mass Spectra of Purines and Pyrimidines. Metabolites 2022, 12, 68. https://doi.org/10.3390/metabo12010068
Lee J, Kind T, Tantillo DJ, Wang L-P, Fiehn O. Evaluating the Accuracy of the QCEIMS Approach for Computational Prediction of Electron Ionization Mass Spectra of Purines and Pyrimidines. Metabolites. 2022; 12(1):68. https://doi.org/10.3390/metabo12010068
Chicago/Turabian StyleLee, Jesi, Tobias Kind, Dean Joseph Tantillo, Lee-Ping Wang, and Oliver Fiehn. 2022. "Evaluating the Accuracy of the QCEIMS Approach for Computational Prediction of Electron Ionization Mass Spectra of Purines and Pyrimidines" Metabolites 12, no. 1: 68. https://doi.org/10.3390/metabo12010068
APA StyleLee, J., Kind, T., Tantillo, D. J., Wang, L.-P., & Fiehn, O. (2022). Evaluating the Accuracy of the QCEIMS Approach for Computational Prediction of Electron Ionization Mass Spectra of Purines and Pyrimidines. Metabolites, 12(1), 68. https://doi.org/10.3390/metabo12010068