
Inside the Insulin Secretory Granule




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
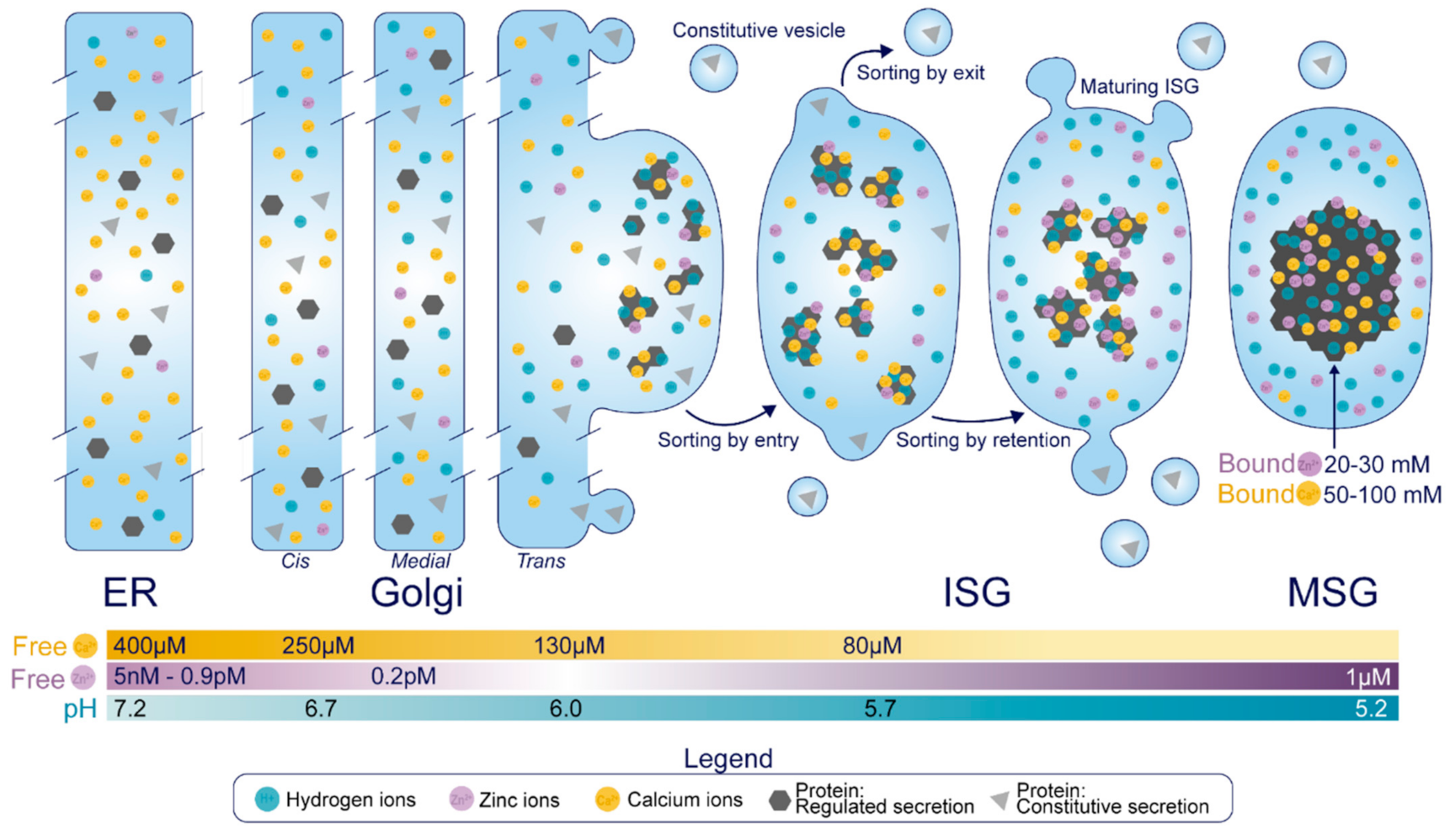
Historical Overview of Insulin SG Formation
2. Luminal Components of the Insulin Secretory Granule
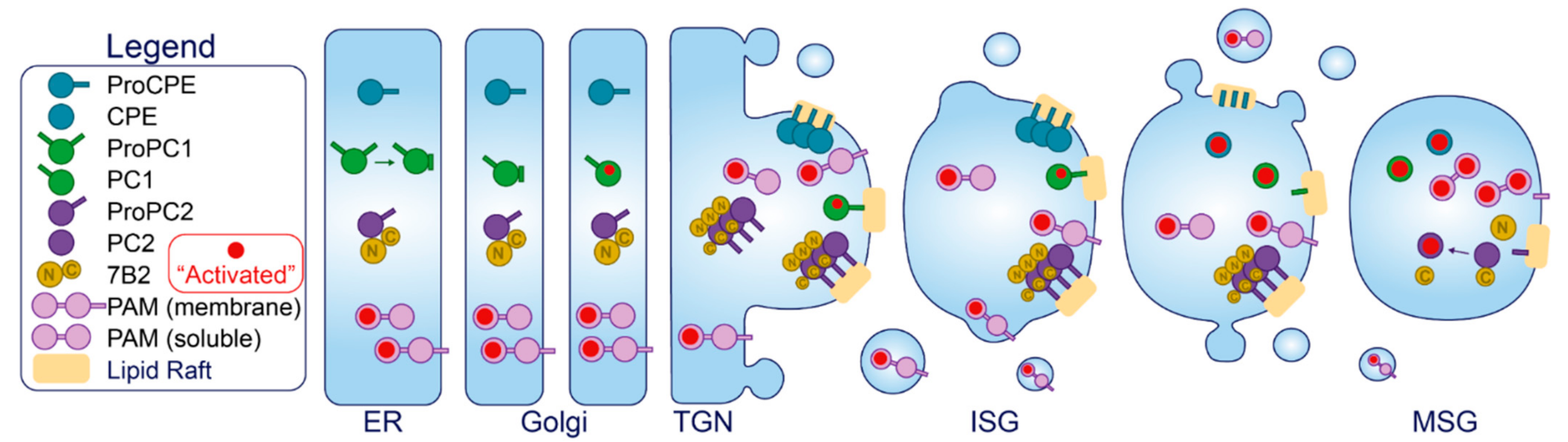
2.1. Cargo Molecules
2.2. Luminal Enzymes and Chaperones
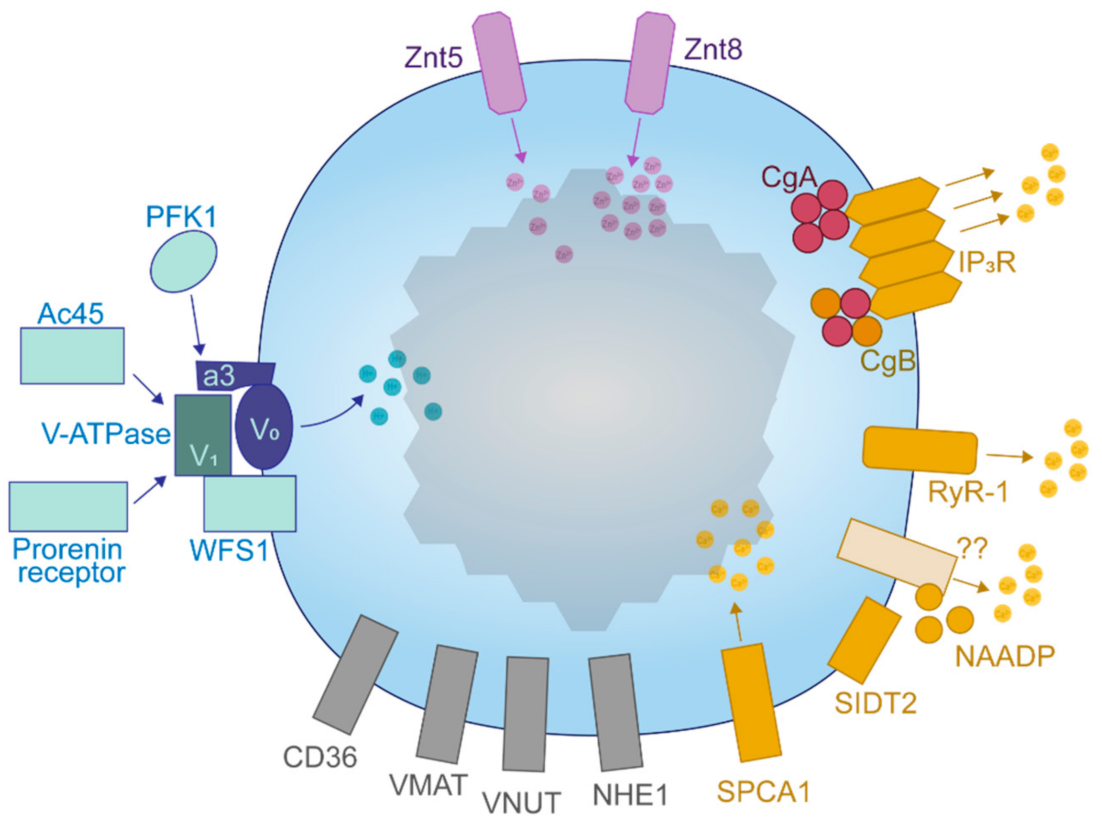
2.3. Ions, Transporters, and Channels
2.4. Sorting Receptors
3. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Orci, L.; Halban, P.; Amherdt, M.; Ravazzola, M.; Vassalli, J.D.; Perrelet, A. Nonconverted, Amino Acid Analog-Modified Proinsulin Stays in a Golgi-Derived Clathrin-Coated Membrane Compartment. J. Cell Biol. 1984, 99, 2187–2192. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Halban, P.A. Newly Synthesized Proinsulin/insulin and Stored Insulin Are Released from Pancreatic B Cells Predominantly via a Regulated, rather than a Constitutive, Pathway. J. Cell Biol. 1987, 105, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.; Carroll, R.; Swift, H.H.; Steiner, D.F. Studies on the Molecular Organization of Rat Insulin Secretory Granules. J. Biol. Chem. 1987, 262, 16531–16535. [Google Scholar] [CrossRef]
- Davidson, H.W.; Rhodes, C.J.; Hutton, J.C. Intraorganellar Calcium and pH Control Proinsulin Cleavage in the Pancreatic Beta Cell via Two Distinct Site-Specific Endopeptidases. Nature 1988, 333, 93–96. [Google Scholar] [CrossRef]
- Smeekens, S.P.; Montag, A.G.; Thomas, G.; Albiges-Rizo, C.; Carroll, R.; Benig, M.; Phillips, L.A.; Martin, S.; Ohagi, S.; Gardner, P. Proinsulin Processing by the Subtilisin-Related Proprotein Convertases Furin, PC2, and PC3. Proc. Natl. Acad. Sci. USA 1992, 89, 8822–8826. [Google Scholar] [CrossRef]
- Orci, L.; Ravazzola, M.; Storch, M.J.; Anderson, R.G.; Vassalli, J.D.; Perrelet, A. Proteolytic Maturation of Insulin Is a Post-Golgi Event Which Occurs in Acidifying Clathrin-Coated Secretory Vesicles. Cell 1987, 49, 865–868. [Google Scholar] [CrossRef]
- Kuliawat, R.; Klumperman, J.; Ludwig, T.; Arvan, P. Differential Sorting of Lysosomal Enzymes out of the Regulated Secretory Pathway in Pancreatic Beta-Cells. J. Cell Biol. 1997, 137, 595–608. [Google Scholar] [CrossRef]
- Baker, E.N.; Blundell, T.L.; Cutfield, J.F.; Dodson, E.J.; Dodson, G.G.; Hodgkin, D.M.C.; Hubbard, R.E.; Isaacs, N.W.; Reynolds, C.D.; Sakabe, K.; et al. The Structure of 2Zn Pig Insulin Crystals at 1.5 Å Resolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1988, 319, 369–456. [Google Scholar]
- Halban, P.A. Inhibition of Proinsulin to Insulin Conversion in Rat Islets Using Arginine and Lysine Analogs. Lack of Effect on Rate of Release of Modified Products. J. Biol. Chem. 1982, 257, 13177–13180. [Google Scholar] [CrossRef]
- Seaquist, E.R.; Kahn, S.E.; Clark, P.M.; Hales, C.N.; Porte, D., Jr.; Robertson, R.P. Hyperproinsulinemia Is Associated with Increased Beta Cell Demand after Hemipancreatectomy in Humans. J. Clin. Investig. 1996, 97, 455–460. [Google Scholar] [CrossRef]
- Mezza, T.; Ferraro, P.M.; Sun, V.A.; Moffa, S.; Cefalo, C.M.A.; Quero, G.; Cinti, F.; Sorice, G.P.; Pontecorvi, A.; Folli, F.; et al. Increased β-Cell Workload Modulates Proinsulin-to-Insulin Ratio in Humans. Diabetes 2018, 67, 2389–2396. [Google Scholar] [CrossRef]
- Alarcon, C.; Boland, B.B.; Uchizono, Y.; Moore, P.C.; Peterson, B.; Rajan, S.; Rhodes, O.S.; Noske, A.B.; Haataja, L.; Arvan, P.; et al. Pancreatic β-Cell Adaptive Plasticity in Obesity Increases Insulin Production but Adversely Affects Secretory Function. Diabetes 2016, 65, 438–450. [Google Scholar] [CrossRef]
- Ward, W.K.; LaCava, E.C.; Paquette, T.L.; Beard, J.C.; Wallum, B.J.; Porte, D., Jr. Disproportionate Elevation of Immunoreactive Proinsulin in Type 2 (non-Insulin-Dependent) Diabetes Mellitus and in Experimental Insulin Resistance. Diabetologia 1987, 30, 698–702. [Google Scholar] [CrossRef]
- Yoshino, H.; Kawakami, K.; Yoshino, G.; Hirose, T. Age-Related Changes of Proinsulin Processing in Diabetic and Non-Diabetic Japanese Individuals. Geriatr. Gerontol. Int. 2018, 18, 1046–1050. [Google Scholar] [CrossRef]
- Pfützner, A.; Standl, E.; Hohberg, C.; Konrad, T.; Strotmann, H.-J.; Lübben, G.; Langenfeld, M.R.; Schulze, J.; Forst, T. IRIS II Study: Intact Proinsulin Is Confirmed as a Highly Specific Indicator for Insulin Resistance in a Large Cross-Sectional Study Design. Diabetes Technol. Ther. 2005, 7, 478–486. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Meigs, J.B.; Rifai, N.; Buring, J.E.; Liu, S.; Ridker, P.M. Insulin, Proinsulin, Proinsulin:insulin Ratio, and the Risk of Developing Type 2 Diabetes Mellitus in Women. Am. J. Med. 2003, 114, 438–444. [Google Scholar] [CrossRef]
- Nijpels, G.; Popp-Snijders, C.; Kostense, P.J.; Bouter, L.M.; Heine, R.J. Fasting Proinsulin and 2-H Post-Load Glucose Levels Predict the Conversion to NIDDM in Subjects with Impaired Glucose Tolerance: The Hoorn Study. Diabetologia 1996, 39, 113–118. [Google Scholar]
- Vangipurapu, J.; Stančáková, A.; Kuulasmaa, T.; Kuusisto, J.; Laakso, M. Both Fasting and Glucose-Stimulated Proinsulin Levels Predict Hyperglycemia and Incident Type 2 Diabetes: A Population-Based Study of 9,396 Finnish Men. PLoS ONE 2015, 10, e0124028. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, M.; Tong, J.; Dong, Z.; Deng, M.; Ren, X.; Li, H.; Yang, J.; Meng, Z.; Sun, J.; et al. Impaired Glucose-Stimulated Proinsulin Secretion Is an Early Marker of β-Cell Impairment Before Prediabetes Stage. J. Clin. Endocrinol. Metab. 2019, 104, 4341–4346. [Google Scholar] [CrossRef]
- Porte, D., Jr.; Kahn, S.E. Hyperproinsulinemia and Amyloid in NIDDM. Clues to Etiology of Islet Beta-Cell Dysfunction? Diabetes 1989, 38, 1333–1336. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Alarcón, C. What Beta-Cell Defect Could Lead to Hyperproinsulinemia in NIDDM? Some Clues from Recent Advances Made in Understanding the Proinsulin-Processing Mechanism. Diabetes 1994, 43, 511–517. [Google Scholar] [CrossRef]
- Ohsawa, H.; Kanatsuka, A.; Yamaguchi, T.; Makino, H.; Yoshida, S. Islet Amyloid Polypeptide Inhibits Glucose-Stimulated Insulin Secretion from Isolated Rat Pancreatic Islets. Biochem. Biophys. Res. Commun. 1989, 160, 961–967. [Google Scholar] [CrossRef]
- Natori, S.; Huttner, W.B. Chromogranin B (secretogranin I) Promotes Sorting to the Regulated Secretory Pathway of Processing Intermediates Derived from a Peptide Hormone Precursor. Proc. Natl. Acad. Sci. USA 1996, 93, 4431–4436. [Google Scholar] [CrossRef] [PubMed]
- Carmon, O.; Laguerre, F.; Riachy, L.; Delestre-Delacour, C.; Wang, Q.; Tanguy, E.; Jeandel, L.; Cartier, D.; Thahouly, T.; Haeberlé, A.-M.; et al. Chromogranin A Preferential Interaction with Golgi Phosphatidic Acid Induces Membrane Deformation and Contributes to Secretory Granule Biogenesis. FASEB J. 2020. [Google Scholar] [CrossRef]
- Bartolomucci, A.; Possenti, R.; Mahata, S.K.; Fischer-Colbrie, R.; Loh, Y.P.; Salton, S.R.J. The Extended Granin Family: Structure, Function, and Biomedical Implications. Endocr. Rev. 2011, 32, 755–797. [Google Scholar] [CrossRef]
- Hutton, J.C.; Peshavaria, M. Proton-Translocating Mg2+-Dependent ATPase Activity in Insulin-Secretory Granules. Biochem. J. 1982, 204, 161–170. [Google Scholar] [CrossRef]
- Schoonderwoert, V.T.; Holthuis, J.C.; Tanaka, S.; Tooze, S.A.; Martens, G.J. Inhibition of the Vacuolar H+-ATPase Perturbs the Transport, Sorting, Processing and Release of Regulated Secretory Proteins. Eur. J. Biochem. 2000, 267, 5646–5654. [Google Scholar] [CrossRef]
- Lemaire, K.; Chimienti, F.; Schuit, F. Zinc Transporters and Their Role in the Pancreatic β-Cell. J. Diabetes Investig. 2012, 3, 202–211. [Google Scholar] [CrossRef]
- Hutton, J.C.; Penn, E.J.; Peshavaria, M. Low-Molecular-Weight Constituents of Isolated Insulin-Secretory Granules. Bivalent Cations, Adenine Nucleotides and Inorganic Phosphate. Biochem. J. 1983, 210, 297–305. [Google Scholar] [CrossRef]
- Hur, Y.S.; Yoo, S.H. Distribution Profile of Inositol 1,4,5-Trisphosphate Receptor/Ca2+ Channels in α and β Cells of Pancreas: Dominant Localization in Secretory Granules and Common Error in Identification of Secretory Granule Membranes. Pancreas 2015, 44, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.J.; Pinton, P.; Varadi, A.; Tacchetti, C.; Ainscow, E.K.; Pozzan, T.; Rizzuto, R.; Rutter, G.A. Dense Core Secretory Vesicles Revealed as a Dynamic Ca(2+) Store in Neuroendocrine Cells with a Vesicle-Associated Membrane Protein Aequorin Chimaera. J. Cell Biol. 2001, 155, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Klumperman, J.; Kuliawat, R.; Griffith, J.M.; Geuze, H.J.; Arvan, P. Mannose 6-Phosphate Receptors Are Sorted from Immature Secretory Granules via Adaptor Protein AP-1, Clathrin, and Syntaxin 6-Positive Vesicles. J. Cell Biol. 1998, 141, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Orci, L.; Ravazzola, M.; Perrelet, A. (Pro)insulin Associates with Golgi Membranes of Pancreatic B Cells. Proc. Natl. Acad. Sci. USA 1984, 81, 6743–6746. [Google Scholar] [CrossRef]
- Kahn, S.E.; Halban, P.A. Release of Incompletely Processed Proinsulin Is the Cause of the Disproportionate Proinsulinemia of NIDDM. Diabetes 1997, 46, 1725–1732. [Google Scholar] [CrossRef]
- Alarcón, C.; Leahy, J.L.; Schuppin, G.T.; Rhodes, C.J. Increased Secretory Demand rather than a Defect in the Proinsulin Conversion Mechanism Causes Hyperproinsulinemia in a Glucose-Infusion Rat Model of Non-Insulin-Dependent Diabetes Mellitus. J. Clin. Investig. 1995, 95, 1032–1039. [Google Scholar] [CrossRef][Green Version]
- Omar-Hmeadi, M.; Idevall-Hagren, O. Insulin Granule Biogenesis and Exocytosis. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Thurmond, D.C.; Gaisano, H.Y. Recent Insights into Beta-Cell Exocytosis in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1310–1325. [Google Scholar] [CrossRef]
- Norris, N.; Yau, B.; Kebede, M.A. Isolation and Proteomics of the Insulin Secretory Granule. Metabolites 2021, 11, 288. [Google Scholar] [CrossRef]
- Magro, M.G.; Solimena, M. Regulation of β-Cell Function by RNA-Binding Proteins. Mol. Metab. 2013, 2, 348–355. [Google Scholar] [CrossRef]
- Arvan, P.; Halban, P.A. Sorting Ourselves out: Seeking Consensus on Trafficking in the Beta-Cell. Traffic 2004, 5, 53–61. [Google Scholar] [CrossRef]
- Arvan, P.; Kuliawat, R.; Prabakaran, D.; Zavacki, A.M.; Elahi, D.; Wang, S.; Pilkey, D. Protein Discharge from Immature Secretory Granules Displays Both Regulated and Constitutive Characteristics. J. Biol. Chem. 1991, 266, 14171–14174. [Google Scholar] [CrossRef]
- Kuliawat, R.; Arvan, P. Protein Targeting via the “Constitutive-like” Secretory Pathway in Isolated Pancreatic Islets: Passive Sorting in the Immature Granule Compartment. J. Cell Biol. 1992, 118, 521–529. [Google Scholar] [CrossRef]
- Kuliawat, R.; Arvan, P. Distinct Molecular Mechanisms for Protein Sorting within Immature Secretory Granules of Pancreatic Beta-Cells. J. Cell Biol. 1994, 126, 77–86. [Google Scholar] [CrossRef]
- Arvan, P.; Castle, D. Sorting and Storage during Secretory Granule Biogenesis: Looking Backward and Looking Forward. Biochem. J. 1998, 332 Pt 3, 593–610. [Google Scholar] [CrossRef]
- Molinete, M.; Irminger, J.C.; Tooze, S.A.; Halban, P.A. Trafficking/sorting and Granule Biogenesis in the Beta-Cell. Semin. Cell Dev. Biol. 2000, 11, 243–251. [Google Scholar] [CrossRef]
- Hummer, B.H.; Maslar, D.; Soltero-Gutierrez, M.; de Leeuw, N.F.; Asensio, C.S. Differential Sorting Behavior for Soluble and Transmembrane Cargoes at the Trans-Golgi Network in Endocrine Cells. Mol. Biol. Cell 2020, 31, 157–166. [Google Scholar] [CrossRef]
- Ericson, L.E.; Håkanson, R.; Lundquist, I. Accumulation of Dopamine in Mouse Pancreatic B-Cells Following Injection of L-DOPA. Localization to Secretory Granules and Inhibition of Insulin Secretion. Diabetologia 1977, 13, 117–124. [Google Scholar] [CrossRef]
- Ekholm, R.; Ericson, L.E.; Lundquist, I. Monoamines in the Pancreatic Islets of the Mouse. Subcellular Localization of 5-Hydroxytryptamine by Electron Microscopic Autoradiography. Diabetologia 1971, 7, 339–348. [Google Scholar] [CrossRef]
- Lundquist, I.; Ekholm, R.; Ericson, L.E. Monoamines in the Pancreatic Islets of the Mouse. 5-Hydroxytryptamine as an Intracellular Modifier of Insulin Secretion, and the Hypoglycaemic Action of Monoamine Oxidase Inhibitors. Diabetologia 1971, 7, 414–422. [Google Scholar] [CrossRef]
- Sakamoto, S.; Miyaji, T.; Hiasa, M.; Ichikawa, R.; Uematsu, A.; Iwatsuki, K.; Shibata, A.; Uneyama, H.; Takayanagi, R.; Yamamoto, A.; et al. Impairment of Vesicular ATP Release Affects Glucose Metabolism and Increases Insulin Sensitivity. Sci. Rep. 2014, 4, 6689. [Google Scholar] [CrossRef]
- Henquin, J.-C. Paracrine and Autocrine Control of Insulin Secretion in Human Islets: Evidence and Pending Questions. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E78–E86. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Lara-Lemus, R.; Shan, S.-O.; Wright, J.; Haataja, L.; Barbetti, F.; Guo, H.; Larkin, D.; Arvan, P. Impaired Cleavage of Preproinsulin Signal Peptide Linked to Autosomal-Dominant Diabetes. Diabetes 2012, 61, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.F.; Arvan, P. Intracellular Transport of Proinsulin in Pancreatic Beta-Cells. Structural Maturation Probed by Disulfide Accessibility. J. Biol. Chem. 1995, 270, 20417–20423. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Snapp, E.; Wright, J.; Liu, M.; Hardy, A.B.; Wheeler, M.B.; Markwardt, M.L.; Rizzo, M.; Arvan, P. Proinsulin Intermolecular Interactions during Secretory Trafficking in Pancreatic β Cells. J. Biol. Chem. 2013, 288, 1896–1906. [Google Scholar] [CrossRef]
- Frank, B.H.; Veros, A.J. Interaction of Zinc with Proinsulin. Biochem. Biophys. Res. Commun. 1970, 38, 284–289. [Google Scholar] [CrossRef]
- Kiselar, J.G.; Datt, M.; Chance, M.R.; Weiss, M.A. Structural Analysis of Proinsulin Hexamer Assembly by Hydroxyl Radical Footprinting and Computational Modeling. J. Biol. Chem. 2011, 286, 43710–43716. [Google Scholar] [CrossRef]
- Bailyes, E.M.; Bennett, D.L.; Hutton, J.C. Proprotein-Processing Endopeptidases of the Insulin Secretory Granule. Enzyme 1991, 45, 301–313. [Google Scholar] [CrossRef]
- Davidson, H.W.; Hutton, J.C. The Insulin-Secretory-Granule Carboxypeptidase H. Purification and Demonstration of Involvement in Proinsulin Processing. Biochem. J. 1987, 245, 575–582. [Google Scholar] [CrossRef]
- Weiss, M.; Steiner, D.F.; Philipson, L.H. Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2014. [Google Scholar]
- Norrman, M.; Schluckebier, G. Crystallographic Characterization of Two Novel Crystal Forms of Human Insulin Induced by Chaotropic Agents and a Shift in pH. BMC Struct. Biol. 2007, 7, 83. [Google Scholar] [CrossRef]
- Steiner, D.F. Cocrystallization of Proinsulin and Insulin. Nature 1973, 243, 528–530. [Google Scholar] [CrossRef]
- Landreh, M.; Alvelius, G.; Willander, H.; Stukenborg, J.-B.; Söder, O.; Johansson, J.; Jörnvall, H. Insulin Solubility Transitions by pH-Dependent Interactions with Proinsulin C-Peptide. FEBS J. 2012, 279, 4589–4597. [Google Scholar] [CrossRef]
- Verchere, C.B.; Paoletta, M.; Neerman-Arbez, M.; Rose, K.; Irminger, J.C.; Gingerich, R.L.; Kahn, S.E.; Halban, P.A. Des-(27–31)C-Peptide. A Novel Secretory Product of the Rat Pancreatic Beta Cell Produced by Truncation of Proinsulin Connecting Peptide in Secretory Granules. J. Biol. Chem. 1996, 271, 27475–27481. [Google Scholar] [CrossRef]
- Gold, G.; Grodsky, G.M. Kinetic Aspects of Compartmental Storage and Secretion of Insulin and Zinc. Experientia 1984, 40, 1105–1114. [Google Scholar] [CrossRef]
- Chan, S.J.; Seino, S.; Gruppuso, P.A.; Schwartz, R.; Steiner, D.F. A Mutation in the B Chain Coding Region Is Associated with Impaired Proinsulin Conversion in a Family with Hyperproinsulinemia. Proc. Natl. Acad. Sci. USA 1987, 84, 2194–2197. [Google Scholar] [CrossRef]
- Carroll, R.J.; Hammer, R.E.; Chan, S.J.; Swift, H.H.; Rubenstein, A.H.; Steiner, D.F. A Mutant Human Proinsulin Is Secreted from Islets of Langerhans in Increased Amounts via an Unregulated Pathway. Proc. Natl. Acad. Sci. USA 1988, 85, 8943–8947. [Google Scholar] [CrossRef]
- Halban, P.A.; Irminger, J.-C. Mutant Proinsulin That Cannot Be Converted Is Secreted Efficiently from Primary Rat Beta-Cells via the Regulated Pathway. Mol. Biol. Cell 2003, 14, 1195–1203. [Google Scholar] [CrossRef]
- Sizonenko, S.; Irminger, J.C.; Buhler, L.; Deng, S.; Morel, P.; Halban, P.A. Kinetics of Proinsulin Conversion in Human Islets. Diabetes 1993, 42, 933–936. [Google Scholar] [CrossRef]
- Sizonenko, S.V.; Halban, P.A. Differential Rates of Conversion of Rat Proinsulins I and II. Evidence for Slow Cleavage at the B-chain/C-Peptide Junction of Proinsulin II. Biochem. J. 1991, 278, 621–625. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the Size of the Active Site in Proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Kaufmann, J.E.; Irminger, J.C.; Halban, P.A. Sequence Requirements for Proinsulin Processing at the B-chain/C-Peptide Junction. Biochem. J. 1995, 310 Pt 3, 869–874. [Google Scholar] [CrossRef]
- Halban, P.A. Proinsulin Processing in the Regulated and the Constitutive Secretory Pathway. Diabetologia 1994, 37 (Suppl. 2), S65–S72. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Lincoln, B.; Shoelson, S.E. Preferential Cleavage of Des-31,32-Proinsulin over Intact Proinsulin by the Insulin Secretory Granule Type II Endopeptidase. Implication of a Favored Route for Prohormone Processing. J. Biol. Chem. 1992, 267, 22719–22727. [Google Scholar] [CrossRef]
- Kaufmann, J.E.; Irminger, J.C.; Mungall, J.; Halban, P.A. Proinsulin Conversion in GH3 Cells after Coexpression of Human Proinsulin with the Endoproteases PC2 And/or PC3. Diabetes 1997, 46, 978–982. [Google Scholar] [CrossRef] [PubMed]
- Irminger, J.C.; Vollenweider, F.M.; Neerman-Arbez, M.; Halban, P.A. Human Proinsulin Conversion in the Regulated and the Constitutive Pathways of Transfected AtT20 Cells. J. Biol. Chem. 1994, 269, 1756–1762. [Google Scholar] [CrossRef]
- Neerman-Arbez, M.; Sizonenko, S.V.; Halban, P.A. Slow Cleavage at the Proinsulin B-Chain/connecting Peptide Junction Associated with Low Levels of Endoprotease PC1/3 in Transformed Beta Cells. J. Biol. Chem. 1993, 268, 16098–16100. [Google Scholar] [CrossRef]
- Neerman-Arbez, M.; Cirulli, V.; Halban, P.A. Levels of the Conversion Endoproteases PC1 (PC3) and PC2 Distinguish between Insulin-Producing Pancreatic Islet Beta Cells and Non-Beta Cells. Biochem. J. 1994, 300 Pt 1, 57–61. [Google Scholar] [CrossRef]
- Zhu, X.; Orci, L.; Carroll, R.; Norrbom, C.; Ravazzola, M.; Steiner, D.F. Severe Block in Processing of Proinsulin to Insulin Accompanied by Elevation of Des-64,65 Proinsulin Intermediates in Islets of Mice Lacking Prohormone Convertase 1/3. Proc. Natl. Acad. Sci. USA 2002, 99, 10299–10304. [Google Scholar] [CrossRef]
- Furuta, M.; Carroll, R.; Martin, S.; Swift, H.H.; Ravazzola, M.; Orci, L.; Steiner, D.F. Incomplete Processing of Proinsulin to Insulin Accompanied by Elevation of Des-31,32 Proinsulin Intermediates in Islets of Mice Lacking Active PC2. J. Biol. Chem. 1998, 273, 3431–3437. [Google Scholar] [CrossRef]
- Ramzy, A.; Asadi, A.; Kieffer, T.J. Revisiting Proinsulin Processing: Evidence That Human β-Cells Process Proinsulin with Prohormone Convertase (PC) 1/3 but Not PC2. Diabetes 2020, 69, 1451–1462. [Google Scholar] [CrossRef]
- Irminger, J.C.; Meyer, K.; Halban, P. Proinsulin Processing in the Rat Insulinoma Cell Line INS after Overexpression of the Endoproteases PC2 or PC3 by Recombinant Adenovirus. Biochem. J. 1996, 320 Pt 1, 11–15. [Google Scholar] [CrossRef]
- Shennan, K.I.; Taylor, N.A.; Jermany, J.L.; Matthews, G.; Docherty, K. Differences in pH Optima and Calcium Requirements for Maturation of the Prohormone Convertases PC2 and PC3 Indicates Different Intracellular Locations for These Events. J. Biol. Chem. 1995, 270, 1402–1407. [Google Scholar] [CrossRef]
- Muller, L.; Zhu, X.; Lindberg, I. Mechanism of the Facilitation of PC2 Maturation by 7B2: Involvement in ProPC2 Transport and Activation but Not Folding. J. Cell Biol. 1997, 139, 625–638. [Google Scholar] [CrossRef]
- Zhu, X.; Rouille, Y.; Lamango, N.S.; Steiner, D.F.; Lindberg, I. Internal Cleavage of the Inhibitory 7B2 Carboxyl-Terminal Peptide by PC2: A Potential Mechanism for Its Inactivation. Proc. Natl. Acad. Sci. USA 1996, 93, 4919–4924. [Google Scholar] [CrossRef]
- Hwang, J.R.; Lindberg, I. Inactivation of the 7B2 Inhibitory CT Peptide Depends on a Functional Furin Cleavage Site. J. Neurochem. 2001, 79, 437–444. [Google Scholar] [CrossRef]
- Lamango, N.S.; Apletalina, E.; Liu, J.; Lindberg, I. The Proteolytic Maturation of Prohormone Convertase 2 (PC2) Is a pH-Driven Process. Arch. Biochem. Biophys. 1999, 362, 275–282. [Google Scholar] [CrossRef]
- Ostrega, D.; Polonsky, K.; Nagi, D.; Yudkin, J.; Cox, L.J.; Clark, P.M.; Hales, C.N. Measurement of Proinsulin and Intermediates. Validation of Immunoassay Methods by High-Performance Liquid Chromatography. Diabetes 1995, 44, 437–440. [Google Scholar] [CrossRef]
- Clark, P.M.; Levy, J.C.; Cox, L.; Burnett, M.; Turner, R.C.; Hales, C.N. Immunoradiometric Assay of Insulin, Intact Proinsulin and 32–33 Split Proinsulin and Radioimmunoassay of Insulin in Diet-Treated Type 2 (non-Insulin-Dependent) Diabetic Subjects. Diabetologia 1992, 35, 469–474. [Google Scholar] [CrossRef]
- Lukinius, A.; Wilander, E.; Westermark, G.T.; Engström, U.; Westermark, P. Co-Localization of Islet Amyloid Polypeptide and Insulin in the B Cell Secretory Granules of the Human Pancreatic Islets. Diabetologia 1989, 32, 240–244. [Google Scholar] [CrossRef]
- Nakazato, M.; Miyazato, M.; Asai, J.; Mitsukawa, T.; Kangawa, K.; Matsuo, H.; Matsukura, S. Islet Amyloid Polypeptide, a Novel Pancreatic Peptide, Is a Circulating Hormone Secreted under Glucose Stimulation. Biochem. Biophys. Res. Commun. 1990, 169, 713–718. [Google Scholar] [CrossRef]
- Stridsberg, M.; Sandler, S.; Wilander, E. Cosecretion of Islet Amyloid Polypeptide (IAPP) and Insulin from Isolated Rat Pancreatic Islets Following Stimulation or Inhibition of Beta-Cell Function. Regul. Pept. 1993, 45, 363–370. [Google Scholar] [CrossRef]
- Lutz, T.A. The Role of Amylin in the Control of Energy Homeostasis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1475–R1484. [Google Scholar] [CrossRef] [PubMed]
- Hull, R.L.; Westermark, G.T.; Westermark, P.; Kahn, S.E. Islet Amyloid: A Critical Entity in the Pathogenesis of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 3629–3643. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Grimelius, L. The Pancreatic Islet Cells in Insular Amyloidosis in Human Diabetic and Non-Diabetic Adults. Acta Pathol. Microbiol. Scand. A 1973, 81, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Wilander, E. The Influence of Amyloid Deposits on the Islet Volume in Maturity Onset Diabetes Mellitus. Diabetologia 1978, 15, 417–421. [Google Scholar] [CrossRef]
- Zhao, H.-L.; Lai, F.M.M.; Tong, P.C.Y.; Zhong, D.-R.; Yang, D.; Tomlinson, B.; Chan, J.C.N. Prevalence and Clinicopathological Characteristics of Islet Amyloid in Chinese Patients with Type 2 Diabetes. Diabetes 2003, 52, 2759–2766. [Google Scholar] [CrossRef]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet Amyloid Polypeptide, Islet Amyloid, and Diabetes Mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef]
- Raleigh, D.; Zhang, X.; Hastoy, B.; Clark, A. The β-Cell Assassin: IAPP Cytotoxicity. J. Mol. Endocrinol. 2017, 59, R121–R140. [Google Scholar] [CrossRef]
- Wang, J.; Xu, J.; Finnerty, J.; Furuta, M.; Steiner, D.F.; Verchere, C.B. The Prohormone Convertase Enzyme 2 (PC2) Is Essential for Processing pro-Islet Amyloid Polypeptide at the NH2-Terminal Cleavage Site. Diabetes 2001, 50, 534–539. [Google Scholar] [CrossRef]
- Marzban, L.; Soukhatcheva, G.; Verchere, C.B. Role of Carboxypeptidase E in Processing of Pro-Islet Amyloid Polypeptide in β-Cells. Endocrinology 2005, 146, 1808–1817. [Google Scholar] [CrossRef]
- Marzban, L.; Trigo-Gonzalez, G.; Verchere, C.B. Processing of pro-Islet Amyloid Polypeptide in the Constitutive and Regulated Secretory Pathways of Beta Cells. Mol. Endocrinol. 2005, 19, 2154–2163. [Google Scholar] [CrossRef]
- Marzban, L.; Trigo-Gonzalez, G.; Zhu, X.; Rhodes, C.J.; Halban, P.A.; Steiner, D.F.; Verchere, C.B. Role of Beta-Cell Prohormone Convertase (PC)1/3 in Processing of pro-Islet Amyloid Polypeptide. Diabetes 2004, 53, 141–148. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Taylor, A.J.; Verchere, C.B. Islet Prohormone Processing in Health and Disease. Diabetes Obes. Metab. 2018, 20 (Suppl. 2), 64–76. [Google Scholar] [CrossRef]
- Rulifson, I.C.; Cao, P.; Miao, L.; Kopecky, D.; Huang, L.; White, R.D.; Samayoa, K.; Gardner, J.; Wu, X.; Chen, K.; et al. Identification of Human Islet Amyloid Polypeptide as a BACE2 Substrate. PLoS ONE 2016, 11, e0147254. [Google Scholar] [CrossRef]
- Courtade, J.A.; Wang, E.Y.; Yen, P.; Dai, D.L.; Soukhatcheva, G.; Orban, P.C.; Verchere, C.B. Loss of Prohormone Convertase 2 Promotes Beta Cell Dysfunction in a Rodent Transplant Model Expressing Human pro-Islet Amyloid Polypeptide. Diabetologia 2017, 60, 453–463. [Google Scholar] [CrossRef]
- Westermark, P.; Engström, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet Amyloid Polypeptide: Pinpointing Amino Acid Residues Linked to Amyloid Fibril Formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef]
- Yonemoto, I.T.; Kroon, G.J.A.; Dyson, H.J.; Balch, W.E.; Kelly, J.W. Amylin Proprotein Processing Generates Progressively More Amyloidogenic Peptides That Initially Sample the Helical State. Biochemistry 2008, 47, 9900–9910. [Google Scholar] [CrossRef]
- Jaikaran, E.T.A.S.; Nilsson, M.R.; Clark, A. Pancreatic Beta-Cell Granule Peptides Form Heteromolecular Complexes Which Inhibit Islet Amyloid Polypeptide Fibril Formation. Biochem. J. 2004, 377, 709–716. [Google Scholar] [CrossRef]
- Westermark, P.; Li, Z.C.; Westermark, G.T.; Leckström, A.; Steiner, D.F. Effects of Beta Cell Granule Components on Human Islet Amyloid Polypeptide Fibril Formation. FEBS Lett. 1996, 379, 203–206. [Google Scholar] [CrossRef]
- Brender, J.R.; Hartman, K.; Nanga, R.P.R.; Popovych, N.; de la Salud Bea, R.; Vivekanandan, S.; Marsh, E.N.G.; Ramamoorthy, A. Role of Zinc in Human Islet Amyloid Polypeptide Aggregation. J. Am. Chem. Soc. 2010, 132, 8973–8983. [Google Scholar] [CrossRef]
- Khemtémourian, L.; Doménech, E.; Doux, J.P.F.; Koorengevel, M.C.; Killian, J.A. Low pH Acts as Inhibitor of Membrane Damage Induced by Human Islet Amyloid Polypeptide. J. Am. Chem. Soc. 2011, 133, 15598–15604. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.; Eriksson, S.; Carlemalm, E.; Ahrén, B. B Cell Granule Peptides Affect Human Islet Amyloid Polypeptide (IAPP) Fibril Formation in Vitro. Biochem. Biophys. Res. Commun. 1997, 236, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Kudva, Y.C.; Mueske, C.; Butler, P.C.; Eberhardt, N.L. A Novel Assay in Vitro of Human Islet Amyloid Polypeptide Amyloidogenesis and Effects of Insulin Secretory Vesicle Peptides on Amyloid Formation. Biochem. J. 1998, 331 Pt 3, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.H.; O’Brien, T.D.; Jordan, K.; Westermark, P. Impaired Glucose Tolerance Is Associated with Increased Islet Amyloid Polypeptide (IAPP) Immunoreactivity in Pancreatic Beta Cells. Am. J. Pathol. 1989, 135, 245–250. [Google Scholar]
- Gurlo, T.; Ryazantsev, S.; Huang, C.-J.; Yeh, M.W.; Reber, H.A.; Hines, O.J.; O’Brien, T.D.; Glabe, C.G.; Butler, P.C. Evidence for Proteotoxicity in Beta Cells in Type 2 Diabetes: Toxic Islet Amyloid Polypeptide Oligomers Form Intracellularly in the Secretory Pathway. Am. J. Pathol. 2010, 176, 861–869. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Gurlo, T.; Kayed, R.; Butler, A.E.; Haataja, L.; Glabe, C.G.; Butler, P.C. Toxic Human Islet Amyloid Polypeptide (h-IAPP) Oligomers Are Intracellular, and Vaccination to Induce Anti-Toxic Oligomer Antibodies Does Not Prevent H-IAPP-Induced Beta-Cell Apoptosis in H-IAPP Transgenic Mice. Diabetes 2007, 56, 1324–1332. [Google Scholar] [CrossRef]
- Yagui, K.; Yamaguchi, T.; Kanatsuka, A.; Shimada, F.; Huang, C.I.; Tokuyama, Y.; Ohsawa, H.; Yamamura, K.; Miyazaki, J.; Mikata, A. Formation of Islet Amyloid Fibrils in Beta-Secretory Granules of Transgenic Mice Expressing Human Islet Amyloid Polypeptide/amylin. Eur. J. Endocrinol. 1995, 132, 487–496. [Google Scholar] [CrossRef]
- Wang, H.; Raleigh, D.P. The Ability of Insulin to Inhibit the Formation of Amyloid by pro-Islet Amyloid Polypeptide Processing Intermediates Is Significantly Reduced in the Presence of Sulfated Glycosaminoglycans. Biochemistry 2014, 53, 2605–2614. [Google Scholar] [CrossRef]
- Janson, J.; Ashley, R.H.; Harrison, D.; McIntyre, S.; Butler, P.C. The Mechanism of Islet Amyloid Polypeptide Toxicity Is Membrane Disruption by Intermediate-Sized Toxic Amyloid Particles. Diabetes 1999, 48, 491–498. [Google Scholar] [CrossRef]
- Brender, J.R.; Lee, E.L.; Cavitt, M.A.; Gafni, A.; Steel, D.G.; Ramamoorthy, A. Amyloid Fiber Formation and Membrane Disruption Are Separate Processes Localized in Two Distinct Regions of IAPP, the Type-2-Diabetes-Related Peptide. J. Am. Chem. Soc. 2008, 130, 6424–6429. [Google Scholar] [CrossRef]
- Brender, J.R.; Salamekh, S.; Ramamoorthy, A. Membrane Disruption and Early Events in the Aggregation of the Diabetes Related Peptide IAPP from a Molecular Perspective. Acc. Chem. Res. 2012, 45, 454–462. [Google Scholar] [CrossRef]
- Westermark, P.; Engström, U.; Westermark, G.T.; Johnson, K.H.; Permerth, J.; Betsholtz, C. Islet Amyloid Polypeptide (IAPP) and pro-IAPP Immunoreactivity in Human Islets of Langerhans. Diabetes Res. Clin. Pract. 1989, 7, 219–226. [Google Scholar] [CrossRef]
- Westermark, G.T.; Steiner, D.F.; Gebre-Medhin, S.; Engström, U.; Westermark, P. Pro Islet Amyloid Polypeptide (ProIAPP) Immunoreactivity in the Islets of Langerhans. Ups. J. Med. Sci. 2000, 105, 97–106. [Google Scholar] [CrossRef]
- Zheng, X.; Ren, W.; Zhang, S.; Liu, J.; Li, S.; Li, J.; Yang, P.; He, J.; Su, S.; Li, P. Serum Levels of Proamylin and Amylin in Normal Subjects and Patients with Impaired Glucose Regulation and Type 2 Diabetes Mellitus. Acta Diabetol. 2010, 47, 265–270. [Google Scholar] [CrossRef]
- Xu, J.; Wijesekara, N.; Regeenes, R.; Rijjal, D.A.; Piro, A.L.; Song, Y.; Wu, A.; Bhattacharjee, A.; Liu, Y.; Marzban, L.; et al. Pancreatic β Cell-Selective Zinc Transporter 8 Insufficiency Accelerates Diabetes Associated with Islet Amyloidosis. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Elias, S.; Delestre, C.; Ory, S.; Marais, S.; Courel, M.; Vazquez-Martinez, R.; Bernard, S.; Coquet, L.; Malagon, M.M.; Driouich, A.; et al. Chromogranin A Induces the Biogenesis of Granules with Calcium- and Actin-Dependent Dynamics and Exocytosis in Constitutively Secreting Cells. Endocrinology 2012, 153, 4444–4456. [Google Scholar] [CrossRef]
- Montero-Hadjadje, M.; Elias, S.; Chevalier, L.; Benard, M.; Tanguy, Y.; Turquier, V.; Galas, L.; Yon, L.; Malagon, M.M.; Driouich, A.; et al. Chromogranin A Promotes Peptide Hormone Sorting to Mobile Granules in Constitutively and Regulated Secreting Cells: Role of Conserved N- and C-Terminal Peptides. J. Biol. Chem. 2009, 284, 12420–12431. [Google Scholar] [CrossRef]
- Inomoto, C.; Umemura, S.; Egashira, N.; Minematsu, T.; Takekoshi, S.; Itoh, Y.; Itoh, J.; Taupenot, L.; O’Connor, D.T.; Osamura, R.Y. Granulogenesis in Non-Neuroendocrine COS-7 Cells Induced by EGFP-Tagged Chromogranin A Gene Transfection: Identical and Distinct Distribution of CgA and EGFP. J. Histochem. Cytochem. 2007, 55, 487–493. [Google Scholar] [CrossRef]
- Huh, Y.H.; Jeon, S.H.; Yoo, S.H. Chromogranin B-Induced Secretory Granule Biogenesis: Comparison with the Similar Role of Chromogranin A. J. Biol. Chem. 2003, 278, 40581–40589. [Google Scholar] [CrossRef]
- Chanat, E.; Huttner, W.B. Milieu-Induced, Selective Aggregation of Regulated Secretory Proteins in the Trans-Golgi Network. J. Cell Biol. 1991, 115, 1505–1519. [Google Scholar] [CrossRef]
- Yoo, S.H.; Albanesi, J.P. High Capacity, Low Affinity Ca2+ Binding of Chromogranin A. Relationship between the pH-Induced Conformational Change and Ca2+ Binding Property. J. Biol. Chem. 1991, 266, 7740–7745. [Google Scholar] [CrossRef]
- Sun-Wada, G.-H.; Toyomura, T.; Murata, Y.; Yamamoto, A.; Futai, M.; Wada, Y. The a3 Isoform of V-ATPase Regulates Insulin Secretion from Pancreatic Beta-Cells. J. Cell Sci. 2006, 119 Pt 21, 4531–4540. [Google Scholar] [CrossRef]
- Taupenot, L.; Harper, K.L.; O’Connor, D.T. Role of H+-ATPase-Mediated Acidification in Sorting and Release of the Regulated Secretory Protein Chromogranin A: Evidence for a Vesiculogenic Function. J. Biol. Chem. 2005, 280, 3885–3897. [Google Scholar] [CrossRef]
- Lissandron, V.; Podini, P.; Pizzo, P.; Pozzan, T. Unique Characteristics of Ca2+ Homeostasis of the Trans-Golgi Compartment. Proc. Natl. Acad. Sci. USA 2010, 107, 9198–9203. [Google Scholar] [CrossRef]
- Klemm, R.W.; Ejsing, C.S.; Surma, M.A.; Kaiser, H.-J.; Gerl, M.J.; Sampaio, J.L.; de Robillard, Q.; Ferguson, C.; Proszynski, T.J.; Shevchenko, A.; et al. Segregation of Sphingolipids and Sterols during Formation of Secretory Vesicles at the Trans-Golgi Network. J. Cell Biol. 2009, 185, 601–612. [Google Scholar] [CrossRef]
- Orci, L.; Montesano, R.; Meda, P.; Malaisse-Lagae, F.; Brown, D.; Perrelet, A.; Vassalli, P. Heterogeneous Distribution of Filipin--Cholesterol Complexes across the Cisternae of the Golgi Apparatus. Proc. Natl. Acad. Sci. USA 1981, 78, 293–297. [Google Scholar] [CrossRef]
- von Blume, J.; Hausser, A. Lipid-Dependent Coupling of Secretory Cargo Sorting and Trafficking at the Trans-Golgi Network. FEBS Lett. 2019, 593, 2412–2427. [Google Scholar] [CrossRef]
- Wang, Y.; Thiele, C.; Huttner, W.B. Cholesterol Is Required for the Formation of Regulated and Constitutive Secretory Vesicles from the Trans-Golgi Network. Traffic 2000, 1, 952–962. [Google Scholar] [CrossRef]
- Wang, T.Y.; Silvius, J.R. Different Sphingolipids Show Differential Partitioning into Sphingolipid/cholesterol-Rich Domains in Lipid Bilayers. Biophys. J. 2000, 79, 1478–1489. [Google Scholar] [CrossRef]
- Kreutzberger, A.J.B.; Kiessling, V.; Doyle, C.A.; Schenk, N.; Upchurch, C.M.; Elmer-Dixon, M.; Ward, A.E.; Preobraschenski, J.; Hussein, S.S.; Tomaka, W.; et al. Distinct Insulin Granule Subpopulations Implicated in the Secretory Pathology of Diabetes Types 1 and 2. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.S.; Harris, M.T.; Kreutzberger, A.J.B.; Inouye, C.M.; Doyle, C.A.; Castle, A.M.; Arvan, P.; Castle, J.D. Control of Insulin Granule Formation and Function by the ABC Transporters ABCG1 and ABCA1 and by Oxysterol Binding Protein OSBP. Mol. Biol. Cell 2018, 29, 1238–1257. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Hosaka, M.; Moriguchi, T.; Zhang, S.; Suda, M.; Yokota-Hashimoto, H.; Shinozuka, K.; Takeuchi, T. Cholesterol Biosynthesis Pathway Intermediates and Inhibitors Regulate Glucose-Stimulated Insulin Secretion and Secretory Granule Formation in Pancreatic Beta-Cells. Endocrinology 2010, 151, 4705–4716. [Google Scholar] [CrossRef]
- Bogan, J.S.; Xu, Y.; Hao, M. Cholesterol Accumulation Increases Insulin Granule Size and Impairs Membrane Trafficking. Traffic 2012, 13, 1466–1480. [Google Scholar] [CrossRef] [PubMed]
- Payet, L.-A.; Pineau, L.; Snyder, E.C.R.; Colas, J.; Moussa, A.; Vannier, B.; Bigay, J.; Clarhaut, J.; Becq, F.; Berjeaud, J.-M.; et al. Saturated Fatty Acids Alter the Late Secretory Pathway by Modulating Membrane Properties. Traffic 2013, 14, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Oh, Y.S.; Kang, M.K.; Huh, Y.H.; So, S.H.; Park, H.S.; Park, H.Y. Localization of Three Types of the Inositol 1,4,5-Trisphosphate Receptor/Ca2+ Channel in the Secretory Granules and Coupling with the Ca2+ Storage Proteins Chromogranins A and B*. J. Biol. Chem. 2001, 276, 45806–45812. [Google Scholar] [CrossRef]
- Yoo, S.H.; Lewis, M.S. Thermodynamic Study of the pH-Dependent Interaction of Chromogranin A with an Intraluminal Loop Peptide of the Inositol 1,4,5-Trisphosphate Receptor. Biochemistry 1995, 34, 632–638. [Google Scholar] [CrossRef]
- Hosaka, M.; Suda, M.; Sakai, Y.; Izumi, T.; Watanabe, T.; Takeuchi, T. Secretogranin III Binds to Cholesterol in the Secretory Granule Membrane as an Adapter for Chromogranin A. J. Biol. Chem. 2004, 279, 3627–3634. [Google Scholar] [CrossRef]
- Hosaka, M.; Watanabe, T.; Sakai, Y.; Uchiyama, Y.; Takeuchi, T. Identification of a Chromogranin A Domain That Mediates Binding to Secretogranin III and Targeting to Secretory Granules in Pituitary Cells and Pancreatic Beta-Cells. Mol. Biol. Cell 2002, 13, 3388–3399. [Google Scholar] [CrossRef]
- Han, L.; Suda, M.; Tsuzuki, K.; Wang, R.; Ohe, Y.; Hirai, H.; Watanabe, T.; Takeuchi, T.; Hosaka, M. A Large Form of Secretogranin III Functions as a Sorting Receptor for Chromogranin a Aggregates in PC12 Cells. Mol. Endocrinol. 2008, 22, 1935–1949. [Google Scholar] [CrossRef]
- Courel, M.; Vasquez, M.S.; Hook, V.Y.; Mahata, S.K.; Taupenot, L. Sorting of the Neuroendocrine Secretory Protein Secretogranin II into the Regulated Secretory Pathway: Role of N- and C-Terminal Alpha-Helical Domains. J. Biol. Chem. 2008, 283, 11807–11822. [Google Scholar] [CrossRef]
- Sun, M.; Watanabe, T.; Bochimoto, H.; Sakai, Y.; Torii, S.; Takeuchi, T.; Hosaka, M. Multiple Sorting Systems for Secretory Granules Ensure the Regulated Secretion of Peptide Hormones. Traffic 2013, 14, 205–218. [Google Scholar] [CrossRef]
- Hotta, K.; Hosaka, M.; Tanabe, A.; Takeuchi, T. Secretogranin II Binds to Secretogranin III and Forms Secretory Granules with Orexin, Neuropeptide Y, and POMC. J. Endocrinol. 2009, 202, 111–121. [Google Scholar]
- Courel, M.; Soler-Jover, A.; Rodriguez-Flores, J.L.; Mahata, S.K.; Elias, S.; Montero-Hadjadje, M.; Anouar, Y.; Giuly, R.J.; O’Connor, D.T.; Taupenot, L. Pro-Hormone Secretogranin II Regulates Dense Core Secretory Granule Biogenesis in Catecholaminergic Cells. J. Biol. Chem. 2010, 285, 10030–10043. [Google Scholar] [CrossRef]
- Glombik, M.M.; Krömer, A.; Salm, T.; Huttner, W.B.; Gerdes, H.H. The Disulfide-Bonded Loop of Chromogranin B Mediates Membrane Binding and Directs Sorting from the Trans-Golgi Network to Secretory Granules. EMBO J. 1999, 18, 1059–1070. [Google Scholar] [CrossRef]
- Pimplikar, S.W.; Huttner, W.B. Chromogranin B (secretogranin I), a Secretory Protein of the Regulated Pathway, Is Also Present in a Tightly Membrane-Associated Form in PC12 Cells. J. Biol. Chem. 1992, 267, 4110–4118. [Google Scholar] [CrossRef]
- Giordano, T.; Brigatti, C.; Podini, P.; Bonifacio, E.; Meldolesi, J.; Malosio, M.L. Beta Cell Chromogranin B Is Partially Segregated in Distinct Granules and Can Be Released Separately from Insulin in Response to Stimulation. Diabetologia 2008, 51, 997–1007. [Google Scholar] [CrossRef]
- Yoo, S.H.; Chu, S.Y.; Kim, K.D.; Huh, Y.H. Presence of Secretogranin II and High-Capacity, Low-Affinity Ca2+ Storage Role in Nucleoplasmic Ca2+ Store Vesicles. Biochemistry 2007, 46, 14663–14671. [Google Scholar] [CrossRef]
- Bearrows, S.C.; Bauchle, C.J.; Becker, M.; Haldeman, J.M.; Swaminathan, S.; Stephens, S.B. Chromogranin B Regulates Early-Stage Insulin Granule Trafficking from the Golgi in Pancreatic Islet β-Cells. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Yoo, S.H.; Lewis, M.S. Effects of pH and Ca2+ on Heterodimer and Heterotetramer Formation by Chromogranin A and Chromogranin B. J. Biol. Chem. 1996, 271, 17041–17046. [Google Scholar] [CrossRef]
- Garcia, A.L.; Han, S.-K.; Janssen, W.G.; Khaing, Z.Z.; Ito, T.; Glucksman, M.J.; Benson, D.L.; Salton, S.R.J. A Prohormone Convertase Cleavage Site within a Predicted α-Helix Mediates Sorting of the Neuronal and Endocrine Polypeptide VGF into the Regulated Secretory Pathway *. J. Biol. Chem. 2005, 280, 41595–41608. [Google Scholar] [CrossRef]
- Obermüller, S.; Calegari, F.; King, A.; Lindqvist, A.; Lundquist, I.; Salehi, A.; Francolini, M.; Rosa, P.; Rorsman, P.; Huttner, W.B.; et al. Defective Secretion of Islet Hormones in Chromogranin-B Deficient Mice. PLoS ONE 2010, 5, e8936. [Google Scholar] [CrossRef] [PubMed]
- Wollam, J.; Mahata, S.; Riopel, M.; Hernandez-Carretero, A.; Biswas, A.; Bandyopadhyay, G.K.; Chi, N.-W.; Eiden, L.E.; Mahapatra, N.R.; Corti, A.; et al. Chromogranin A Regulates Vesicle Storage and Mitochondrial Dynamics to Influence Insulin Secretion. Cell Tissue Res. 2017, 368, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, S.K.; Raimondo, A.; Hastoy, B.; Sengupta, S.; Dai, X.-Q.; Bautista, A.; Censin, J.; Payne, A.J.; Umapathysivam, M.M.; Spigelman, A.F.; et al. Type 2 Diabetes Risk Alleles in PAM Impact Insulin Release from Human Pancreatic β-Cells. Nat. Genet. 2018, 50, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Kudo, S.; Tsushima, K.; Sato, E.; Kubota, C.; Kayamori, A.; Bochimoto, H.; Koga, D.; Torii, S.; Gomi, H.; et al. Impaired Processing of Prohormones in Secretogranin III-Null Mice Causes Maladaptation to an Inadequate Diet and Stress. Endocrinology 2018, 159, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Stephens, S.B.; Edwards, R.J.; Sadahiro, M.; Lin, W.-J.; Jiang, C.; Salton, S.R.; Newgard, C.B. The Prohormone VGF Regulates β Cell Function via Insulin Secretory Granule Biogenesis. Cell Rep. 2017, 20, 2480–2489. [Google Scholar] [CrossRef]
- Dittié, A.S.; Thomas, L.; Thomas, G.; Tooze, S.A. Interaction of Furin in Immature Secretory Granules from Neuroendocrine Cells with the AP-1 Adaptor Complex Is Modulated by Casein Kinase II Phosphorylation. EMBO J. 1997, 16, 4859–4870. [Google Scholar] [CrossRef]
- Finzi, G.; Franzi, F.; Placidi, C.; Acquati, F.; Palumbo, E.; Russo, A.; Taramelli, R.; Sessa, F.; La Rosa, S. BACE2 Is Stored in Secretory Granules of Mouse and Rat Pancreatic Beta Cells. Ultrastruct. Pathol. 2008, 32, 246–251. [Google Scholar] [CrossRef]
- Arias, A.E.; Vélez-Granell, C.S.; Mayer, G.; Bendayan, M. Colocalization of Chaperone Cpn60, Proinsulin and Convertase PC1 within Immature Secretory Granules of Insulin-Secreting Cells Suggests a Role for Cpn60 in Insulin Processing. J. Cell Sci. 2000, 113 Pt 11, 2075–2083. [Google Scholar] [CrossRef]
- Docherty, K.; Hutton, J.C. Carboxypeptidase Activity in the Insulin Secretory Granule. FEBS Lett. 1983, 162, 137–141. [Google Scholar] [CrossRef]
- Dhanvantari, S.; Arnaoutova, I.; Snell, C.R.; Steinbach, P.J.; Hammond, K.; Caputo, G.A.; London, E.; Loh, Y.P. Carboxypeptidase E, a Prohormone Sorting Receptor, Is Anchored to Secretory Granules via a C-Terminal Transmembrane Insertion. Biochemistry 2002, 41, 52–60. [Google Scholar] [CrossRef]
- Fricker, L.D.; Das, B.; Angeletti, R.H. Identification of the pH-Dependent Membrane Anchor of Carboxypeptidase E (EC 3.4.17.10). J. Biol. Chem. 1990, 265, 2476–2482. [Google Scholar] [CrossRef]
- Demaurex, N.; Furuya, W.; D’Souza, S.; Bonifacino, J.S.; Grinstein, S. Mechanism of Acidification of the Trans-Golgi Network (TGN). In Situ Measurements of pH Using Retrieval of TGN38 and Furin from the Cell Surface. J. Biol. Chem. 1998, 273, 2044–2051. [Google Scholar] [CrossRef]
- Song, L.; Fricker, L.D. Calcium- and pH-Dependent Aggregation of Carboxypeptidase E. J. Biol. Chem. 1995, 270, 7963–7967. [Google Scholar] [CrossRef]
- Rindler, M.J. Carboxypeptidase E, a Peripheral Membrane Protein Implicated in the Targeting of Hormones to Secretory Granules, Co-Aggregates with Granule Content Proteins at Acidic pH. J. Biol. Chem. 1998, 273, 31180–31185. [Google Scholar] [CrossRef][Green Version]
- Hosaka, M.; Watanabe, T.; Sakai, Y.; Kato, T.; Takeuchi, T. Interaction between Secretogranin III and Carboxypeptidase E Facilitates Prohormone Sorting within Secretory Granules. J. Cell Sci. 2005, 118 Pt 20, 4785–4795. [Google Scholar] [CrossRef]
- Guest, P.C.; Arden, S.D.; Rutherford, N.G.; Hutton, J.C. The Post-Translational Processing and Intracellular Sorting of Carboxypeptidase H in the Islets of Langerhans. Mol. Cell. Endocrinol. 1995, 113, 99–108. [Google Scholar] [CrossRef]
- Chu, K.Y.; Briggs, M.J.L.; Albrecht, T.; Drain, P.F.; Johnson, J.D. Differential Regulation and Localization of Carboxypeptidase D and Carboxypeptidase E in Human and Mouse β-Cells. Islets 2011, 3, 155–165. [Google Scholar] [CrossRef]
- Greene, D.; Das, B.; Fricker, L.D. Regulation of Carboxypeptidase E. Effect of pH, Temperature and Co2+ on Kinetic Parameters of Substrate Hydrolysis. Biochem. J. 1992, 2, 613–618. [Google Scholar] [CrossRef]
- Benjannet, S.; Rondeau, N.; Paquet, L.; Boudreault, A.; Lazure, C.; Chrétien, M.; Seidah, N.G. Comparative Biosynthesis, Covalent Post-Translational Modifications and Efficiency of Prosegment Cleavage of the Prohormone Convertases PC1 and PC2: Glycosylation, Sulphation and Identification of the Intracellular Site of Prosegment Cleavage of PC1 and PC2. Biochem. J. 1993, 294 Pt 3, 735–743. [Google Scholar]
- Zhou, A.; Mains, R.E. Endoproteolytic Processing of Proopiomelanocortin and Prohormone Convertases 1 and 2 in Neuroendocrine Cells Overexpressing Prohormone Convertases 1 or 2. J. Biol. Chem. 1994, 269, 17440–17447. [Google Scholar] [CrossRef]
- Boudreault, A.; Gauthier, D.; Lazure, C. Proprotein Convertase PC1/3-Related Peptides Are Potent Slow Tight-Binding Inhibitors of Murine PC1/3 and Hfurin*. J. Biol. Chem. 1998, 273, 31574–31580. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jutras, I.; Seidah, N.G.; Reudelhuber, T.L.; Brechler, V. Two Activation States of the Prohormone Convertase PC1 in the Secretory Pathway. J. Biol. Chem. 1997, 272, 15184–15188. [Google Scholar] [CrossRef] [PubMed]
- Rabah, N.; Gauthier, D.; Dikeakos, J.D.; Reudelhuber, T.L.; Lazure, C. The C-Terminal Region of the Proprotein Convertase 1/3 (PC1/3) Exerts a Bimodal Regulation of the Enzyme Activity in Vitro. FEBS J. 2007, 274, 3482–3491. [Google Scholar] [CrossRef] [PubMed]
- Benjannet, S.; Rondeau, N.; Day, R.; Chrétien, M.; Seidah, N.G. PC1 and PC2 Are Proprotein Convertases Capable of Cleaving Proopiomelanocortin at Distinct Pairs of Basic Residues. Proc. Natl. Acad. Sci. USA 1991, 88, 3564–3568. [Google Scholar] [CrossRef]
- Thomas, L.; Leduc, R.; Thorne, B.A.; Smeekens, S.P.; Steiner, D.F.; Thomas, G. Kex2-like Endoproteases PC2 and PC3 Accurately Cleave a Model Prohormone in Mammalian Cells: Evidence for a Common Core of Neuroendocrine Processing Enzymes. Proc. Natl. Acad. Sci. USA 1991, 88, 5297–5301. [Google Scholar] [CrossRef]
- Jutras, I.; Seidah, N.G.; Reudelhuber, T.L. A Predicted Alpha -Helix Mediates Targeting of the Proprotein Convertase PC1 to the Regulated Secretory Pathway. J. Biol. Chem. 2000, 275, 40337–40343. [Google Scholar] [CrossRef]
- Blázquez, M.; Docherty, K.; Shennan, K.I. Association of Prohormone Convertase 3 with Membrane Lipid Rafts. J. Mol. Endocrinol. 2001, 27, 107–116. [Google Scholar] [CrossRef][Green Version]
- Zhou, Y.; Lindberg, I. Enzymatic Properties of Carboxyl-Terminally Truncated Prohormone Convertase 1 (PC1/SPC3) and Evidence for Autocatalytic Conversion. J. Biol. Chem. 1994, 269, 18408–18413. [Google Scholar] [CrossRef]
- Zhou, Y.; Lindberg, I. Purification and Characterization of the Prohormone Convertase PC1(PC3). J. Biol. Chem. 1993, 268, 5615–5623. [Google Scholar] [CrossRef]
- Muller, L.; Cameron, A.; Fortenberry, Y.; Apletalina, E.V.; Lindberg, I. Processing and Sorting of the Prohormone Convertase 2 Propeptide *. J. Biol. Chem. 2000, 275, 39213–39222. [Google Scholar] [CrossRef]
- Lee, S.-N.; Lindberg, I. 7B2 Prevents Unfolding and Aggregation of Prohormone Convertase 2. Endocrinology 2008, 149, 4116–4127. [Google Scholar] [CrossRef]
- Benjannet, S.; Savaria, D.; Chrétien, M.; Seidah, N.G. 7B2 Is a Specific Intracellular Binding Protein of the Prohormone Convertase PC2. J. Neurochem. 1995, 64, 2303–2311. [Google Scholar] [CrossRef]
- Benjannet, S.; Mamarbachi, A.M.; Hamelin, J.; Savaria, D.; Munzer, J.S.; Chrétien, M.; Seidah, N.G. Residues Unique to the pro-Hormone Convertase PC2 Modulate Its Autoactivation, Binding to 7B2 and Enzymatic Activity. FEBS Lett. 1998, 428, 37–42. [Google Scholar] [CrossRef][Green Version]
- Paquet, L.; Bergeron, F.; Boudreault, A.; Seidah, N.G.; Chrétien, M.; Mbikay, M.; Lazure, C. The Neuroendocrine Precursor 7B2 Is a Sulfated Protein Proteolytically Processed by a Ubiquitous Furin-like Convertase. J. Biol. Chem. 1994, 269, 19279–19285. [Google Scholar] [CrossRef]
- Braks, J.A.; Van Horssen, A.M.; Martens, G.J. Dissociation of the Complex between the Neuroendocrine Chaperone 7B2 and Prohormone Convertase PC2 Is Not Associated with proPC2 Maturation. Eur. J. Biochem. 1996, 238, 505–510. [Google Scholar] [CrossRef]
- Shennan, K.I.; Taylor, N.A.; Docherty, K. Calcium- and pH-Dependent Aggregation and Membrane Association of the Precursor of the Prohormone Convertase PC2. J. Biol. Chem. 1994, 269, 18646–18650. [Google Scholar] [CrossRef]
- Linard, C.G.; Tadros, H.; Sirois, F.; Mbikay, M. Calcium-Induced Aggregation of Neuroendocrine Protein 7B2 in Vitro and Its Modulation by ATP. Mol. Cell. Biochem. 1995, 151, 39–47. [Google Scholar] [CrossRef]
- Blázquez, M.; Thiele, C.; Huttner, W.B.; Docherty, K.; Shennan, K.I. Involvement of the Membrane Lipid Bilayer in Sorting Prohormone Convertase 2 into the Regulated Secretory Pathway. Biochem. J. 2000, 349 Pt 3, 843–852. [Google Scholar] [CrossRef]
- Rovère, C.; Mort, J.S.; Chrétien, M.; Seidah, N.G. Cathepsin-B Fusion Proteins Misroute Secretory Protein Partners such as the Proprotein Convertase PC2-7B2 Complex toward the Lysosomal Degradation Pathways. Biochem. Biophys. Res. Commun. 2000, 276, 594–599. [Google Scholar] [CrossRef]
- Barbero, P.; Kitabgi, P. Protein 7B2 Is Essential for the Targeting and Activation of PC2 into the Regulated Secretory Pathway of rMTC 6-23 Cells. Biochem. Biophys. Res. Commun. 1999, 257, 473–479. [Google Scholar] [CrossRef]
- Perkins, S.N.; Husten, E.J.; Eipper, B.A. The 108-kDA Peptidylglycine Alpha-Amidating Monooxygenase Precursor Contains Two Separable Enzymatic Activities Involved in Peptide Amidation. Biochem. Biophys. Res. Commun. 1990, 171, 926–932. [Google Scholar] [CrossRef]
- Bradbury, A.F.; Finnie, M.D.; Smyth, D.G. Mechanism of C-Terminal Amide Formation by Pituitary Enzymes. Nature 1982, 298, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Merkler, D.J. C-Terminal Amidated Peptides: Production by the in Vitro Enzymatic Amidation of Glycine-Extended Peptides and the Importance of the Amide to Bioactivity. Enzym. Microb. Technol. 1994, 16, 450–456. [Google Scholar] [CrossRef]
- Bousquet-Moore, D.; Mains, R.E.; Eipper, B.A. Peptidylgycine α-Amidating Monooxygenase and Copper: A Gene-Nutrient Interaction Critical to Nervous System Function. J. Neurosci. Res. 2010, 88, 2535–2545. [Google Scholar] [CrossRef]
- Eipper, B.A.; Perkins, S.N.; Husten, E.J.; Johnson, R.C.; Keutmann, H.T.; Mains, R.E. Peptidyl-Alpha-Hydroxyglycine Alpha-Amidating Lyase. Purification, Characterization, and Expression. J. Biol. Chem. 1991, 266, 7827–7833. [Google Scholar] [CrossRef]
- Bundgaard, H.; Kahns, A.H. Chemical Stability and Plasma-Catalyzed Dealkylation of Peptidyl-α-Hydroxyglycine derivatives—Intermediates in Peptide α-Amidation. Peptides 1991, 12, 745–748. [Google Scholar] [CrossRef]
- Kyun-Hwan, K.; Baik, L.S. Peptide Amidation: Production of Peptide Hormones in Vivo And in Vitro. Biotechnol. Bioproce. Eng. 2001, 6, 244–251. [Google Scholar]
- Chen, Y.-C.; Mains, R.E.; Eipper, B.A.; Hoffman, B.G.; Czyzyk, T.A.; Pintar, J.E.; Verchere, C.B. PAM Haploinsufficiency Does Not Accelerate the Development of Diet- and Human IAPP-Induced Diabetes in Mice. Diabetologia 2020, 63, 561–576. [Google Scholar] [CrossRef]
- Kumar, D.; Mains, R.E.; Eipper, B.A. 60 YEARS OF POMC: From POMC and α-MSH to PAM, Molecular Oxygen, Copper, and Vitamin C. J. Mol. Endocrinol. 2016, 56, T63–T76. [Google Scholar] [CrossRef]
- Oyarce, A.M.; Eipper, B.A. Neurosecretory Vesicles Contain Soluble and Membrane-Associated Monofunctional and Bifunctional Peptidylglycine Alpha-Amidating Monooxygenase Proteins. J. Neurochem. 1993, 60, 1105–1114. [Google Scholar] [CrossRef]
- Husten, E.J.; Eipper, B.A. Purification and Characterization of PAM-1, an Integral Membrane Protein Involved in Peptide Processing. Arch. Biochem. Biophys. 1994, 312, 487–492. [Google Scholar] [CrossRef]
- Milgram, S.L.; Eipper, B.A.; Mains, R.E. Differential Trafficking of Soluble and Integral Membrane Secretory Granule-Associated Proteins. J. Cell Biol. 1994, 124, 33–41. [Google Scholar] [CrossRef]
- Milgram, S.L.; Johnson, R.C.; Mains, R.E. Expression of Individual Forms of Peptidylglycine Alpha-Amidating Monooxygenase in AtT-20 Cells: Endoproteolytic Processing and Routing to Secretory Granules. J. Cell Biol. 1992, 117, 717–728. [Google Scholar] [CrossRef]
- Milgram, S.L.; Mains, R.E.; Eipper, B.A. COOH-Terminal Signals Mediate the Trafficking of a Peptide Processing Enzyme in Endocrine Cells. J. Cell Biol. 1993, 121, 23–36. [Google Scholar] [CrossRef]
- Milgram, S.L.; Mains, R.E.; Eipper, B.A. Identification of Routing Determinants in the Cytosolic Domain of a Secretory Granule-Associated Integral Membrane Protein. J. Biol. Chem. 1996, 271, 17526–17535. [Google Scholar] [CrossRef]
- El Meskini, R.; Galano, G.J.; Marx, R.; Mains, R.E.; Eipper, B.A. Targeting of Membrane Proteins to the Regulated Secretory Pathway in Anterior Pituitary Endocrine Cells. J. Biol. Chem. 2001, 276, 3384–3393. [Google Scholar] [CrossRef]
- Marx, R.; Mains, R.E. Routing of Membrane Proteins to Large Dense Core Vesicles in PC12 Cells. J. Mol. Neurosci. 2002, 18, 113–127. [Google Scholar] [CrossRef]
- Chufán, E.E.; De, M.; Eipper, B.A.; Mains, R.E.; Amzel, L.M. Amidation of Bioactive Peptides: The Structure of the Lyase Domain of the Amidating Enzyme. Structure 2009, 17, 965–973. [Google Scholar] [CrossRef]
- Kulathila, R.; Consalvo, A.P.; Fitzpatrick, P.F.; Freeman, J.C.; Snyder, L.M.; Villafranca, J.J.; Merkler, D.J. Bifunctional Peptidylglcine Alpha-Amidating Enzyme Requires Two Copper Atoms for Maximum Activity. Arch. Biochem. Biophys. 1994, 311, 191–195. [Google Scholar] [CrossRef]
- Hutton, J.C. The Internal pH and Membrane Potential of the Insulin-Secretory Granule. Biochem. J. 1982, 204, 171–178. [Google Scholar] [CrossRef]
- Barg, S.; Huang, P.; Eliasson, L.; Nelson, D.J.; Obermüller, S.; Rorsman, P.; Thévenod, F.; Renström, E. Priming of Insulin Granules for Exocytosis by Granular Cl(-) Uptake and Acidification. J. Cell Sci. 2001, 114, 2145–2154. [Google Scholar] [CrossRef]
- Deriy, L.V.; Gomez, E.A.; Jacobson, D.A.; Wang, X.; Hopson, J.A.; Liu, X.Y.; Zhang, G.; Bindokas, V.P.; Philipson, L.H.; Nelson, D.J. The Granular Chloride Channel ClC-3 Is Permissive for Insulin Secretion. Cell Metab. 2009, 10, 316–323. [Google Scholar] [CrossRef]
- Li, D.-Q.; Jing, X.; Salehi, A.; Collins, S.C.; Hoppa, M.B.; Rosengren, A.H.; Zhang, E.; Lundquist, I.; Olofsson, C.S.; Mörgelin, M.; et al. Suppression of Sulfonylurea- and Glucose-Induced Insulin Secretion in Vitro and in Vivo in Mice Lacking the Chloride Transport Protein ClC-3. Cell Metab. 2009, 10, 309–315. [Google Scholar] [CrossRef]
- Maritzen, T.; Keating, D.J.; Neagoe, I.; Zdebik, A.A.; Jentsch, T.J. Role of the Vesicular Chloride Transporter ClC-3 in Neuroendocrine Tissue. J. Neurosci. 2008, 28, 10587–10598. [Google Scholar] [CrossRef]
- Jentsch, T.J.; Maritzen, T.; Keating, D.J.; Zdebik, A.A.; Thévenod, F. ClC-3--a Granular Anion Transporter Involved in Insulin Secretion? Cell Metab. 2010, 12, 307–308. [Google Scholar] [CrossRef]
- Blackmore, C.G.; Varro, A.; Dimaline, R.; Bishop, L.; Gallacher, D.V.; Dockray, G.J. Measurement of Secretory Vesicle pH Reveals Intravesicular Alkalinization by Vesicular Monoamine Transporter Type 2 Resulting in Inhibition of Prohormone Cleavage. J. Physiol. 2001, 531 Pt 3, 605–617. [Google Scholar] [CrossRef]
- Holliday, L.S. Vacuolar H+-ATPase: An Essential Multitasking Enzyme in Physiology and Pathophysiology. New J. Sci. 2014, 2014. [Google Scholar] [CrossRef]
- Maxson, M.E.; Grinstein, S. The Vacuolar-Type H+-ATPase at a Glance—More than a Proton Pump. J. Cell Sci. 2014, 127 Pt 23, 4987–4993. [Google Scholar] [CrossRef]
- Lafourcade, C.; Sobo, K.; Kieffer-Jaquinod, S.; Garin, J.; van der Goot, F.G. Regulation of the V-ATPase along the Endocytic Pathway Occurs through Reversible Subunit Association and Membrane Localization. PLoS ONE 2008, 3, e2758. [Google Scholar] [CrossRef]
- Kane, P.M. Disassembly and Reassembly of the Yeast Vacuolar H(+)-ATPase in Vivo. J. Biol. Chem. 1995, 270, 17025–17032. [Google Scholar] [CrossRef]
- Chung, J.-H.; Lester, R.L.; Dickson, R.C. Sphingolipid Requirement for Generation of a Functional V1 Component of the Vacuolar ATPase*. J. Biol. Chem. 2003, 278, 28872–28881. [Google Scholar] [CrossRef] [PubMed]
- Kettner, C.; Bertl, A.; Obermeyer, G.; Slayman, C.; Bihler, H. Electrophysiological Analysis of the Yeast V-Type Proton Pump: Variable Coupling Ratio and Proton Shunt. Biophys. J. 2003, 85, 3730–3738. [Google Scholar] [CrossRef]
- Cross, R.L.; Müller, V. The Evolution of A-, F-, and V-Type ATP Synthases and ATPases: Reversals in Function and Changes in the H+/ATP Coupling Ratio. FEBS Lett. 2004, 576, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki-Nishi, S.; Bowers, K.; Nishi, T.; Forgac, M.; Stevens, T.H. The Amino-Terminal Domain of the Vacuolar Proton-Translocating ATPase a Subunit Controls Targeting and in Vivo Dissociation, and the Carboxyl-Terminal Domain Affects Coupling of Proton Transport and ATP Hydrolysis*. J. Biol. Chem. 2001, 276, 47411–47420. [Google Scholar] [CrossRef]
- Su, Y.; Zhou, A.; Al-Lamki, R.S.; Karet, F.E. The a-Subunit of the V-Type H+-ATPase Interacts with Phosphofructokinase-1 in Humans. J. Biol. Chem. 2003, 278, 20013–20018. [Google Scholar] [CrossRef]
- Banerjee, S.; Kane, P.M. Regulation of V-ATPase Activity and Organelle pH by Phosphatidylinositol Phosphate Lipids. Front. Cell Dev. Biol. 2020, 8, 510. [Google Scholar] [CrossRef]
- Stiernet, P.; Guiot, Y.; Gilon, P.; Henquin, J.-C. Glucose Acutely Decreases pH of Secretory Granules in Mouse Pancreatic Islets. Mechanisms and Influence on Insulin Secretion. J. Biol. Chem. 2006, 281, 22142–22151. [Google Scholar] [CrossRef]
- Louagie, E.; Taylor, N.A.; Flamez, D.; Roebroek, A.J.M.; Bright, N.A.; Meulemans, S.; Quintens, R.; Herrera, P.L.; Schuit, F.; Van de Ven, W.J.M.; et al. Role of Furin in Granular Acidification in the Endocrine Pancreas: Identification of the V-ATPase Subunit Ac45 as a Candidate Substrate. Proc. Natl. Acad. Sci. USA 2008, 105, 12319–12324. [Google Scholar] [CrossRef]
- Dai, F.F.; Bhattacharjee, A.; Liu, Y.; Batchuluun, B.; Zhang, M.; Wang, X.S.; Huang, X.; Luu, L.; Zhu, D.; Gaisano, H.; et al. A Novel GLP1 Receptor Interacting Protein ATP6ap2 Regulates Insulin Secretion in Pancreatic Beta Cells. J. Biol. Chem. 2015, 290, 25045–25061. [Google Scholar] [CrossRef]
- Binger, K.J.; Neukam, M.; Tattikota, S.G.; Qadri, F.; Puchkov, D.; Willmes, D.M.; Wurmsee, S.; Geisberger, S.; Dechend, R.; Raile, K.; et al. Atp6ap2 Deletion Causes Extensive Vacuolation That Consumes the Insulin Content of Pancreatic β Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19983–19988. [Google Scholar] [CrossRef]
- Brouwers, B.; Coppola, I.; Vints, K.; Dislich, B.; Jouvet, N.; Van Lommel, L.; Segers, C.; Gounko, N.V.; Thorrez, L.; Schuit, F.; et al. Loss of Furin in β-Cells Induces an mTORC1-ATF4 Anabolic Pathway That Leads to β-Cell Dysfunction. Diabetes 2021, 70, 492–503. [Google Scholar] [CrossRef]
- Hatanaka, M.; Tanabe, K.; Yanai, A.; Ohta, Y.; Kondo, M.; Akiyama, M.; Shinoda, K.; Oka, Y.; Tanizawa, Y. Wolfram Syndrome 1 Gene (WFS1) Product Localizes to Secretory Granules and Determines Granule Acidification in Pancreatic Beta-Cells. Hum. Mol. Genet. 2011, 20, 1274–1284. [Google Scholar] [CrossRef]
- Gharanei, S.; Zatyka, M.; Astuti, D.; Fenton, J.; Sik, A.; Nagy, Z.; Barrett, T.G. Vacuolar-Type H+-ATPase V1A Subunit Is a Molecular Partner of Wolfram Syndrome 1 (WFS1) Protein, Which Regulates Its Expression and Stability. Hum. Mol. Genet. 2013, 22, 203–217. [Google Scholar] [CrossRef]
- Vinkenborg, J.L.; Nicolson, T.J.; Bellomo, E.A.; Koay, M.S.; Rutter, G.A.; Merkx, M. Genetically Encoded FRET Sensors to Monitor Intracellular Zn2+ Homeostasis. Nat. Methods 2009, 6, 737–740. [Google Scholar] [CrossRef]
- Bafaro, E.; Liu, Y.; Xu, Y.; Dempski, R.E. The Emerging Role of Zinc Transporters in Cellular Homeostasis and Cancer. Signal Transd. Target. Ther. 2017, 2. [Google Scholar] [CrossRef]
- Lubag, A.J.M.; De Leon-Rodriguez, L.M.; Burgess, S.C.; Sherry, A.D. Noninvasive MRI of β-Cell Function Using a Zn2+-Responsive Contrast Agent. Proc. Natl. Acad. Sci. USA 2011, 108, 18400–18405. [Google Scholar] [CrossRef]
- Slepchenko, K.G.; Daniels, N.A.; Guo, A.; Li, Y.V. Autocrine Effect of Zn2+ on the Glucose-Stimulated Insulin Secretion. Endocrine 2015, 50, 110–122. [Google Scholar] [CrossRef]
- Slepchenko, K.G.; James, C.B.L.; Li, Y.V. Inhibitory Effect of Zinc on Glucose-Stimulated Zinc/insulin Secretion in an Insulin-Secreting β-Cell Line. Exp. Physiol. 2013, 98, 1301–1311. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, T.; Harmon, J.S.; Bryan, J.; Robertson, R.P. Zinc, Not Insulin, Regulates the Rat Alpha-Cell Response to Hypoglycemia in Vivo. Diabetes 2007, 56, 1107–1112. [Google Scholar] [CrossRef]
- Hardy, A.B.; Serino, A.S.; Wijesekara, N.; Chimienti, F.; Wheeler, M.B. Regulation of Glucagon Secretion by Zinc: Lessons from the β Cell-Specific Znt8 Knockout Mouse Model. Diabetes Obes. Metab. 2011, 13, 112–117. [Google Scholar] [CrossRef]
- Tamaki, M.; Fujitani, Y.; Hara, A.; Uchida, T.; Tamura, Y.; Takeno, K.; Kawaguchi, M.; Watanabe, T.; Ogihara, T.; Fukunaka, A.; et al. The Diabetes-Susceptible Gene SLC30A8/ZnT8 Regulates Hepatic Insulin Clearance. J. Clin. Investig. 2013, 123, 4513–4524. [Google Scholar] [CrossRef]
- Nedumpully-Govindan, P.; Ding, F. Inhibition of IAPP Aggregation by Insulin Depends on the Insulin Oligomeric State Regulated by Zinc Ion Concentration. Sci. Rep. 2015, 5, 8240. [Google Scholar] [CrossRef]
- Banaszak, M.; Górna, I.; Przysławski, J. Zinc and the Innovative Zinc-α2-Glycoprotein Adipokine Play an Important Role in Lipid Metabolism: A Critical Review. Nutrients 2021, 13, 2023. [Google Scholar] [CrossRef]
- Rutter, G.A.; Chabosseau, P.; Bellomo, E.A.; Maret, W.; Mitchell, R.K.; Hodson, D.J.; Solomou, A.; Hu, M. Intracellular Zinc in Insulin Secretion and Action: A Determinant of Diabetes Risk? Proc. Nutr. Soc. 2016, 75, 61–72. [Google Scholar] [CrossRef]
- Cooper-Capetini, V.; de Vasconcelos, D.A.A.; Martins, A.R.; Hirabara, S.M.; Donato, J., Jr.; Carpinelli, A.R.; Abdulkader, F. Zinc Supplementation Improves Glucose Homeostasis in High Fat-Fed Mice by Enhancing Pancreatic β-Cell Function. Nutrients 2017, 9, 1150. [Google Scholar] [CrossRef]
- Daniels, M.J.; Jagielnicki, M.; Yeager, M. Structure/Function Analysis of Human ZnT8 (SLC30A8): A Diabetes Risk Factor and Zinc Transporter. Curr. Res. Struct. Biol. 2020, 2, 144–155. [Google Scholar] [CrossRef]
- Smidt, K.; Larsen, A.; Brønden, A.; Sørensen, K.S.; Nielsen, J.V.; Praetorius, J.; Martensen, P.M.; Rungby, J. The Zinc Transporter ZNT3 Co-Localizes with Insulin in INS-1E Pancreatic Beta Cells and Influences Cell Survival, Insulin Secretion Capacity, and ZNT8 Expression. Biometals 2016, 29, 287–298. [Google Scholar] [CrossRef]
- Bellomo, E.A.; Meur, G.; Rutter, G.A. Glucose Regulates Free Cytosolic Zn2+ Concentration, Slc39 (ZiP), and Metallothionein Gene Expression in Primary Pancreatic Islet β-Cells. J. Biol. Chem. 2011, 286, 25778–25789. [Google Scholar] [CrossRef]
- Barragán-Álvarez, C.P.; Padilla-Camberos, E.; Díaz, N.F.; Cota-Coronado, A.; Hernández-Jiménez, C.; Bravo-Reyna, C.C.; Díaz-Martínez, N.E. Loss of Znt8 Function in Diabetes Mellitus: Risk or Benefit? Mol. Cell. Biochem. 2021. [Google Scholar] [CrossRef]
- Rutter, G.; Chimienti, F. SLC30A8 Mutations in Type 2 Diabetes. Diabetologia 2014, 58, 31–36. [Google Scholar] [CrossRef]
- Davidson, H.W.; Wenzlau, J.M.; O’Brien, R.M. Zinc Transporter 8 (ZnT8) and β Cell Function. Trends Endocrinol. Metab. 2014, 25, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.E.; Kells, D.I.; Yip, C.C. Physical and Biological Properties of Guinea Pig Insulin. Biochem. Biophys. Res. Commun. 1972, 46, 2127–2133. [Google Scholar] [CrossRef]
- O’Halloran, T.V.; Kebede, M.; Philips, S.J.; Attie, A.D. Zinc, Insulin, and the Liver: A Ménage à Trois. J. Clin. Investig. 2013, 123, 4136–4139. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, K.; Ravier, M.A.; Schraenen, A.; Creemers, J.W.M.; Van de Plas, R.; Granvik, M.; Van Lommel, L.; Waelkens, E.; Chimienti, F.; Rutter, G.A.; et al. Insulin Crystallization Depends on Zinc Transporter ZnT8 Expression, but Is Not Required for Normal Glucose Homeostasis in Mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14872–14877. [Google Scholar] [CrossRef]
- Syring, K.E.; Bosma, K.J.; Goleva, S.B.; Singh, K.; Oeser, J.K.; Lopez, C.A.; Skaar, E.P.; McGuinness, O.P.; Davis, L.K.; Powell, D.R.; et al. Potential Positive and Negative Consequences of ZnT8 Inhibition. J. Endocrinol. 2020, 246, 189–205. [Google Scholar] [CrossRef]
- Syring, K.E.; Boortz, K.A.; Oeser, J.K.; Ustione, A.; Platt, K.A.; Shadoan, M.K.; McGuinness, O.P.; Piston, D.W.; Powell, D.R.; O’Brien, R.M. Combined Deletion of Slc30a7 and Slc30a8 Unmasks a Critical Role for ZnT8 in Glucose-Stimulated Insulin Secretion. Endocrinology 2016, 157, 4534–4541. [Google Scholar] [CrossRef]
- Merriman, C.; Huang, Q.; Rutter, G.A.; Fu, D. Lipid-Tuned Zinc Transport Activity of Human ZnT8 Protein Correlates with Risk for Type-2 Diabetes. J. Biol. Chem. 2016, 291, 26950–26957. [Google Scholar] [CrossRef]
- Kirchhoff, K.; Machicao, F.; Haupt, A.; Schäfer, S.A.; Tschritter, O.; Staiger, H.; Stefan, N.; Häring, H.-U.; Fritsche, A. Polymorphisms in the TCF7L2, CDKAL1 and SLC30A8 Genes Are Associated with Impaired Proinsulin Conversion. Diabetologia 2008, 51, 597–601. [Google Scholar] [CrossRef]
- Tumarada, N.; Li, L.; Bai, S.; Sheline, C.T. hZnT8 (Slc30a8) Transgenic Mice That Overexpress the R325W Polymorph Have Reduced Islet Zn2+ and Proinsulin Levels, Increased Glucose Tolerance After a High-Fat Diet, and Altered Levels of Pancreatic Zinc Binding Proteins. Diabetes 2017, 66, 551–559. [Google Scholar] [CrossRef]
- Dwivedi, O.P.; Lehtovirta, M.; Hastoy, B.; Chandra, V.; Krentz, N.A.J.; Kleiner, S.; Jain, D.; Richard, A.-M.; Abaitua, F.; Beer, N.L.; et al. Loss of ZnT8 Function Protects against Diabetes by Enhanced Insulin Secretion. Nat. Genet. 2019, 51, 1596–1606. [Google Scholar] [CrossRef]
- Kim, I.; Kang, E.S.; Yim, Y.S.; Ko, S.J.; Jeong, S.-H.; Rim, J.H.; Kim, Y.S.; Ahn, C.W.; Cha, B.S.; Lee, H.C.; et al. A Low-Risk ZnT-8 Allele (W325) for Post-Transplantation Diabetes Mellitus Is Protective against Cyclosporin A-Induced Impairment of Insulin Secretion. Pharm. J. 2011, 11, 191–198. [Google Scholar] [CrossRef]
- Cauchi, S.; Nead, K.T.; Choquet, H.; Horber, F.; Potoczna, N.; Balkau, B.; Marre, M.; Charpentier, G.; Froguel, P.; Meyre, D. The Genetic Susceptibility to Type 2 Diabetes May Be Modulated by Obesity Status: Implications for Association Studies. BMC Med. Genet. 2008, 9, 45. [Google Scholar] [CrossRef]
- Gerdes, H.H.; Rosa, P.; Phillips, E.; Baeuerle, P.A.; Frank, R.; Argos, P.; Huttner, W.B. The Primary Structure of Human Secretogranin II, a Widespread Tyrosine-Sulfated Secretory Granule Protein That Exhibits Low pH- and Calcium-Induced Aggregation. J. Biol. Chem. 1989, 264, 12009–12015. [Google Scholar] [CrossRef]
- Scheenen, W.J.; Wollheim, C.B.; Pozzan, T.; Fasolato, C. Ca2+ Depletion from Granules Inhibits Exocytosis. A Study with Insulin-Secreting Cells. J. Biol. Chem. 1998, 273, 19002–19008. [Google Scholar] [CrossRef]
- Mitchell, K.J.; Lai, F.A.; Rutter, G.A. Ryanodine Receptor Type I and Nicotinic Acid Adenine Dinucleotide Phosphate Receptors Mediate Ca2+ Release from Insulin-Containing Vesicles in Living Pancreatic Beta-Cells (MIN6). J. Biol. Chem. 2003, 278, 11057–11064. [Google Scholar] [CrossRef]
- von Blume, J.; Alleaume, A.-M.; Kienzle, C.; Carreras-Sureda, A.; Valverde, M.; Malhotra, V. Cab45 Is Required for Ca(2+)-Dependent Secretory Cargo Sorting at the Trans-Golgi Network. J. Cell Biol. 2012, 199, 1057–1066. [Google Scholar] [CrossRef]
- von Blume, J.; Alleaume, A.-M.; Cantero-Recasens, G.; Curwin, A.; Carreras-Sureda, A.; Zimmermann, T.; van Galen, J.; Wakana, Y.; Valverde, M.A.; Malhotra, V. ADF/cofilin Regulates Secretory Cargo Sorting at the TGN via the Ca2+ ATPase SPCA1. Dev. Cell 2011, 20, 652–662. [Google Scholar] [CrossRef]
- Mitchell, K.J.; Tsuboi, T.; Rutter, G.A. Role for Plasma Membrane-Related Ca2+-ATPase-1 (ATP2C1) in Pancreatic Beta-Cell Ca2+ Homeostasis Revealed by RNA Silencing. Diabetes 2004, 53, 393–400. [Google Scholar] [CrossRef]
- Masgrau, R.; Churchill, G.C.; Morgan, A.J.; Ashcroft, S.J.H.; Galione, A. NAADP: A New Second Messenger for Glucose-Induced Ca2+ Responses in Clonal Pancreatic Beta Cells. Curr. Biol. 2003, 13, 247–251. [Google Scholar] [CrossRef]
- Chang, G.; Yang, R.; Cao, Y.; Nie, A.; Gu, X.; Zhang, H. SIDT2 Is Involved in the NAADP-Mediated Release of Calcium from Insulin Secretory Granules. J. Mol. Endocrinol. 2016, 56, 249–259. [Google Scholar] [CrossRef]
- Gao, J.; Gu, X.; Mahuran, D.J.; Wang, Z.; Zhang, H. Impaired Glucose Tolerance in a Mouse Model of sidt2 Deficiency. PLoS ONE 2013, 8, e66139. [Google Scholar] [CrossRef]
- Blondel, O.; Moody, M.M.; Depaoli, A.M.; Sharp, A.H.; Ross, C.A.; Swift, H.; Bell, G.I. Localization of Inositol Trisphosphate Receptor Subtype 3 to Insulin and Somatostatin Secretory Granules and Regulation of Expression in Islets and Insulinoma Cells. Proc. Natl. Acad. Sci. USA 1994, 91, 7777–7781. [Google Scholar] [CrossRef]
- Ravazzola, M.; Halban, P.A.; Orci, L. Inositol 1,4,5-Trisphosphate Receptor Subtype 3 in Pancreatic Islet Cell Secretory Granules Revisited. Proc. Natl. Acad. Sci. USA 1996, 93, 2745–2748. [Google Scholar] [CrossRef]
- Taylor, C.W.; Konieczny, V. IP3 Receptors: Take Four IP3 to Open. Sci. Signal. 2016, 9, e1. [Google Scholar] [CrossRef]
- Taylor, C.W.; Tovey, S.C. IP(3) Receptors: Toward Understanding Their Activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a004010. [Google Scholar] [CrossRef]
- Huh, Y.H.; Kim, K.D.; Yoo, S.H. Comparison of and Chromogranin Effect on Inositol 1,4,5-Trisphosphate Sensitivity of Cytoplasmic and Nucleoplasmic Inositol 1,4,5-Trisphosphate receptor/Ca2+ Channels. Biochemistry 2007, 46, 14032–14043. [Google Scholar] [CrossRef]
- Marshall, I.C.; Taylor, C.W. Biphasic Effects of Cytosolic Ca2+ on Ins(1,4,5)P3-Stimulated Ca2+ Mobilization in Hepatocytes. J. Biol. Chem. 1993, 268, 13214–13220. [Google Scholar] [CrossRef]
- Storto, M.; Capobianco, L.; Battaglia, G.; Molinaro, G.; Gradini, R.; Riozzi, B.; Di Mambro, A.; Mitchell, K.J.; Bruno, V.; Vairetti, M.P.; et al. Insulin Secretion Is Controlled by mGlu5 Metabotropic Glutamate Receptors. Mol. Pharmacol. 2006, 69, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Yokoi, N.; Seino, S. Glutamate as Intracellular and Extracellular Signals in Pancreatic Islet Functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Lewis, M.S. Interaction of Chromogranin B and the near N-Terminal Region of Chromogranin B with an Intraluminal Loop Peptide of the Inositol 1,4, 5-Trisphosphate Receptor. J. Biol. Chem. 2000, 275, 30293–30300. [Google Scholar] [CrossRef]
- Yoo, S.H.; Jeon, C.J. Inositol 1,4,5-Trisphosphate receptor/Ca2+ Channel Modulatory Role of Chromogranin A, a Ca2+ Storage Protein of Secretory Granules. J. Biol. Chem. 2000, 275, 15067–15073. [Google Scholar] [CrossRef]
- Yoo, S.H.; So, S.H.; Kweon, H.S.; Lee, J.S.; Kang, M.K.; Jeon, C.J. Coupling of the Inositol 1,4,5-Trisphosphate Receptor and Chromogranins A and B in Secretory Granules. J. Biol. Chem. 2000, 275, 12553–12559. [Google Scholar] [CrossRef][Green Version]
- Yoo, S.H. Secretory Granules in Inositol 1,4,5-Trisphosphate-Dependent Ca2+ Signaling in the Cytoplasm of Neuroendocrine Cells. FASEB J. 2010, 24, 653–664. [Google Scholar] [CrossRef]
- Yoo, S.H. Coupling of the IP3 receptor/Ca2+ Channel with Ca2+ Storage Proteins Chromogranins A and B in Secretory Granules. Trends Neurosci. 2000, 23, 424–428. [Google Scholar] [CrossRef]
- Moulin, P.; Guiot, Y.; Jonas, J.-C.; Rahier, J.; Devuyst, O.; Henquin, J.-C. Identification and Subcellular Localization of the Na+/H+ Exchanger and a Novel Related Protein in the Endocrine Pancreas and Adrenal Medulla. J. Mol. Endocrinol. 2007, 38, 409–422. [Google Scholar] [CrossRef]
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty Acid Translocase (FAT/CD36) Is Localized on Insulin-Containing Granules in Human Pancreatic Beta-Cells and Mediates Fatty Acid Effects on Insulin Secretion. Diabetes 2005, 54, 472–481. [Google Scholar] [CrossRef]
- Nagao, M.; Esguerra, J.L.S.; Asai, A.; Ofori, J.K.; Edlund, A.; Wendt, A.; Sugihara, H.; Wollheim, C.B.; Oikawa, S.; Eliasson, L. Potential Protection Against Type 2 Diabetes in Obesity Through Lower CD36 Expression and Improved Exocytosis in β-Cells. Diabetes 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Geisler, J.C.; Corbin, K.L.; Li, Q.; Feranchak, A.P.; Nunemaker, C.S.; Li, C. Vesicular Nucleotide Transporter-Mediated ATP Release Regulates Insulin Secretion. Endocrinology 2013, 154, 675–684. [Google Scholar] [CrossRef]
- Su, X.; Abumrad, N.A. Cellular Fatty Acid Uptake: A Pathway under Construction. Trends Endocrinol. Metab. 2009, 20, 72–77. [Google Scholar] [CrossRef]
- Manialawy, Y.; Khan, S.R.; Bhattacharjee, A.; Wheeler, M.B. The Magnesium Transporter NIPAL1 Is a Pancreatic Islet-Expressed Protein That Conditionally Impacts Insulin Secretion. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Cool, D.R.; Normant, E.; Shen, F.; Chen, H.C.; Pannell, L.; Zhang, Y.; Loh, Y.P. Carboxypeptidase E Is a Regulated Secretory Pathway Sorting Receptor: Genetic Obliteration Leads to Endocrine Disorders in Cpe(fat) Mice. Cell 1997, 88, 73–83. [Google Scholar] [CrossRef]
- Normant, E.; Loh, Y.P. Depletion of Carboxypeptidase E, a Regulated Secretory Pathway Sorting Receptor, Causes Misrouting and Constitutive Secretion of Proinsulin and Proenkephalin, but Not Chromogranin A. Endocrinology 1998, 139, 2137–2145. [Google Scholar] [CrossRef]
- Irminger, J.C.; Verchere, C.B.; Meyer, K.; Halban, P.A. Proinsulin Targeting to the Regulated Pathway Is Not Impaired in Carboxypeptidase E-Deficient Cpefat/Cpefat Mice. J. Biol. Chem. 1997, 272, 27532–27534. [Google Scholar] [CrossRef]
- Klumperman, J.; Hille, A.; Veenendaal, T.; Oorschot, V.; Stoorvogel, W.; von Figura, K.; Geuze, H.J. Differences in the Endosomal Distributions of the Two Mannose 6-Phosphate Receptors. J. Cell Biol. 1993, 121, 997–1010. [Google Scholar] [CrossRef]
- Dittié, A.S.; Klumperman, J.; Tooze, S.A. Differential Distribution of Mannose-6-Phosphate Receptors and Furin in Immature Secretory Granules. J. Cell Sci. 1999, 112 Pt 22, 3955–3966. [Google Scholar] [CrossRef]
- Puertollano, R.; Aguilar, R.C.; Gorshkova, I.; Crouch, R.J.; Bonifacino, J.S. Sorting of Mannose 6-Phosphate Receptors Mediated by the GGAs. Science 2001, 292, 1712–1716. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Germanos, M.; Gao, A.; Taper, M.; Yau, B.; Kebede, M.A. Inside the Insulin Secretory Granule. Metabolites 2021, 11, 515. https://doi.org/10.3390/metabo11080515
Germanos M, Gao A, Taper M, Yau B, Kebede MA. Inside the Insulin Secretory Granule. Metabolites. 2021; 11(8):515. https://doi.org/10.3390/metabo11080515
Chicago/Turabian StyleGermanos, Mark, Andy Gao, Matthew Taper, Belinda Yau, and Melkam A. Kebede. 2021. "Inside the Insulin Secretory Granule" Metabolites 11, no. 8: 515. https://doi.org/10.3390/metabo11080515
APA StyleGermanos, M., Gao, A., Taper, M., Yau, B., & Kebede, M. A. (2021). Inside the Insulin Secretory Granule. Metabolites, 11(8), 515. https://doi.org/10.3390/metabo11080515