
The Role of Gut Microbiota and Its Produced Metabolites in Obesity, Dyslipidemia, Adipocyte Dysfunction, and Its Interventions


Abstract
1. Introduction
2. Obesity and Dyslipidemia
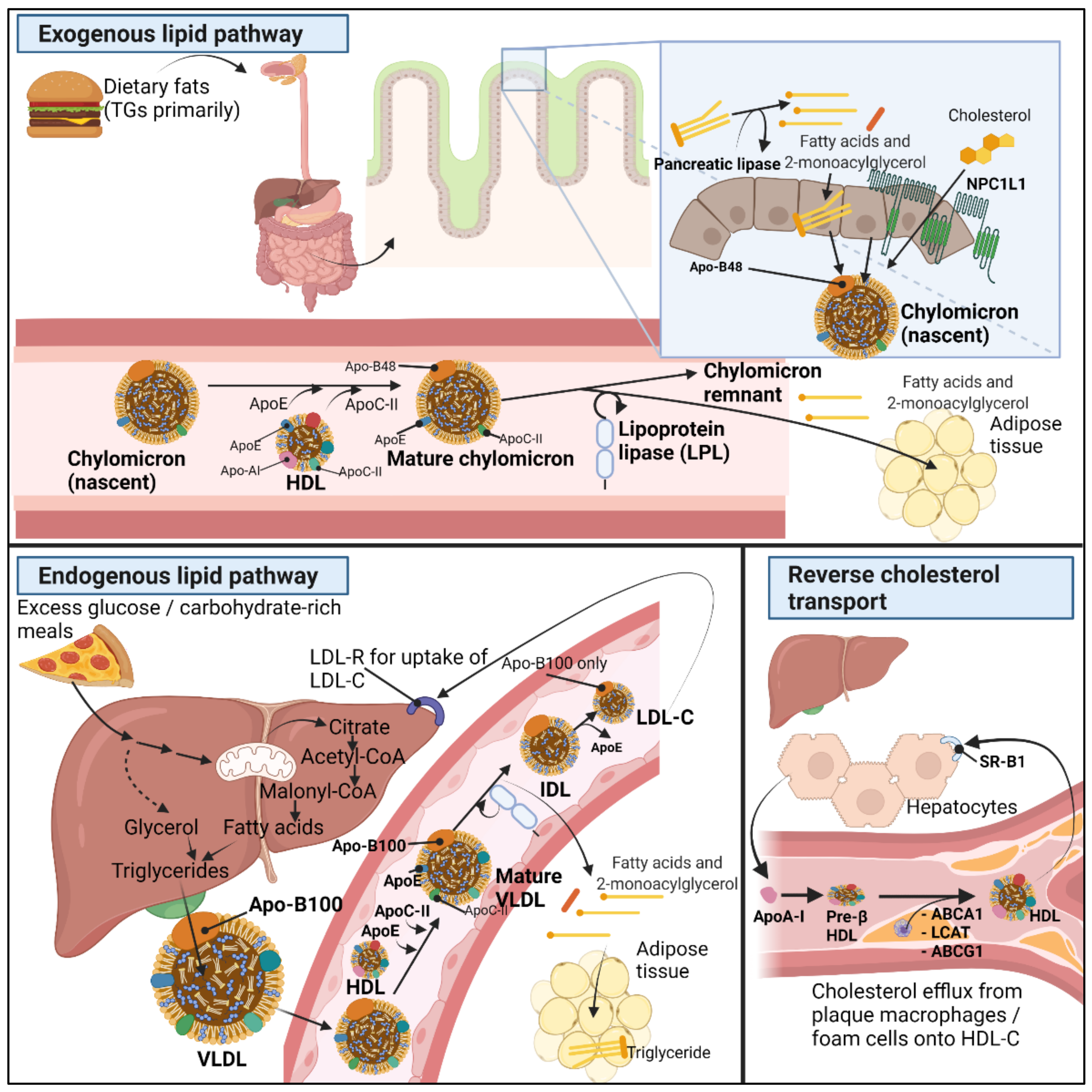
2.1. Normal Lipid Metabolism
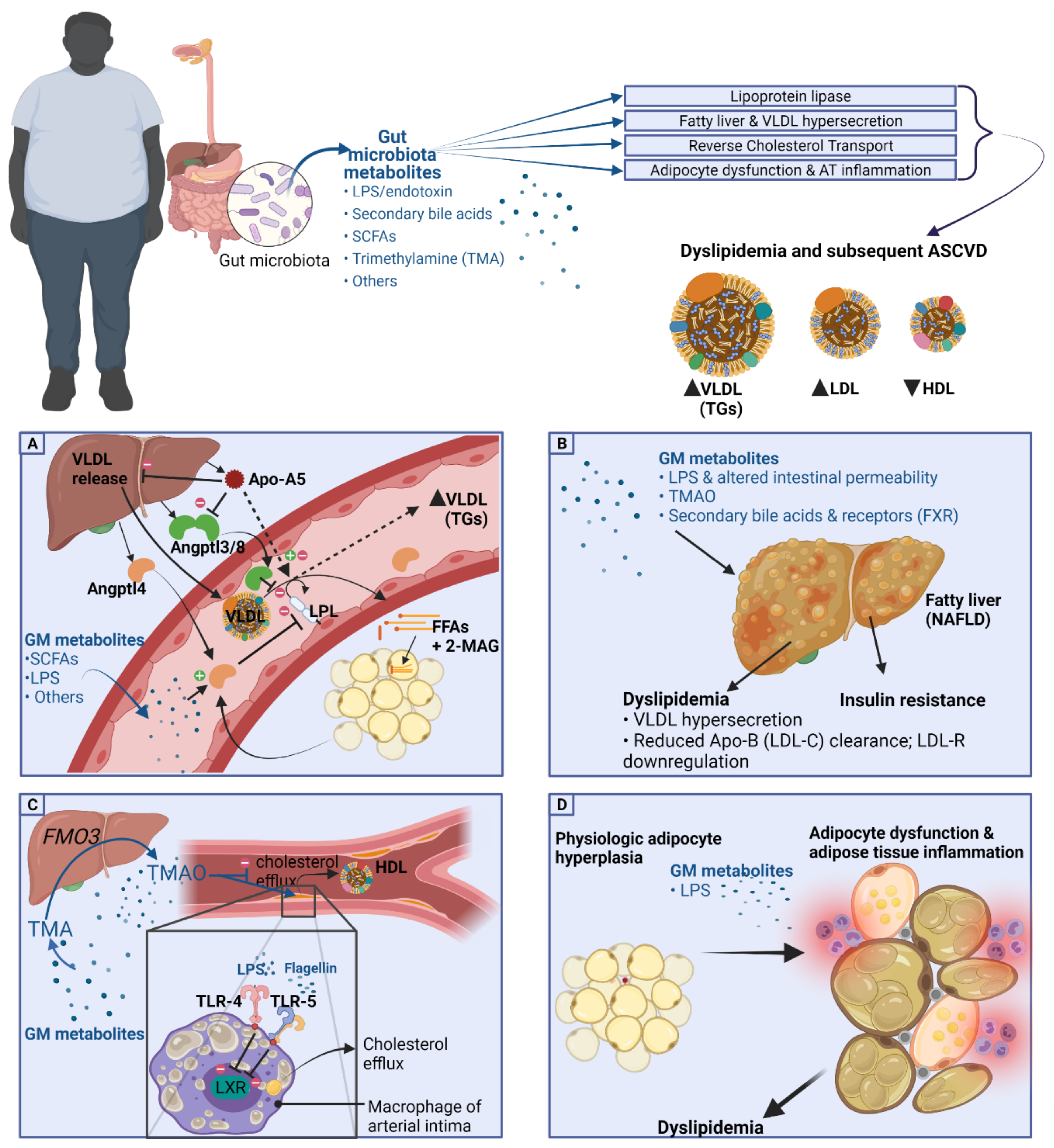
2.2. Basic Pathogenetic Mechanisms of Dyslipidemia in the Obese Population
3. Gut Microbiota Composition in Obesity and Dyslipidemia
3.1. Gut Microbiota Composition and Dyslipidemia
3.2. Coprostanol
4. Gut-Microbiota-Derived Metabolites in Dyslipidemia and Obesity
4.1. Lipoprotein Lipase and GM Metabolites
4.1.1. Angptl4 and Angptl3
4.1.2. Apo-A5
4.2. Fatty Liver and VLDL Hypersecretion Association with GM Metabolites
4.3. HDL-C, Reverse Cholesterol Transport, and Cholesterol Transfer (CETP) Association with GM Metabolites
4.4. Gut Microbiota and Bile Acids in Dyslipidemia
4.4.1. FXR
4.4.2. GPBAR-1
4.5. Short Chain Fatty Acids and Dyslipidemia
4.6. TMAO and Lipid Metabolism
5. Adipocyte Dysfunction and Gut Metabolites
5.1. GM Metabolites and Adipose Tissue Dysfunction
5.2. GM Metabolites in Adipose Tissue Inflammation
6. Gut Microbiota Metabolites in Dietary, Weight Loss, and Pharmacologic Interventions
6.1. Dietary Interventions and Gut Microbiota
6.2. Weight Loss Surgery and Gut Microbiota
6.3. Lipid Lowering Agents and GM
6.4. Supplementation and GM
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 6 January 2021).
- World Health Organization. Noncommunicable Disease Risk Factors. In World Health Statistics 2021: Monitoring Health for the SDGs, Sustainable Development Goals; World Health Statistics; World Health Organization: Geneva, Switzerland, 2021; pp. 38–39. ISBN 978-92-4-002706-0. [Google Scholar]
- Seidell, J.C.; Halberstadt, J. The Global Burden of Obesity and the Challenges of Prevention. Ann. Nutr. Metab. 2015, 66, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Finer, N. Medical Consequences of Obesity. Medicine 2015, 43, 88–93. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-Density Lipoproteins Cause Atherosclerotic Cardiovascular Disease. 1. Evidence from Genetic, Epidemiologic, and Clinical Studies. A Consensus Statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; Backer, G.G.D.; Delgado, V.; Ference, B.A.; Graham, I.M.; et al. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Atherosclerosis 2020, 290, 140–205. [Google Scholar] [CrossRef]
- Kopin, L.; Lowenstein, C.J. Dyslipidemia. Ann. Intern. Med. 2017, 167, ITC81. [Google Scholar] [CrossRef]
- Townsend, N.; Wilson, L.; Bhatnagar, P.; Wickramasinghe, K.; Rayner, M.; Nichols, M. Cardiovascular Disease in Europe: Epidemiological Update 2016. Eur. Heart J. 2016, 37, 3232–3245. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Mittendorfer, B.; Klein, S. Metabolically Healthy Obesity: Facts and Fantasies. J. Clin. Investig. 2019, 129, 3978–3989. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Warmbrunn, M.V.; Herrema, H.; Aron-Wisnewsky, J.; Soeters, M.R.; Van Raalte, D.H.; Nieuwdorp, M. Gut Microbiota: A Promising Target against Cardiometabolic Diseases. Expert Rev. Endocrinol. Metab. 2020, 15, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Helkin, A.; Stein, J.J.; Lin, S.; Siddiqui, S.; Maier, K.G.; Gahtan, V. Dyslipidemia Part 1—Review of Lipid Metabolism and Vascular Cell Physiology. Vasc. Endovasc. Surg. 2016, 50, 107–118. [Google Scholar] [CrossRef]
- Kindel, T.; Lee, D.M.; Tso, P. The Mechanism of the Formation and Secretion of Chylomicrons. Atheroscler. Suppl. 2010, 11, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Stahel, P.; Nahmias, A.; Lewis, G.F. Emerging Role of Lymphatics in the Regulation of Intestinal Lipid Mobilization. Front. Physiol. 2019, 10, 1604. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.W.; Davis, H.R.; Zhu, L.-J.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.N.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 Protein Is Critical for Intestinal Cholesterol Absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef]
- Cooper, A.D. Hepatic Uptake of Chylomicron Remnants. J. Lipid Res. 1997, 38, 2173–2192. [Google Scholar] [CrossRef]
- Schönfeld, P.; Wojtczak, L. Short- and Medium-Chain Fatty Acids in Energy Metabolism: The Cellular Perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef]
- Sanders, F.W.B.; Griffin, J.L. De Novo Lipogenesis in the Liver in Health and Disease: More than Just a Shunting Yard for Glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De Novo Fatty-Acid Synthesis and Related Pathways as Molecular Targets for Cancer Therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef]
- Foster, D.W. Malonyl-CoA: The Regulator of Fatty Acid Synthesis and Oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP Pathway: Regulation of Cholesterol Metabolism by Proteolysis of a Membrane-Bound Transcription Factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Sparks, J.D.; Sparks, C.E.; Adeli, K. Selective Hepatic Insulin Resistance, VLDL Overproduction, and Hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2104–2112. [Google Scholar] [CrossRef]
- Wang, H.; Eckel, R.H. Lipoprotein Lipase: From Gene to Obesity. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E271–E288. [Google Scholar] [CrossRef] [PubMed]
- Wolska, A.; Dunbar, R.L.; Freeman, L.A.; Ueda, M.; Amar, M.J.; Sviridov, D.O.; Remaley, A.T. Apolipoprotein C-II: New Findings Related to Genetics, Biochemistry, and Role in Triglyceride Metabolism. Atherosclerosis 2017, 267, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Kei, A.A.; Filippatos, T.D.; Tsimihodimos, V.; Elisaf, M.S. A Review of the Role of Apolipoprotein C-II in Lipoprotein Metabolism and Cardiovascular Disease. Metabolism 2012, 61, 906–921. [Google Scholar] [CrossRef]
- Sadur, C.N.; Eckel, R.H. Insulin Stimulation of Adipose Tissue Lipoprotein Lipase. Use of the Euglycemic Clamp Technique. J. Clin. Investig. 1982, 69, 1119–1125. [Google Scholar] [CrossRef]
- Tall, A.R. Plasma Cholesteryl Ester Transfer Protein. J. Lipid Res. 1993, 34, 1255–1274. [Google Scholar] [CrossRef]
- Barter, P.J.; Brewer, H.B.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl Ester Transfer Protein. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 160–167. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.-C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol Efflux and Atheroprotection: Advancing the Concept of Reverse Cholesterol Transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L.; Terasaka, N.; Pagler, T.; Wang, N. HDL, ABC Transporters, and Cholesterol Efflux: Implications for the Treatment of Atherosclerosis. Cell Metab. 2008, 7, 365–375. [Google Scholar] [CrossRef]
- März, W.; Kleber, M.E.; Scharnagl, H.; Speer, T.; Zewinger, S.; Ritsch, A.; Parhofer, K.G.; von Eckardstein, A.; Landmesser, U.; Laufs, U. HDL Cholesterol: Reappraisal of Its Clinical Relevance. Clin. Res. Cardiol. 2017, 106, 663–675. [Google Scholar] [CrossRef]
- Gillard, B.K.; Rosales, C.; Xu, B.; Gotto, A.M.; Pownall, H.J. Rethinking Reverse Cholesterol Transport and Dysfunctional High-Density Lipoproteins. J. Clin. Lipidol. 2018, 12, 849–856. [Google Scholar] [CrossRef]
- Zannis, V.I.; Chroni, A.; Krieger, M. Role of ApoA-I, ABCA1, LCAT, and SR-BI in the Biogenesis of HDL. J. Mol. Med. 2006, 84, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Toth, P.P.; Kris-Etherton, P.M.; Abate, N.; Aronne, L.J.; Brown, W.V.; Gonzalez-Campoy, J.M.; Jones, S.R.; Kumar, R.; La Forge, R.; et al. Obesity, Adiposity, and Dyslipidemia: A Consensus Statement from the National Lipid Association. J. Clin. Lipidol. 2013, 7, 304–383. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E. “Sick Fat,” Metabolic Disease, and Atherosclerosis. Am. J. Med. 2009, 122, S26–S37. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Häring, H.-U.; Hu, F.B.; Schulze, M.B. Metabolically Healthy Obesity: Epidemiology, Mechanisms, and Clinical Implications. Lancet Diabetes Endocrinol. 2013, 1, 152–162. [Google Scholar] [CrossRef]
- Lorenzo, A.D. New Obesity Classification Criteria as a Tool for Bariatric Surgery Indication. WJG 2016, 22, 681. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, A.S.; Tousoulis, D. The Molecular Mechanisms of Obesity Paradox. Cardiovasc. Res. 2017, 113, 1074–1086. [Google Scholar] [CrossRef]
- Mathew, H.; Farr, O.M.; Mantzoros, C.S. Metabolic Health and Weight: Understanding Metabolically Unhealthy Normal Weight or Metabolically Healthy Obese Patients. Metabolism 2016, 65, 73–80. [Google Scholar] [CrossRef]
- Wildman, R.P.; Muntner, P.; Reynolds, K.; McGinn, A.P.; Rajpathak, S.; Wylie-Rosett, J.; Sowers, M.R. The Obese without Cardiometabolic Risk Factor Clustering and the Normal Weight with Cardiometabolic Risk Factor Clustering: Prevalence and Correlates of 2 Phenotypes among the US Population (NHANES 1999-2004). Arch. Intern. Med. 2008, 168, 1617–1624. [Google Scholar] [CrossRef]
- Blüher, M. Mechanisms in endocrinology: Are Metabolically Healthy Obese Individuals Really Healthy? Eur. J. Endocrinol. 2014, 171, R209–R219. [Google Scholar] [CrossRef]
- Hubert, H.B.; Feinleib, M.; McNamara, P.M.; Castelli, W.P. Obesity as an Independent Risk Factor for Cardiovascular Disease: A 26-Year Follow-up of Participants in the Framingham Heart Study. Circulation 1983, 67, 968–977. [Google Scholar] [CrossRef]
- Kramer, C.K.; Zinman, B.; Retnakaran, R. Are Metabolically Healthy Overweight and Obesity Benign Conditions?: A Systematic Review and Meta-Analysis. Ann. Intern. Med. 2013, 159, 758. [Google Scholar] [CrossRef]
- Caleyachetty, R.; Thomas, G.N.; Toulis, K.A.; Mohammed, N.; Gokhale, K.M.; Balachandran, K.; Nirantharakumar, K. Metabolically Healthy Obese and Incident Cardiovascular Disease Events Among 3.5 Million Men and Women. J. Am. Coll. Cardiol. 2017, 70, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Appleton, S.L.; Seaborn, C.J.; Visvanathan, R.; Hill, C.L.; Gill, T.K.; Taylor, A.W.; Adams, R.J. North West Adelaide Health Study Team Diabetes and Cardiovascular Disease Outcomes in the Metabolically Healthy Obese Phenotype: A Cohort Study. Diabetes Care 2013, 36, 2388–2394. [Google Scholar] [CrossRef] [PubMed]
- Klop, B.; Elte, J.W.F.; Castro Cabezas, M. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D.; Bartha, P.; Zinder, O.; Kerner, A.; Markiewicz, W.; Avizohar, O.; Brook, G.J.; Levy, Y. Obesity Is the Major Determinant of Elevated C-Reactive Protein in Subjects with the Metabolic Syndrome. Int. J. Obes. 2004, 28, 674–679. [Google Scholar] [CrossRef]
- Rader, D.J.; Kathiresan, S. Disorders of Lipoprotein Metabolism. In Harrison’s Principles of Internal Medicine, 20th ed.; McGraw-Hill Education: New York, NY, USA, 2018; Volume 2, pp. 2889–2902. ISBN 978-1-259-64404-7. [Google Scholar]
- Björntorp, P.; Bergman, H.; Varnauskas, E. Plasma Free Fatty Acid Turnover Rate in obesity. Acta Med. Scand. 1969, 185, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Rydén, M. Fatty Acids, Obesity and Insulin Resistance. Obes. Facts 2015, 8, 147–155. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The Metabolic Syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and Nonalcoholic Fatty Liver Disease: Biochemical, Metabolic, and Clinical Implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Chatrath, H.; Vuppalanchi, R.; Chalasani, N. Dyslipidemia in Patients with Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2012, 32, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Safari, Z.; Gérard, P. The Links between the Gut Microbiome and Non-Alcoholic Fatty Liver Disease (NAFLD). Cell. Mol. Life Sci. 2019, 76, 1541–1558. [Google Scholar] [CrossRef]
- Kersten, S. Physiological Regulation of Lipoprotein Lipase. Biochim. Et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 919–933. [Google Scholar] [CrossRef]
- Nilsson-Ehle, P. Impaired Regulation of Adipose Tissue Lipoprotein Lipase in Obesity. Int. J. Obes. 1981, 5, 695–699. [Google Scholar]
- Arai, T.; Yamashita, S.; Hirano, K.; Sakai, N.; Kotani, K.; Fujioka, S.; Nozaki, S.; Keno, Y.; Yamane, M.; Shinohara, E. Increased Plasma Cholesteryl Ester Transfer Protein in Obese Subjects. A Possible Mechanism for the Reduction of Serum HDL Cholesterol Levels in Obesity. Arter. Thromb. 1994, 14, 1129–1136. [Google Scholar] [CrossRef]
- MacLean, P.S.; Vadlamudi, S.; MacDonald, K.G.; Pories, W.J.; Barakat, H.A. Suppression of Hepatic Cholesteryl Ester Transfer Protein Expression in Obese Humans with the Development of Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2005, 90, 2250–2258. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The Gut Microbiota as an Environmental Factor That Regulates Fat Storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.; Gasbarrini, A.; Mele, M. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Leeming, E.R.; Johnson, A.J.; Spector, T.D.; Le Roy, C.I. Effect of Diet on the Gut Microbiota: Rethinking Intervention Duration. Nutrients 2019, 11, 2862. [Google Scholar] [CrossRef]
- Nicolas, S.; Blasco-Baque, V.; Fournel, A.; Gilleron, J.; Klopp, P.; Waget, A.; Ceppo, F.; Marlin, A.; Padmanabhan, R.; Iacovoni, J.S.; et al. Transfer of Dysbiotic Gut Microbiota Has Beneficial Effects on Host Liver Metabolism. Mol. Syst. Biol. 2017, 13, 921. [Google Scholar] [CrossRef]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-Balance Studies Reveal Associations between Gut Microbes, Caloric Load, and Nutrient Absorption in Humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut Dysbiosis Is Linked to Hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human Gut Microbes Associated with Obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Bäckhed, F.; Landmesser, U.; Hazen, S.L. Intestinal Microbiota in Cardiovascular Health and Disease. J. Am. Coll. Cardiol. 2019, 73, 2089–2105. [Google Scholar] [CrossRef]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.M.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo, C.; Cuevas, A.; Zambrano, T.; Acuña, J.J.; Jorquera, M.A.; Saavedra, K.; Martínez, C.; Lanas, F.; Serón, P.; Salazar, L.A.; et al. Bacterial Community Profile of the Gut Microbiota Differs between Hypercholesterolemic Subjects and Controls. Biomed. Res. Int. 2017, 2017, 8127814. [Google Scholar] [CrossRef] [PubMed]
- Vojinovic, D.; Radjabzadeh, D.; Kurilshikov, A.; Amin, N.; Wijmenga, C.; Franke, L.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Relationship between Gut Microbiota and Circulating Metabolites in Population-Based Cohorts. Nat. Commun. 2019, 10, 5813. [Google Scholar] [CrossRef]
- Le Roy, T.; Lécuyer, E.; Chassaing, B.; Rhimi, M.; Lhomme, M.; Boudebbouze, S.; Ichou, F.; Haro Barceló, J.; Huby, T.; Guerin, M.; et al. The Intestinal Microbiota Regulates Host Cholesterol Homeostasis. BMC Biol. 2019, 17, 94. [Google Scholar] [CrossRef]
- Yun, K.E.; Kim, J.; Kim, M.; Park, E.; Kim, H.-L.; Chang, Y.; Ryu, S.; Kim, H.-N. Major Lipids, Apolipoproteins, and Alterations of Gut Microbiota. JCM 2020, 9, 1589. [Google Scholar] [CrossRef]
- Dam, H. The Formation of Coprosterol in the Intestine1: The Action of Intestinal Bacteria on Cholesterol. Biochem. J. 1934, 28, 820–825. [Google Scholar] [CrossRef]
- Veiga, P.; Juste, C.; Lepercq, P.; Saunier, K.; Béguet, F.; Gérard, P. Correlation between Faecal Microbial Community Structure and Cholesterol-to-Coprostanol Conversion in the Human Gut. FEMS Microbiol. Lett. 2005, 242, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Kriaa, A.; Bourgin, M.; Mkaouar, H.; Jablaoui, A.; Akermi, N.; Soussou, S.; Maguin, E.; Rhimi, M. Microbial Reduction of Cholesterol to Coprostanol: An Old Concept and New Insights. Catalysts 2019, 9, 167. [Google Scholar] [CrossRef]
- Kenny, D.J.; Plichta, D.R.; Shungin, D.; Koppel, N.; Hall, A.B.; Fu, B.; Vasan, R.S.; Shaw, S.Y.; Vlamakis, H.; Balskus, E.P.; et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 2020, 28, 245–257.e6. [Google Scholar] [CrossRef] [PubMed]
- Matysik, S.; Krautbauer, S.; Liebisch, G.; Schött, H.-F.; Kjølbaek, L.; Astrup, A.; Blachier, F.; Beaumont, M.; Nieuwdorp, M.; Hartstra, A.; et al. Short-Chain Fatty Acids and Bile Acids in Human Faeces Are Associated with the Intestinal Cholesterol Conversion Status. Br. J. Pharm. 2021, 178, 3342–3353. [Google Scholar] [CrossRef]
- Leenders, J.; Grootveld, M.; Percival, B.; Gibson, M.; Casanova, F.; Wilson, P.B. Benchtop Low-Frequency 60 MHz NMR Analysis of Urine: A Comparative Metabolomics Investigation. Metabolites 2020, 10, 155. [Google Scholar] [CrossRef] [PubMed]
- Percival, B.C.; Grootveld, M.; Gibson, M.; Osman, Y.; Molinari, M.; Jafari, F.; Sahota, T.; Martin, M.; Casanova, F.; Mather, M.L.; et al. Low-Field, Benchtop NMR Spectroscopy as a Potential Tool for Point-of-Care Diagnostics of Metabolic Conditions: Validation, Protocols and Computational Models. High-Throughput 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Pottanat, T.G.; Zhen, E.Y.; Siegel, R.W.; Ehsani, M.; Qian, Y.-W.; Konrad, R.J. ApoA5 Lowers Triglyceride Levels via Suppression of ANGPTL3/8-Mediated LPL Inhibition. J. Lipid Res. 2021, 62, 100068. [Google Scholar] [CrossRef]
- Korecka, A.; de Wouters, T.; Cultrone, A.; Lapaque, N.; Pettersson, S.; Doré, J.; Blottière, H.M.; Arulampalam, V. ANGPTL4 Expression Induced by Butyrate and Rosiglitazone in Human Intestinal Epithelial Cells Utilizes Independent Pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1025–G1037. [Google Scholar] [CrossRef] [PubMed]
- Köster, A.; Chao, Y.B.; Mosior, M.; Ford, A.; Gonzalez-DeWhitt, P.A.; Hale, J.E.; Li, D.; Qiu, Y.; Fraser, C.C.; Yang, D.D.; et al. Transgenic Angiopoietin-Like (Angptl)4 Overexpression and Targeted Disruption of Angptl4 and Angptl3: Regulation of Triglyceride Metabolism. Endocrinology 2005, 146, 4943–4950. [Google Scholar] [CrossRef]
- Mandard, S.; Zandbergen, F.; van Straten, E.; Wahli, W.; Kuipers, F.; Müller, M.; Kersten, S. The Fasting-Induced Adipose Factor/Angiopoietin-like Protein 4 Is Physically Associated with Lipoproteins and Governs Plasma Lipid Levels and Adiposity. J. Biol. Chem. 2006, 281, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Shimizugawa, T.; Ono, M.; Furukawa, H. Angiopoietin-like Protein 4 Is a Potent Hyperlipidemia-Inducing Factor in Mice and Inhibitor of Lipoprotein Lipase. J. Lipid Res. 2002, 43, 1770–1772. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, L.; Berbée, J.F.P.; van Dijk, S.J.; van Dijk, K.W.; Bensadoun, A.; Kema, I.P.; Voshol, P.J.; Müller, M.; Rensen, P.C.N.; Kersten, S. Angptl4 Upregulates Cholesterol Synthesis in Liver via Inhibition of LPL- and HL-Dependent Hepatic Cholesterol Uptake. ATVB 2007, 27, 2420–2427. [Google Scholar] [CrossRef]
- Kersten, S. Regulation of Lipid Metabolism via Angiopoietin-like Proteins. Biochem. Soc. Trans. 2005, 33, 4. [Google Scholar] [CrossRef]
- Kersten, S.; Lichtenstein, L.; Steenbergen, E.; Mudde, K.; Hendriks, H.F.J.; Hesselink, M.K.; Schrauwen, P.; Müller, M. Caloric Restriction and Exercise Increase Plasma ANGPTL4 Levels in Humans via Elevated Free Fatty Acids. ATVB 2009, 29, 969–974. [Google Scholar] [CrossRef]
- Joosen, A.M.C.P.; Bakker, A.H.F.; Kersten, S.; Westerterp, K.R. The PPARγ Ligand Rosiglitazone Influences Triacylglycerol Metabolism in Non-Obese Males, without Increasing the Transcriptional Activity of PPARγ in the Subcutaneous Adipose Tissue. Br. J. Nutr. 2008, 99, 487–493. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smart-Halajko, M.C.; Robciuc, M.R.; Cooper, J.A.; Jauhiainen, M.; Kumari, M.; Kivimaki, M.; Khaw, K.-T.; Boekholdt, S.M.; Wareham, N.J.; Gaunt, T.R.; et al. The Relationship Between Plasma Angiopoietin-like Protein 4 Levels, Angiopoietin-like Protein 4 Genotype, and Coronary Heart Disease Risk. ATVB 2010, 30, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; O’Dushlaine, C.; Gottesman, O.; Trejos, J.; Hunt, C.; Van Hout, C.V.; Habegger, L.; Buckler, D.; Lai, K.-M.V.; et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Mattijssen, F.; Alex, S.; Swarts, H.J.; Groen, A.K.; van Schothorst, E.M.; Kersten, S. Angptl4 Serves as an Endogenous Inhibitor of Intestinal Lipid Digestion. Mol. Metab. 2013, 3, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-Chain Fatty Acids Stimulate Angiopoietin-like 4 Synthesis in Human Colon Adenocarcinoma Cells by Activating Peroxisome Proliferator-Activated Receptor γ. Mol. Cell Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef]
- Blædel, T.; Holm, J.B.; Sundekilde, U.K.; Schmedes, M.S.; Hess, A.L.; Lorenzen, J.K.; Kristiansen, K.; Dalsgaard, T.K.; Astrup, A.; Larsen, L.H. A Randomised, Controlled, Crossover Study of the Effect of Diet on Angiopoietin-like Protein 4 (ANGPTL4) through Modification of the Gut Microbiome. J. Nutr. Sci. 2016, 5, e45. [Google Scholar] [CrossRef] [PubMed]
- Akoumianakis, I.; Zvintzou, E.; Kypreos, K.; Filippatos, T.D. ANGPTL3 and Apolipoprotein C-III as Novel Lipid-Lowering Targets. Curr. Atheroscler. Rep. 2021, 23, 20. [Google Scholar] [CrossRef]
- Su, X.; Peng, D. New Insights into ANGPLT3 in Controlling Lipoprotein Metabolism and Risk of Cardiovascular Diseases. Lipids Health Dis. 2018, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Moser, A.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K.R. The Acute Phase Response Stimulates the Expression of Angiopoietin like Protein 4. Biochem. Biophys. Res. Commun. 2010, 391, 1737–1741. [Google Scholar] [CrossRef]
- Guardiola, M.; Ribalta, J. Update on APOA5 Genetics: Toward a Better Understanding of Its Physiological Impact. Curr. Atheroscler. Rep. 2017, 19, 30. [Google Scholar] [CrossRef]
- Guardiola, M.; Alvaro, A.; Vallvé, J.C.; Rosales, R.; Solà, R.; Girona, J.; Serra, N.; Duran, P.; Esteve, E.; Masana, L.; et al. APOA5 Gene Expression in the Human Intestinal Tissue and Its Response to in Vitro Exposure to Fatty Acid and Fibrate. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 756–762. [Google Scholar] [CrossRef]
- Lim, M.Y.; You, H.J.; Yoon, H.S.; Kwon, B.; Lee, J.Y.; Lee, S.; Song, Y.-M.; Lee, K.; Sung, J.; Ko, G. The Effect of Heritability and Host Genetics on the Gut Microbiota and Metabolic Syndrome. Gut 2017, 66, 1031–1038. [Google Scholar] [CrossRef]
- Paul, A. Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2002, 346, 11. [Google Scholar]
- Geisler, C.E.; Renquist, B.J. Hepatic Lipid Accumulation: Cause and Consequence of Dysregulated Glucoregulatory Hormones. J. Endocrinol. 2017, 234, R1–R21. [Google Scholar] [CrossRef] [PubMed]
- Perseghin, G. Lipids in the Wrong Place: Visceral Fat and Nonalcoholic Steatohepatitis. Diabetes Care 2011, 34, S367–S370. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A Vicious Circle between Insulin Resistance and Inflammation in Nonalcoholic Fatty Liver Disease. Lipids Health Dis. 2017, 16, 203. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut Microbiota and Human NAFLD: Disentangling Microbial Signatures from Metabolic Disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Hoozemans, J.; de Brauw, M.; Nieuwdorp, M.; Gerdes, V. Gut Microbiome and Metabolites in Patients with NAFLD and after Bariatric Surgery: A Comprehensive Review. Metabolites 2021, 11, 353. [Google Scholar] [CrossRef]
- Baranowski, M. Biological Role of Liver X Receptors. J. Physiol. Pharm. 2008, 59 (Suppl. 7), 31–55. [Google Scholar]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and Toll-like Receptor Signaling Mediates Bacterial and Viral Antagonism of Cholesterol Metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef]
- Breevoort, S.R.; Angdisen, J.; Schulman, I.G. Macrophage-Independent Regulation of Reverse Cholesterol Transport by Liver X Receptors. Arter. Thromb. Vasc. Biol. 2014, 34, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal Microbiota Metabolism of L-Carnitine, a Nutrient in Red Meat, Promotes Atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef]
- Bordoni, L.; Samulak, J.J.; Sawicka, A.K.; Pelikant-Malecka, I.; Radulska, A.; Lewicki, L.; Kalinowski, L.; Gabbianelli, R.; Olek, R.A. Trimethylamine N-Oxide and the Reverse Cholesterol Transport in Cardiovascular Disease: A Cross-Sectional Study. Sci. Rep. 2020, 10, 18675. [Google Scholar] [CrossRef]
- Yiu, J.H.C.; Chan, K.-S.; Cheung, J.; Li, J.; Liu, Y.; Wang, Y.; Fung, W.W.L.; Cai, J.; Cheung, S.W.M.; Dorweiler, B.; et al. Gut Microbiota-Associated Activation of TLR5 Induces Apolipoprotein A1 Production in the Liver. Circ. Res. 2020, 127, 1236–1252. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Šarenac, T.M.; Mikov, M. Bile Acid Synthesis: From Nature to the Chemical Modification and Synthesis and Their Applications as Drugs and Nutrients. Front. Pharm. 2018, 9, 939. [Google Scholar] [CrossRef]
- Devlin, A.S.; Fischbach, M.A. A Biosynthetic Pathway for a Prominent Class of Microbiota-Derived Bile Acids. Nat. Chem. Biol. 2015, 11, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile Acids Lower Triglyceride Levels via a Pathway Involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.-C.; Staels, B. Bile Acids Induce the Expression of the Human Peroxisome Proliferator-Activated Receptor Alpha Gene via Activation of the Farnesoid X Receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Hirokane, H.; Nakahara, M.; Tachibana, S.; Shimizu, M.; Sato, R. Bile Acid Reduces the Secretion of Very Low Density Lipoprotein by Repressing Microsomal Triglyceride Transfer Protein Gene Expression Mediated by Hepatocyte Nuclear Factor-4. J. Biol. Chem. 2004, 279, 45685–45692. [Google Scholar] [CrossRef]
- Kast, H.R.; Nguyen, C.M.; Sinal, C.J.; Jones, S.A.; Laffitte, B.A.; Reue, K.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Farnesoid X-Activated Receptor Induces Apolipoprotein C-II Transcription: A Molecular Mechanism Linking Plasma Triglyceride Levels to Bile Acids. Mol. Endocrinol. 2001, 15, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted Disruption of the Nuclear Receptor FXR/BAR Impairs Bile Acid and Lipid Homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Colin, S.; Briand, O.; Touche, V.; Wouters, K.; Baron, M.; Pattou, F.; Hanf, R.; Tailleux, A.; Chinetti, G.; Staels, B.; et al. Activation of Intestinal Peroxisome Proliferator-Activated Receptor-α Increases High-Density Lipoprotein Production. Eur. Heart J. 2013, 34, 2566–2574. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X Nuclear Receptor Ligand Obeticholic Acid for Non-Cirrhotic, Non-Alcoholic Steatohepatitis (FLINT): A Multicentre, Randomised, Placebo-Controlled Trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Gege, C.; Hambruch, E.; Hambruch, N.; Kinzel, O.; Kremoser, C. Nonsteroidal FXR Ligands: Current Status and Clinical Applications. Handb. Exp. Pharm. 2019, 256, 167–205. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Di Ciaula, A.; Garruti, G.; Vacca, M.; De Angelis, M.; Wang, D.Q.-H. Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients 2020, 12, 3709. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Cupisti, K.; Ullmer, C.; Knoefel, W.T.; Kubitz, R.; Häussinger, D. The Membrane-Bound Bile Acid Receptor TGR5 Is Localized in the Epithelium of Human Gallbladders. Hepatology 2009, 50, 861–870. [Google Scholar] [CrossRef]
- Vassileva, G.; Hu, W.; Hoos, L.; Tetzloff, G.; Yang, S.; Liu, L.; Kang, L.; Davis, H.R.; Hedrick, J.A.; Lan, H.; et al. Gender-Dependent Effect of Gpbar1 Genetic Deletion on the Metabolic Profiles of Diet-Induced Obese Mice. J. Endocrinol. 2010, 205, 225–232. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short Chain Fatty Acids in Human Large Intestine, Portal, Hepatic and Venous Blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The Role of Short-Chain Fatty Acids in the Interplay between Diet, Gut Microbiota, and Host Energy Metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in Obesity and Inflammation - Protective or Causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Layden, B.T.; Angueira, A.R.; Brodsky, M.; Durai, V.; Lowe, W.L. Short Chain Fatty Acids and Their Receptors: New Metabolic Targets. Transl. Res. 2013, 161, 131–140. [Google Scholar] [CrossRef]
- Van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-Chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- Ge, H.; Li, X.; Weiszmann, J.; Wang, P.; Baribault, H.; Chen, J.-L.; Tian, H.; Li, Y. Activation of G Protein-Coupled Receptor 43 in Adipocytes Leads to Inhibition of Lipolysis and Suppression of Plasma Free Fatty Acids. Endocrinology 2008, 149, 4519–4526. [Google Scholar] [CrossRef]
- Hong, Y.-H.; Nishimura, Y.; Hishikawa, D.; Tsuzuki, H.; Miyahara, H.; Gotoh, C.; Choi, K.-C.; Feng, D.D.; Chen, C.; Lee, H.-G.; et al. Acetate and Propionate Short Chain Fatty Acids Stimulate Adipogenesis via GPCR43. Endocrinology 2005, 146, 5092–5099. [Google Scholar] [CrossRef] [PubMed]
- Jocken, J.W.E.; González Hernández, M.A.; Hoebers, N.T.H.; van der Beek, C.M.; Essers, Y.P.G.; Blaak, E.E.; Canfora, E.E. Short-Chain Fatty Acids Differentially Affect Intracellular Lipolysis in a Human White Adipocyte Model. Front. Endocrinol. 2017, 8, 372. [Google Scholar] [CrossRef]
- Hara, H.; Haga, S.; Aoyama, Y.; Kiriyama, S. Short-Chain Fatty Acids Suppress Cholesterol Synthesis in Rat Liver and Intestine. J. Nutr. 1999, 129, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.S.; Anderson, J.W.; Bridges, S.R. Propionate Inhibits Hepatocyte Lipid Synthesis. Proc. Soc. Exp. Biol. Med. 1990, 195, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Fushimi, T.; Suruga, K.; Oshima, Y.; Fukiharu, M.; Tsukamoto, Y.; Goda, T. Dietary Acetic Acid Reduces Serum Cholesterol and Triacylglycerols in Rats Fed a Cholesterol-Rich Diet. Br. J. Nutr. 2006, 95, 916–924. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, J.; Hao, W.; Zhu, H.; Liang, N.; He, Z.; Ma, K.Y.; Chen, Z.-Y. Structure-Specific Effects of Short-Chain Fatty Acids on Plasma Cholesterol Concentration in Male Syrian Hamsters. J. Agric. Food Chem. 2017, 65, 10984–10992. [Google Scholar] [CrossRef]
- Ding, L.; Chang, M.; Guo, Y.; Zhang, L.; Xue, C.; Yanagita, T.; Zhang, T.; Wang, Y. Trimethylamine-N-Oxide (TMAO)-Induced Atherosclerosis Is Associated with Bile Acid Metabolism. Lipids Health Dis. 2018, 17, 286. [Google Scholar] [CrossRef]
- Zhao, Z.-H.; Xin, F.-Z.; Zhou, D.; Xue, Y.-Q.; Liu, X.-L.; Yang, R.-X.; Pan, Q.; Fan, J.-G. Trimethylamine N-Oxide Attenuates High-Fat High-Cholesterol Diet-Induced Steatohepatitis by Reducing Hepatic Cholesterol Overload in Rats. WJG 2019, 25, 2450–2462. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Q.; Jiang, H. Gut Microbiota in Atherosclerosis: Focus on Trimethylamine N-oxide. APMIS 2020, 128, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and Metabolic Health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Liu, F.; He, J.; Wang, H.; Zhu, D.; Bi, Y. Adipose Morphology: A Critical Factor in Regulation of Human Metabolic Diseases and Adipose Tissue Dysfunction. Obes. Surg. 2020, 30, 5086–5100. [Google Scholar] [CrossRef]
- Baldini, F.; Fabbri, R.; Eberhagen, C.; Voci, A.; Portincasa, P.; Zischka, H.; Vergani, L. Adipocyte Hypertrophy Parallels Alterations of Mitochondrial Status in a Cell Model for Adipose Tissue Dysfunction in Obesity. Life Sci. 2021, 265, 118812. [Google Scholar] [CrossRef]
- DeBari, M.K.; Abbott, R.D. Adipose Tissue Fibrosis: Mechanisms, Models, and Importance. Int. J. Mol. Sci. 2020, 21, 6030. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired Adipogenesis and Dysfunctional Adipose Tissue in Human Hypertrophic Obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Guo, J.; Liu, T.; Mullen, S.; Hall, K.D.; Cushman, S.W.; Periwal, V. Hypertrophy-Driven Adipocyte Death Overwhelms Recruitment under Prolonged Weight Gain. Biophys. J. 2010, 99, 3535–3544. [Google Scholar] [CrossRef][Green Version]
- Alexopoulos, N.; Katritsis, D.; Raggi, P. Visceral Adipose Tissue as a Source of Inflammation and Promoter of Atherosclerosis. Atherosclerosis 2014, 233, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Foley, J.E.; Bogardus, C.; Tataranni, P.A.; Pratley, R.E. Enlarged Subcutaneous Abdominal Adipocyte Size, but Not Obesity Itself, Predicts Type II Diabetes Independent of Insulin Resistance. Diabetologia 2000, 43, 1498–1506. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity Induces a Phenotypic Switch in Adipose Tissue Macrophage Polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between Adipocyte Size and Adipokine Expression and Secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; González-Campoy, J.M.; Bray, G.A.; Kitabchi, A.E.; Bergman, D.A.; Schorr, A.B.; Rodbard, H.W.; Henry, R.R. Pathogenic Potential of Adipose Tissue and Metabolic Consequences of Adipocyte Hypertrophy and Increased Visceral Adiposity. Expert Rev. Cardiovasc. Ther. 2008, 6, 343–368. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, X. Effect of Lipopolysaccharides on Adipogenic Potential and Premature Senescence of Adipocyte Progenitors. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E334–E344. [Google Scholar] [CrossRef]
- Virtue, A.T.; McCright, S.J.; Wright, J.M.; Jimenez, M.T.; Mowel, W.K.; Kotzin, J.J.; Joannas, L.; Basavappa, M.G.; Spencer, S.P.; Clark, M.L.; et al. The Gut Microbiota Regulates White Adipose Tissue Inflammation and Obesity via a Family of MicroRNAs. Sci. Transl. Med. 2019, 11, eaav1892. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The Endocannabinoid System Links Gut Microbiota to Adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef]
- Khan, R.N.; Maner-Smith, K.; Owens, J.A.; Barbian, M.E.; Jones, R.M.; Naudin, C.R. At the Heart of Microbial Conversations: Endocannabinoids and the Microbiome in Cardiometabolic Risk. Gut Microbes 2021, 13, 1911572. [Google Scholar] [CrossRef]
- Laakso, M.; Kuusisto, J.; Stančáková, A.; Kuulasmaa, T.; Pajukanta, P.; Lusis, A.J.; Collins, F.S.; Mohlke, K.L.; Boehnke, M. The Metabolic Syndrome in Men Study: A Resource for Studies of Metabolic and Cardiovascular Diseases. J. Lipid Res. 2017, 58, 481–493. [Google Scholar] [CrossRef]
- Das, S.K.; Sharma, N.K.; Zhang, B. Integrative Network Analysis Reveals Different Pathophysiological Mechanisms of Insulin Resistance among Caucasians and African Americans. BMC Med. Genom. 2015, 8, 4. [Google Scholar] [CrossRef]
- Sharma, N.K.; Sajuthi, S.P.; Chou, J.W.; Calles-Escandon, J.; Demons, J.; Rogers, S.; Ma, L.; Palmer, N.D.; McWilliams, D.R.; Beal, J.; et al. Tissue-Specific and Genetic Regulation of Insulin Sensitivity-Associated Transcripts in African Americans. J. Clin. Endocrinol. Metab. 2016, 101, 1455–1468. [Google Scholar] [CrossRef]
- Schugar, R.C.; Shih, D.M.; Warrier, M.; Helsley, R.N.; Burrows, A.; Ferguson, D.; Brown, A.L.; Gromovsky, A.D.; Heine, M.; Chatterjee, A.; et al. The TMAO-Producing Enzyme Flavin-Containing Monooxygenase 3 Regulates Obesity and the Beiging of White Adipose Tissue. Cell Rep. 2017, 19, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige Adipocytes Are a Distinct Type of Thermogenic Fat Cell in Mouse and Human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Ricquier, D. Uncoupling Protein 1 of Brown Adipocytes, the Only Uncoupler: A Historical Perspective. Front. Endocrin. 2011, 2, 85. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, A.; Krause, F.N.; Moran, A.; MacCannell, A.D.V.; Scragg, J.L.; McNally, B.D.; Boateng, E.; Murfitt, S.A.; Virtue, S.; Wright, J.; et al. Brown and Beige Adipose Tissue Regulate Systemic Metabolism through a Metabolite Interorgan Signaling Axis. Nat. Commun. 2021, 12, 1905. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of Human Gut Microbiome Correlates with Metabolic Markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose Expression of Tumor Necrosis Factor-Alpha: Direct Role in Obesity-Linked Insulin Resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Ghanbari, M.; Momen Maragheh, S.; Aghazadeh, A.; Mehrjuyan, S.R.; Hussen, B.M.; Abdoli Shadbad, M.; Dastmalchi, N.; Safaralizadeh, R. Interleukin-1 in Obesity-Related Low-Grade Inflammation: From Molecular Mechanisms to Therapeutic Strategies. Int. Immunopharmacol. 2021, 96, 107765. [Google Scholar] [CrossRef]
- Højbjerre, L.; Sonne, M.P.; Alibegovic, A.C.; Nielsen, N.B.; Dela, F.; Vaag, A.; Bruun, J.M.; Stallknecht, B. Impact of Physical Inactivity on Adipose Tissue Low-Grade Inflammation in First-Degree Relatives of Type 2 Diabetic Patients. Diabetes Care 2011, 34, 2265–2272. [Google Scholar] [CrossRef]
- Otvos, J.D.; Shalaurova, I.; Wolak-Dinsmore, J.; Connelly, M.A.; Mackey, R.H.; Stein, J.H.; Tracy, R.P. GlycA: A Composite Nuclear Magnetic Resonance Biomarker of Systemic Inflammation. Clin. Chem. 2015, 61, 714–723. [Google Scholar] [CrossRef]
- Mokkala, K.; Houttu, N.; Koivuniemi, E.; Sørensen, N.; Nielsen, H.B.; Laitinen, K. GlycA, a Novel Marker for Low Grade Inflammation, Reflects Gut Microbiome Diversity and Is More Accurate than High Sensitive CRP in Reflecting Metabolomic Profile. Metabolomics 2020, 16, 76. [Google Scholar] [CrossRef]
- Silva, F.M.; de Almeida, J.C.; Feoli, A.M. Effect of Diet on Adiponectin Levels in Blood. Nutr. Rev. 2011, 69, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Diep Nguyen, T. Adiponectin: Role in Physiology and Pathophysiology. Int. J. Prev. Med. 2020, 11, 136. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Santos-Alvarez, J.; Goberna, R.; Sánchez-Margalet, V. Human Leptin Stimulates Proliferation and Activation of Human Circulating Monocytes. Cell. Immunol. 1999, 194, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Faggioni, R.; Fantuzzi, G.; Fuller, J.; Dinarello, C.A.; Feingold, K.R.; Grunfeld, C. IL-1 Beta Mediates Leptin Induction during Inflammation. Am. J. Physiol. 1998, 274, R204–R208. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Moludi, J.; Maleki, V.; Jafari-Vayghyan, H.; Vaghef-Mehrabany, E.; Alizadeh, M. Metabolic Endotoxemia and Cardiovascular Disease: A Systematic Review about Potential Roles of Prebiotics and Probiotics. Clin. Exp. Pharmacol. Physiol. 2020, 47, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated with Endotoxemia That Originates from the Gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [PubMed]
- Dalby, M.J.; Aviello, G.; Ross, A.W.; Walker, A.W.; Barrett, P.; Morgan, P.J. Diet Induced Obesity Is Independent of Metabolic Endotoxemia and TLR4 Signalling, but Markedly Increases Hypothalamic Expression of the Acute Phase Protein, SerpinA3N. Sci. Rep. 2018, 8, 15648. [Google Scholar] [CrossRef] [PubMed]
- Creely, S.J.; McTernan, P.G.; Kusminski, C.M.; Fisherff, M.; Da Silva, N.F.; Khanolkar, M.; Evans, M.; Harte, A.L.; Kumar, S. Lipopolysaccharide Activates an Innate Immune System Response in Human Adipose Tissue in Obesity and Type 2 Diabetes. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E740–E747. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; McTernan, P.G.; Harte, A.L.; da Silva, N.F.; Strazzullo, P.; Alberti, K.G.M.M.; Kumar, S.; Cappuccio, F.P. Ethnic and Sex Differences in Circulating Endotoxin Levels: A Novel Marker of Atherosclerotic and Cardiovascular Risk in a British Multi-Ethnic Population. Atherosclerosis 2009, 203, 494–502. [Google Scholar] [CrossRef]
- Murch, O.; Collin, M.; Hinds, C.J.; Thiemermann, C. Lipoproteins in Inflammation and Sepsis. I. Basic Science. Intensive Care Med. 2007, 33, 13–24. [Google Scholar] [CrossRef]
- Lassenius, M.I.; Pietiläinen, K.H.; Kaartinen, K.; Pussinen, P.J.; Syrjänen, J.; Forsblom, C.; Pörsti, I.; Rissanen, A.; Kaprio, J.; Mustonen, J.; et al. Bacterial Endotoxin Activity in Human Serum Is Associated with Dyslipidemia, Insulin Resistance, Obesity, and Chronic Inflammation. Dia Care 2011, 34, 1809–1815. [Google Scholar] [CrossRef]
- Massier, L.; Chakaroun, R.; Tabei, S.; Crane, A.; Didt, K.D.; Fallmann, J.; von Bergen, M.; Haange, S.-B.; Heyne, H.; Stumvoll, M.; et al. Adipose Tissue Derived Bacteria Are Associated with Inflammation in Obesity and Type 2 Diabetes. Gut 2020, 69, 1796–1806. [Google Scholar] [CrossRef]
- Sofi, F.; Abbate, R.; Gensini, G.F.; Casini, A. Accruing Evidence on Benefits of Adherence to the Mediterranean Diet on Health: An Updated Systematic Review and Meta-Analysis. Am. J. Clin. Nutr. 2010, 92, 1189–1196. [Google Scholar] [CrossRef]
- Ge, L.; Sadeghirad, B.; Ball, G.D.C.; da Costa, B.R.; Hitchcock, C.L.; Svendrovski, A.; Kiflen, R.; Quadri, K.; Kwon, H.Y.; Karamouzian, M.; et al. Comparison of Dietary Macronutrient Patterns of 14 Popular Named Dietary Programmes for Weight and Cardiovascular Risk Factor Reduction in Adults: Systematic Review and Network Meta-Analysis of Randomised Trials. BMJ 2020, 369, m696. [Google Scholar] [CrossRef]
- Albenberg, L.G.; Wu, G.D. Diet and the Intestinal Microbiome: Associations, Functions, and Implications for Health and Disease. Gastroenterology 2014, 146, 1564–1572. [Google Scholar] [CrossRef]
- Meslier, V.; Laiola, M.; Roager, H.M.; De Filippis, F.; Roume, H.; Quinquis, B.; Giacco, R.; Mennella, I.; Ferracane, R.; Pons, N.; et al. Mediterranean Diet Intervention in Overweight and Obese Subjects Lowers Plasma Cholesterol and Causes Changes in the Gut Microbiome and Metabolome Independently of Energy Intake. Gut 2020, 69, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Tsigalou, C.; Paraschaki, A.; Karvelas, A.; Kantartzi, K.; Gagali, K.; Tsairidis, D.; Bezirtzoglou, E. Gut Microbiome and Mediterranean Diet in the Context of Obesity. Current Knowledge, Perspectives and Potential Therapeutic Targets. Metab. Open 2021, 9, 100081. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.K.; O’Sullivan, J.M.; Plank, L.D.; Murphy, R. Altered Gut Microbiome after Bariatric Surgery and Its Association with Metabolic Benefits: A Systematic Review. Surg. Obes. Relat. Dis. 2019, 15, 656–665. [Google Scholar] [CrossRef]
- Meijnikman, A.S.; Gerdes, V.E.; Nieuwdorp, M.; Herrema, H. Evaluating Causality of Gut Microbiota in Obesity and Diabetes in Humans. Endocr. Rev. 2018, 39, 133–153. [Google Scholar] [CrossRef]
- Zlabek, J.A.; Grimm, M.S.; Larson, C.J.; Mathiason, M.A.; Lambert, P.J.; Kothari, S.N. The Effect of Laparoscopic Gastric Bypass Surgery on Dyslipidemia in Severely Obese Patients. Surg. Obes. Relat. Dis. 2005, 1, 537–542. [Google Scholar] [CrossRef]
- Spivak, H.; Sakran, N.; Dicker, D.; Rubin, M.; Raz, I.; Shohat, T.; Blumenfeld, O. Different Effects of Bariatric Surgical Procedures on Dyslipidemia: A Registry-Based Analysis. Surg. Obes. Relat. Dis. 2017, 13, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.; Kothari, S.N.; Azagury, D.E.; Morton, J.M.; Nguyen, N.T.; Jones, P.H.; Jacobson, T.A.; Cohen, D.E.; Orringer, C.; Westman, E.C.; et al. Lipids and Bariatric Procedures Part 2 of 2: Scientific Statement from the American Society for Metabolic and Bariatric Surgery (ASMBS), the National Lipid Association (NLA), and Obesity Medicine Association (OMA). Surg. Obes. Relat. Dis. 2016, 12, 468–495. [Google Scholar] [CrossRef]
- Palmisano, S.; Campisciano, G.; Silvestri, M.; Guerra, M.; Giuricin, M.; Casagranda, B.; Comar, M.; de Manzini, N. Changes in Gut Microbiota Composition after Bariatric Surgery: A New Balance to Decode. J. Gastrointest. Surg. 2020, 24, 1736–1746. [Google Scholar] [CrossRef]
- Gutiérrez-Repiso, C.; Moreno-Indias, I.; de Hollanda, A.; Martín-Núñez, G.M.; Vidal, J.; Tinahones, F.J. Gut Microbiota Specific Signatures Are Related to the Successful Rate of Bariatric Surgery. Am. J. Transl. Res. 2019, 11, 942–952. [Google Scholar] [PubMed]
- Chakaroun, R.M.; Massier, L.; Heintz-Buschart, A.; Said, N.; Fallmann, J.; Crane, A.; Schütz, T.; Dietrich, A.; Blüher, M.; Stumvoll, M.; et al. Circulating Bacterial Signature Is Linked to Metabolic Disease and Shifts with Metabolic Alleviation after Bariatric Surgery. Genome Med. 2021, 13, 105. [Google Scholar] [CrossRef]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the Evidence for the Efficacy and Safety of Statin Therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef]
- Stancu, C.; Sima, A. Statins: Mechanism of Action and Effects. J. Cell Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Baillie, R.A.; Zhu, H.; Zeng, Z.-B.; Wiest, M.M.; Nguyen, U.T.; Wojnoonski, K.; Watkins, S.M.; Trupp, M.; Krauss, R.M. Enteric Microbiome Metabolites Correlate with Response to Simvastatin Treatment. PLoS ONE 2011, 6, e25482. [Google Scholar] [CrossRef] [PubMed]
- Vieira-Silva, S.; Falony, G.; Belda, E.; Nielsen, T.; Aron-Wisnewsky, J.; Chakaroun, R.; Forslund, S.K.; Assmann, K.; Valles-Colomer, M.; Nguyen, T.T.D.; et al. Statin Therapy Is Associated with Lower Prevalence of Gut Microbiota Dysbiosis. Nature 2020, 581, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.B. Improved Gut Microbiota Profile in Individuals with Obesity Taking Statins. Nat. Rev. Cardiol. 2020, 17, 385. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.J.; Ahmed, Y.M.; Zamzami, M.A.; Siddiqui, A.M.; Khan, I.; Baothman, O.A.S.; Mehanna, M.G.; Kuerban, A.; Kaleemuddin, M.; Yasir, M. Atorvastatin Treatment Modulates the Gut Microbiota of the Hypercholesterolemic Patients. OMICS 2018, 22, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Caparrós-Martín, J.A.; Lareu, R.R.; Ramsay, J.P.; Peplies, J.; Reen, F.J.; Headlam, H.A.; Ward, N.C.; Croft, K.D.; Newsholme, P.; Hughes, J.D.; et al. Statin Therapy Causes Gut Dysbiosis in Mice through a PXR-Dependent Mechanism. Microbiome 2017, 5, 95. [Google Scholar] [CrossRef]
- Hegele, R.A. 7—Lipoprotein and Lipid Metabolism. In Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics, 7th ed.; Pyeritz, R.E., Korf, B.R., Grody, W.W., Eds.; Academic Press: London, UK, 2021; pp. 235–278. ISBN 978-0-12-812535-9. [Google Scholar]
- Kondo, T.; Kishi, M.; Fushimi, T.; Kaga, T. Acetic Acid Upregulates the Expression of Genes for Fatty Acid Oxidation Enzymes in Liver to Suppress Body Fat Accumulation. J. Agric. Food Chem. 2009, 57, 5982–5986. [Google Scholar] [CrossRef]
- Nihei, N.; Okamoto, H.; Furune, T.; Ikuta, N.; Sasaki, K.; Rimbach, G.; Yoshikawa, Y.; Terao, K. Dietary α-Cyclodextrin Modifies Gut Microbiota and Reduces Fat Accumulation in High-Fat-Diet-Fed Obese Mice. Biofactors 2018. [Google Scholar] [CrossRef]
- Huang, J.; Xiao, N.; Sun, Y.; Wu, S.; Tian, W.; Lai, Y.; Li, P.; Du, B. Supplementation of Bacillus Sp. DU-106 Reduces Hypercholesterolemia and Ameliorates Gut Dysbiosis in High-Fat Diet Rats. Appl. Microbiol. Biotechnol. 2021, 105, 287–299. [Google Scholar] [CrossRef]
- Ahn, H.Y.; Kim, M.; Chae, J.S.; Ahn, Y.-T.; Sim, J.-H.; Choi, I.-D.; Lee, S.-H.; Lee, J.H. Supplementation with Two Probiotic Strains, Lactobacillus Curvatus HY7601 and Lactobacillus Plantarum KY1032, Reduces Fasting Triglycerides and Enhances Apolipoprotein A-V Levels in Non-Diabetic Subjects with Hypertriglyceridemia. Atherosclerosis 2015, 241, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, A.S.; Brown, T.J.; Brainard, J.S.; Biswas, P.; Thorpe, G.C.; Moore, H.J.; Deane, K.H.; Summerbell, C.D.; Worthington, H.V.; Song, F.; et al. Omega-3 Fatty Acids for the Primary and Secondary Prevention of Cardiovascular Disease. Cochrane Database Syst. Rev. 2020, 3, CD003177. [Google Scholar] [CrossRef] [PubMed]
- Ferchaud-Roucher, V.; Zair, Y.; Aguesse, A.; Krempf, M.; Ouguerram, K. Omega 3 Improves Both ApoB100-Containing Lipoprotein Turnover and Their Sphingolipid Profile in Hypertriglyceridemia. J. Clin. Endocrinol. Metab. 2020, 105, dgaa459. [Google Scholar] [CrossRef]
- Kastelein, J.J.P.; Maki, K.C.; Susekov, A.; Ezhov, M.; Nordestgaard, B.G.; Machielse, B.N.; Kling, D.; Davidson, M.H. Omega-3 Free Fatty Acids for the Treatment of Severe Hypertriglyceridemia: The EpanoVa fOr Lowering Very High TriglyceridEs (EVOLVE) Trial. J. Clin. Lipidol. 2014, 8, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.Y.; Jacobson, T.A. Effects of Eicosapentaenoic Acid versus Docosahexaenoic Acid on Serum Lipids: A Systematic Review and Meta-Analysis. Curr. Atheroscler. Rep. 2011, 13, 474–483. [Google Scholar] [CrossRef]
- Li, H.; Ruan, X.Z.; Powis, S.H.; Fernando, R.; Mon, W.Y.; Wheeler, D.C.; Moorhead, J.F.; Varghese, Z. EPA and DHA Reduce LPS-Induced Inflammation Responses in HK-2 Cells: Evidence for a PPAR-γ–Dependent Mechanism. Kidney Int. 2005, 67, 867–874. [Google Scholar] [CrossRef]
- Sahebkar, A.; Simental-Mendía, L.E.; Mikhailidis, D.P.; Pirro, M.; Banach, M.; Sirtori, C.R.; Reiner, Ž. Effect of Omega-3 Supplements on Plasma Apolipoprotein C-III Concentrations: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Ann. Med. 2018, 50, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Watson, H.; Mitra, S.; Croden, F.C.; Taylor, M.; Wood, H.M.; Perry, S.L.; Spencer, J.A.; Quirke, P.; Toogood, G.J.; Lawton, C.L.; et al. A Randomised Trial of the Effect of Omega-3 Polyunsaturated Fatty Acid Supplements on the Human Intestinal Microbiota. Gut 2018, 67, 1974–1983. [Google Scholar] [CrossRef]
- Menni, C.; Zierer, J.; Pallister, T.; Jackson, M.A.; Long, T.; Mohney, R.P.; Steves, C.J.; Spector, T.D.; Valdes, A.M. Omega-3 Fatty Acids Correlate with Gut Microbiome Diversity and Production of N-Carbamylglutamate in Middle Aged and Elderly Women. Sci. Rep. 2017, 7, 11079. [Google Scholar] [CrossRef]
- Vijay, A.; Astbury, S.; Le Roy, C.; Spector, T.D.; Valdes, A.M. The Prebiotic Effects of Omega-3 Fatty Acid Supplementation: A Six-Week Randomised Intervention Trial. Gut Microbes 2021, 13, 1–11. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Reference | Study Description | Subjects | Methods | Main Findings |
|---|---|---|---|---|
| Fu et al., 2015 [70] | Large cohort study | 893 participants of the LifeLines-DEEP cohort | Lipid panels of total cholesterol, HDL-C, TGs, LDL-C. Sample genotyping. Fecal sample gut microbiota profiling with 16s rRNA gene sequencing. Operational taxonomic unit (OTU) picking and OTU richness. Microbial Shannon diversity index calculation. Cross validation analysis. | In total, 34 bacterial taxa correlated with plasma lipid levels and BMI. Specifically, family Clostridiaceae/Lachnospiracease was correlated with LDL. Coprococcus (Firmicutes) and Collinsella had strong correlation with TGs. Cross validation analysis revealed that GM explained ≤25.9% of HDL-C variance and that microbiota explained 6% of the variance in TGs. |
| Rebolledo et al., 2017 [71] | Case control study | 30 hypercholesterolemic subjects, 27 controls with normal cholesterol levels | Anthropometric data. Serum fasting glucose and lipid profile. Stool sample analysis of GM by gel electrophoresis with denaturing gradient (DGGE) technique and nonmetric multidimensional scaling (NMDS). Shannon–Weaver, Simpson, and Richness microbial community diversity indices. | DGGE banding profiles differed between case and control, confirmed by 2 separate groups forming on NMDS scaling analysis. Significant (p < 0.05) decrease of all three bacterial DNA indices of microbial diversity. |
| Vojinovic et al., 2019 [72] | Prospective cohort study of two large cohorts. | 2309 individuals from the Rotterdam and the LifeLines-DEEP cohort. | Fasting plasma metabolite profiling with 1H-nuclear magnetic resonance (NMR). Fecal sample gut microbiota profiling with 16s rRNA gene sequencing. | Significant (p < 0.05) associations between 18 families and genera of bacteria with VLDL of different sizes, 22 with HDL, 13 with HDL and VLDL, and 15 with serum triglycerides |
| Le Roy et al., 2019 [73] | Mouse study | Hypercholesterolemic female Apoe−/− and LDLr−/− mice | Depletion of all microbes in hypercholesterolemic mice with a combination of 4 antibiotics. Human feces intestinal microbiota transplantation. Further on mice: Plasma lipid and lipoprotein profile analysis. Bile acid synthesis measurement with labeled (14C) cholesterol dissolved in olive oil. Liver, ileum, and jejunum gene expression with qPCR. Sterol quantification in liver and bile. 16S rRNA gene sequencing of fecal gut microbiota. | Depletion of microbiota in mice raises cholesterol, mainly VLDL and LDL. Depletion also enhances liver uptake of cholesterol. Depletion increases cholesterol de novo synthesis by liver. Cholesterol level is transmissible in mice by microbiota transplantation from humans with altered cholesterol levels to microbiota depleted Apoe−/− mice. |
| Yun et al., 2020 [74] | Cross sectional study | 1141 subjects from the Kangbuk Samsung Health Study in South Korea, divided into dyslipidemic (G0) and normal lipid (G1) groups based on total cholesterol, LDL-C, TG, HDL-C, ApoB and ApoA1 levels. | Anthropometric data. Blood sample after 10 h fasting of total cholesterol, triglycerides, LDL-C, HDL-C, and apoA1 (HDL particle component) and ApoB (LDL-C and other particle component) determination. Gut microbiota 16s rRNA gene sequencing of fecal sample. | The group with high TGs had lower alpha diversity indices (Shannon’s index and Faith’s phylogenetic diversity, both p < 0.001, Pielou’s evenness p < 0.030). Abnormally low ApoA1 group had higher alpha diversity. No association with alpha diversity for other lipid parameters. 12 taxa associated with TGs: The high TGs group had high amount of the genus Fusobacterium and low levels of Oscillospira, which produces butyrate (a SCFA) 10 and 6 taxa were associated with ApoA1 and ApoB levels, respectively. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zwartjes, M.S.Z.; Gerdes, V.E.A.; Nieuwdorp, M. The Role of Gut Microbiota and Its Produced Metabolites in Obesity, Dyslipidemia, Adipocyte Dysfunction, and Its Interventions. Metabolites 2021, 11, 531. https://doi.org/10.3390/metabo11080531
Zwartjes MSZ, Gerdes VEA, Nieuwdorp M. The Role of Gut Microbiota and Its Produced Metabolites in Obesity, Dyslipidemia, Adipocyte Dysfunction, and Its Interventions. Metabolites. 2021; 11(8):531. https://doi.org/10.3390/metabo11080531
Chicago/Turabian StyleZwartjes, Max S. Z., Victor E. A. Gerdes, and Max Nieuwdorp. 2021. "The Role of Gut Microbiota and Its Produced Metabolites in Obesity, Dyslipidemia, Adipocyte Dysfunction, and Its Interventions" Metabolites 11, no. 8: 531. https://doi.org/10.3390/metabo11080531
APA StyleZwartjes, M. S. Z., Gerdes, V. E. A., & Nieuwdorp, M. (2021). The Role of Gut Microbiota and Its Produced Metabolites in Obesity, Dyslipidemia, Adipocyte Dysfunction, and Its Interventions. Metabolites, 11(8), 531. https://doi.org/10.3390/metabo11080531