
Understanding the Long-Lasting Effects of Fetal Nutrient Restriction versus Exposure to an Obesogenic Diet on Islet-Cell Mass and Function


Abstract
1. Introduction
2. An Overview of Animal Models of Restricted Fetal Growth and Exposure to Maternal Caloric Excess
2.1. Animal Models of Restricted Fetal Growth
2.2. Animal Models of Maternal Caloric Excess
3. The Impact of a Compromised Early Life Environment on Beta-Cell Morphology and Function
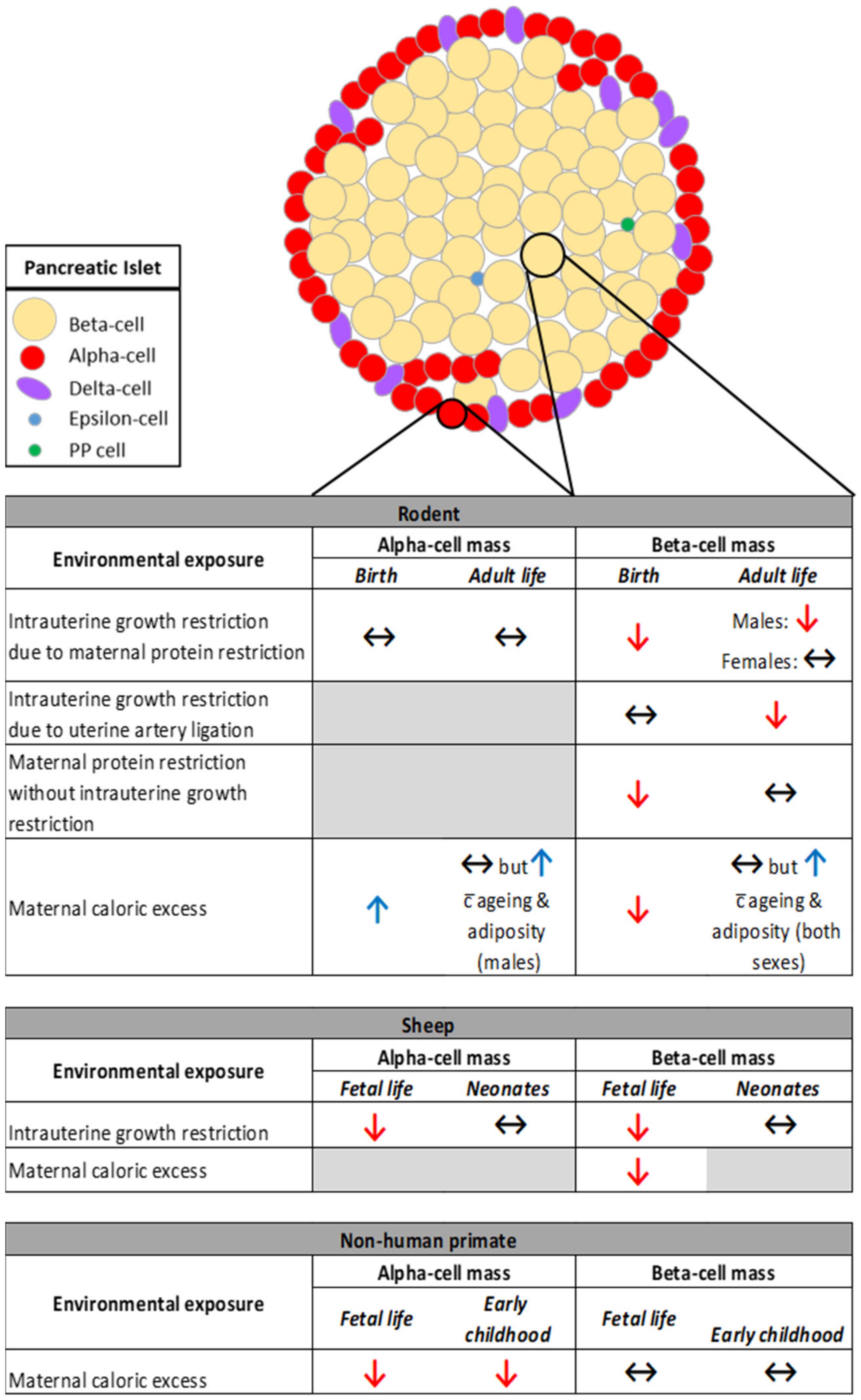
3.1. The Impact of Restricted Nutrition versus Caloric Excess on Islet Morphology
3.2. The Impact of Restricted Nutrition versus Caloric Excess on Beta-Cell Function
4. Similarities and Differences in the Mechanisms Identified in Offspring Exposed to Restricted Nutrition versus Caloric Excess
4.1. The impact of Restricted Nutrition versus Caloric Excess on Pdx1 Expression
4.2. The Impact of Restricted Nutrition versus Caloric Excess on Mitochondrial Metabolism and Oxidative Stress
4.3. Maternal Protein Restriction Leads to Reduced mTOR Signaling in Offspring Islets
4.4. Epigenetic Changes Underlying the Developmental Origins of Pancreatic Islet Dysfunction
4.5. The Impact of Restricted Nutrition on DNA Methylation and Histone Modifications
4.6. The Impact of Restricted Nutrition on miRNA Expression
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Hales, C.N.; Barker, D.J. Type 2 (non-insulin-dependent) diabetes mellitus: The thrifty phenotype hypothesis. Diabetologia 1992, 35, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Harder, T.; Rodekamp, E.; Schellong, K.; Dudenhausen, J.W.; Plagemann, A. Birth weight and subsequent risk of type 2 diabetes: A meta-analysis. Am. J. Epidemiol. 2007, 165, 849–857. [Google Scholar] [CrossRef]
- Phipps, K.; Barker, D.J.; Hales, C.N.; Fall, C.H.; Osmond, C.; Clark, P.M. Fetal growth and impaired glucose tolerance in men and women. Diabetologia 1993, 36, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Rich-Edwards, J.W.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C.; Gillman, M.W.; Hennekens, C.H.; Speizer, F.E.; Manson, J.E. Birthweight and the risk for type 2 diabetes mellitus in adult women. Ann. Intern. Med. 1999, 130, 278–284. [Google Scholar] [CrossRef]
- Stein, A.D.; Obrutu, O.E.; Behere, R.V.; Yajnik, C.S. Developmental undernutrition, offspring obesity and type 2 diabetes. Diabetologia 2019, 62, 1773–1778. [Google Scholar] [CrossRef]
- Hattersley, A.T.; Tooke, J.E. The fetal insulin hypothesis: An alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet 1999, 353, 1789–1792. [Google Scholar] [CrossRef]
- Hughes, A.E.; Hattersley, A.T.; Flanagan, S.E.; Freathy, R.M. Two decades since the fetal insulin hypothesis: What have we learned from genetics? Diabetologia 2021, 64, 717–726. [Google Scholar] [CrossRef]
- Poulsen, P.; Vaag, A.A.; Kyvik, K.O.; Moller Jensen, D.; Beck-Nielsen, H. Low birth weight is associated with niddm in discordant monozygotic and dizygotic twin pairs. Diabetologia 1997, 40, 439–446. [Google Scholar] [CrossRef]
- Knop, M.R.; Geng, T.T.; Gorny, A.W.; Ding, R.; Li, C.; Ley, S.H.; Huang, T. Birth weight and risk of type 2 diabetes mellitus, cardiovascular disease, and hypertension in adults: A meta-analysis of 7 646 267 participants from 135 studies. J. Am. Heart Assoc. 2018, 7, e008870. [Google Scholar] [CrossRef]
- Branum, A.M.; Kirmeyer, S.E.; Gregory, E.C. Prepregnancy body mass index by maternal characteristics and state: Data from the birth certificate, 2014. Natl. Vital Stat. Rep. 2016, 65, 1–11. [Google Scholar]
- Gaudet, L.; Ferraro, Z.M.; Wen, S.W.; Walker, M. Maternal obesity and occurrence of fetal macrosomia: A systematic review and meta-analysis. Biomed Res. Int. 2014, 2014, 640291. [Google Scholar] [CrossRef] [PubMed]
- Lahti-Pulkkinen, M.; Bhattacharya, S.; Wild, S.H.; Lindsay, R.S.; Raikkonen, K.; Norman, J.E.; Bhattacharya, S.; Reynolds, R.M. Consequences of being overweight or obese during pregnancy on diabetes in the offspring: A record linkage study in aberdeen, scotland. Diabetologia 2019, 62, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, J.; Richmond, R.C.; Palmer, T.M.; Feenstra, B.; Rangarajan, J.; Metrustry, S.; Cavadino, A.; Paternoster, L.; Armstrong, L.L.; De Silva, N.M.G.; et al. Genetic evidence for causal relationships between maternal obesity-related traits and birth weight. JAMA 2016, 315, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Richmond, R.C.; Timpson, N.J.; Felix, J.F.; Palmer, T.; Gaillard, R.; McMahon, G.; Davey Smith, G.; Jaddoe, V.W.; Lawlor, D.A. Using genetic variation to explore the causal effect of maternal pregnancy adiposity on future offspring adiposity: A mendelian randomisation study. PLOS Med. 2017, 14, e1002221. [Google Scholar] [CrossRef]
- Cetin, I.; Marconi, A.M.; Bozzetti, P.; Sereni, L.P.; Corbetta, C.; Pardi, G.; Battaglia, F.C. Umbilical amino acid concentrations in appropriate and small for gestational age infants: A biochemical difference present in utero. Am. J. Obstet. Gynecol. 1988, 158, 120–126. [Google Scholar] [CrossRef]
- Dicke, J.M.; Henderson, G.I. Placental amino acid uptake in normal and complicated pregnancies. Am. J. Med Sci. 1988, 295, 223–227. [Google Scholar] [CrossRef] [PubMed]
- King, R.; Hill, J.L.; Saha, B.; Tong, Y.; Strutt, B.J.; Russell, M.A.; Morgan, N.G.; Richardson, S.J.; Hill, D.J. Offspring of mice exposed to a low-protein diet in utero demonstrate changes in mtor signaling in pancreatic islets of langerhans, associated with altered glucagon and insulin expression and a lower β-cell mass. Nutrients 2019, 11, 605. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, E.U.; Jo, S.; Akhaphong, B.; Llacer, P.R.; Gianchandani, M.; Gregg, B.; Parlee, S.D.; MacDougald, O.A.; Bernal-Mizrachi, E. Maternal low-protein diet on the last week of pregnancy contributes to insulin resistance and β-cell dysfunction in the mouse offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 319, R485–R496. [Google Scholar] [CrossRef] [PubMed]
- Alejandro, E.U.; Gregg, B.; Wallen, T.; Kumusoglu, D.; Meister, D.; Chen, A.; Merrins, M.J.; Satin, L.S.; Liu, M.; Arvan, P.; et al. Maternal diet-induced micrornas and mtor underlie β cell dysfunction in offspring. J. Clin. Investig. 2014, 124, 4395–4410. [Google Scholar] [CrossRef]
- Theys, N.; Bouckenooghe, T.; Ahn, M.T.; Remacle, C.; Reusens, B. Maternal low-protein diet alters pancreatic islet mitochondrial function in a sex-specific manner in the adult rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1516–R1525. [Google Scholar] [CrossRef]
- Tarry-Adkins, J.L.; Chen, J.H.; Smith, N.S.; Jones, R.H.; Cherif, H.; Ozanne, S.E. Poor maternal nutrition followed by accelerated postnatal growth leads to telomere shortening and increased markers of cell senescence in rat islets. FASEB J. 2009, 23, 1521–1528. [Google Scholar] [CrossRef]
- Rodríguez-Trejo, A.; Ortiz-López, M.G.; Zambrano, E.; Granados-Silvestre Mde, L.; Méndez, C.; Blondeau, B.; Bréant, B.; Nathanielsz, P.W.; Menjivar, M. Developmental programming of neonatal pancreatic β-cells by a maternal low-protein diet in rats involves a switch from proliferation to differentiation. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1431–E1439. [Google Scholar] [CrossRef]
- Fernandez-Twinn, D.S.; Wayman, A.; Ekizoglou, S.; Martin, M.S.; Hales, C.N.; Ozanne, S.E. Maternal protein restriction leads to hyperinsulinemia and reduced insulin-signaling protein expression in 21-mo-old female rat offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R368–R373. [Google Scholar] [CrossRef]
- Petry, C.J.; Dorling, M.W.; Pawlak, D.B.; Ozanne, S.E.; Hales, C.N. Diabetes in old male offspring of rat dams fed a reduced protein diet. Int. J. Exp. Diabetes Res. 2001, 2, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, W.; Dai, Y.; Zhu, Z.; Liu, Q. Detection of expressional changes induced by intrauterine growth restriction in the developing rat pancreas. Exp. Biol. Med. 2016, 241, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gronert, M.S.; Tarry-Adkins, J.L.; Cripps, R.L.; Chen, J.H.; Ozanne, S.E. Maternal protein restriction leads to early life alterations in the expression of key molecules involved in the aging process in rat offspring. Am. J. Physiol. Integr. Comp. Physiol. 2008, 294, R494–R500. [Google Scholar] [CrossRef][Green Version]
- Tarry-Adkins, J.L.; Fernandez-Twinn, D.S.; Chen, J.H.; Hargreaves, I.P.; Neergheen, V.; Aiken, C.E.; Ozanne, S.E. Poor maternal nutrition and accelerated postnatal growth induces an accelerated aging phenotype and oxidative stress in skeletal muscle of male rats. Dis. Model. Mech. 2016, 9, 1221–1229. [Google Scholar] [CrossRef]
- Chen, J.H.; Martin-Gronert, M.S.; Tarry-Adkins, J.; Ozanne, S.E. Maternal protein restriction affects postnatal growth and the expression of key proteins involved in lifespan regulation in mice. PLoS ONE 2009, 4, e4950. [Google Scholar] [CrossRef] [PubMed]
- Rashid, C.S.; Lien, Y.C.; Bansal, A.; Jaeckle-Santos, L.J.; Li, C.; Won, K.J.; Simmons, R.A. Transcriptomic analysis reveals novel mechanisms mediating islet dysfunction in the intrauterine growth-restricted rat. Endocrinology 2018, 159, 1035–1049. [Google Scholar] [CrossRef]
- Akhaphong, B.; Lockridge, A.; Jo, S.; Mohan, R.; Wilcox, J.A.; Wing, C.R.; Regal, J.F.; Alejandro, E.U. Reduced uterine perfusion pressure causes loss of pancreatic β-cell area but normal function in fetal rat offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R1220–R1231. [Google Scholar] [CrossRef]
- Lien, Y.C.; Wang, P.Z.; Lu, X.M.; Simmons, R.A. Altered transcription factor binding and gene bivalency in islets of intrauterine growth retarded rats. Cells 2020, 9, 1435. [Google Scholar] [CrossRef]
- Stoffers, D.A.; Desai, B.M.; DeLeon, D.D.; Simmons, R.A. Neonatal exendin-4 prevents the development of diabetes in the intrauterine growth retarded rat. Diabetes 2003, 52, 734–740. [Google Scholar] [CrossRef][Green Version]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, R.A. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of pdx1. J. Clin. Investig. 2008, 118, 2316–2324. [Google Scholar] [CrossRef]
- Thompson, R.F.; Fazzari, M.J.; Niu, H.; Barzilai, N.; Simmons, R.A.; Greally, J.M. Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J. Biol. Chem. 2010, 285, 15111–15118. [Google Scholar] [CrossRef] [PubMed]
- Delghingaro-Augusto, V.; Madad, L.; Chandra, A.; Simeonovic, C.J.; Dahlstrom, J.E.; Nolan, C.J. Islet inflammation, hemosiderosis, and fibrosis in intrauterine growth-restricted and high fat-fed sprague-dawley rats. Am. J. Pathol. 2014, 184, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Siebel, A.L.; Gallo, L.A.; Guan, T.C.; Owens, J.A.; Wlodek, M.E. Cross-fostering and improved lactation ameliorates deficits in endocrine pancreatic morphology in growth-restricted adult male rat offspring. J. Dev. Orig. Health Dis. 2010, 1, 234–244. [Google Scholar] [CrossRef]
- Robinson, J.; Kingston, E.; Jones, C.; Thorburn, G. Studies on experimental growth retardation in sheep. The effect of removal of a endometrial caruncles on fetal size and metabolism. J. Dev. Physiol. 1979, 1, 379–398. [Google Scholar]
- Owens, J.A.; Gatford, K.L.; De Blasio, M.J.; Edwards, L.J.; McMillen, I.C.; Fowden, A.L. Restriction of placental growth in sheep impairs insulin secretion but not sensitivity before birth. J. Physiol. 2007, 584, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Leos, R.A.; Anderson, M.J.; Chen, X.; Pugmire, J.; Anderson, K.A.; Limesand, S.W. Chronic exposure to elevated norepinephrine suppresses insulin secretion in fetal sheep with placental insufficiency and intrauterine growth restriction. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E770–E778. [Google Scholar] [CrossRef] [PubMed]
- Rozance, P.J.; Limesand, S.W.; Barry, J.S.; Brown, L.D.; Hay, W.W. Glucose replacement to euglycemia causes hypoxia, acidosis, and decreased insulin secretion in fetal sheep with intrauterine growth restriction. Pediatr. Res. 2009, 65, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.C.; Bidwell, C.A.; McCarthy, F.M.; Taska, D.J.; Anderson, M.J.; Camacho, L.E.; Limesand, S.W. Rna sequencing exposes adaptive and immune responses to intrauterine growth restriction in fetal sheep islets. Endocrinology 2017, 158, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, B.H.; Brown, L.D.; Wesolowski, S.R.; Hay, W.W.; Rozance, P.J. Pulsatile hyperglycemia increases insulin secretion but not pancreatic β-cell mass in intrauterine growth-restricted fetal sheep. J. Dev. Orig. Health Dis. 2018, 9, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Limesand, S.W.; Jensen, J.; Hutton, J.C.; Hay, W.W., Jr. Diminished beta-cell replication contributes to reduced beta-cell mass in fetal sheep with intrauterine growth restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1297–R1305. [Google Scholar] [CrossRef]
- Elsakr, J.M.; Dunn, J.C.; Tennant, K.; Zhao, S.K.; Kroeten, K.; Pasek, R.C.; Takahashi, D.L.; Dean, T.A.; Velez Edwards, D.R.; McCurdy, C.E.; et al. Maternal western-style diet affects offspring islet composition and function in a non-human primate model of maternal over-nutrition. Mol. Metab. 2019, 25, 73–82. [Google Scholar] [CrossRef]
- Comstock, S.M.; Pound, L.D.; Bishop, J.M.; Takahashi, D.L.; Kostrba, A.M.; Smith, M.S.; Grove, K.L. High-fat diet consumption during pregnancy and the early post-natal period leads to decreased α cell plasticity in the nonhuman primate. Mol. Metab. 2012, 2, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Nicol, L.E.; Grant, W.F.; Comstock, S.M.; Nguyen, M.L.; Smith, M.S.; Grove, K.L.; Marks, D.L. Pancreatic inflammation and increased islet macrophages in insulin-resistant juvenile primates. J. Endocrinol. 2013, 217, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Pound, L.D.; Comstock, S.M.; Grove, K.L. Consumption of a western-style diet during pregnancy impairs offspring islet vascularization in a japanese macaque model. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E115–E123. [Google Scholar] [CrossRef]
- Conrad, E.; Dai, C.; Spaeth, J.; Guo, M.; Cyphert, H.A.; Scoville, D.; Carroll, J.; Yu, W.M.; Goodrich, L.V.; Harlan, D.M.; et al. The mafb transcription factor impacts islet α-cell function in rodents and represents a unique signature of primate islet β-cells. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E91–E102. [Google Scholar] [CrossRef]
- Ford, S.P.; Zhang, L.; Zhu, M.; Miller, M.M.; Smith, D.T.; Hess, B.W.; Moss, G.E.; Nathanielsz, P.W.; Nijland, M.J. Maternal obesity accelerates fetal pancreatic beta-cell but not alpha-cell development in sheep: Prenatal consequences. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R835–R843. [Google Scholar] [CrossRef]
- Zhang, L.; Long, N.M.; Hein, S.M.; Ma, Y.; Nathanielsz, P.W.; Ford, S.P. Maternal obesity in ewes results in reduced fetal pancreatic β-cell numbers in late gestation and decreased circulating insulin concentration at term. Domest. Anim. Endocrinol. 2011, 40, 30–39. [Google Scholar] [CrossRef]
- Nicholas, L.M.; Ozanne, S.E. Early life programming in mice by maternal overnutrition: Mechanistic insights and interventional approaches. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2019, 374, 20180116. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Xu, J.; Epstein, P.N.; Liu, Y.Q. Long-term effect of maternal obesity on pancreatic beta cells of offspring: Reduced beta cell adaptation to high glucose and high-fat diet challenges in adult female mouse offspring. Diabetologia 2005, 48, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Duhl, D.M.; Vrieling, H.; Miller, K.A.; Wolff, G.L.; Barsh, G.S. Neomorphic agouti mutations in obese yellow mice. Nat. Genet. 1994, 8, 59–65. [Google Scholar] [CrossRef]
- Li, C.C.; Young, P.E.; Maloney, C.A.; Eaton, S.A.; Cowley, M.J.; Buckland, M.E.; Preiss, T.; Henstridge, D.C.; Cooney, G.J.; Febbraio, M.A.; et al. Maternal obesity and diabetes induces latent metabolic defects and widespread epigenetic changes in isogenic mice. Epigenetics 2013, 8, 602–611. [Google Scholar] [CrossRef]
- Waterland, R.A.; Travisano, M.; Tahiliani, K.G.; Rached, M.T.; Mirza, S. Methyl donor supplementation prevents transgenerational amplification of obesity. Int. J. Obes. 2008, 32, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Duque-Guimaraes, D.E.; Ozanne, S.E. Nutritional programming of insulin resistance: Causes and consequences. Trends Endocrinol. Metab. 2013, 24, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, L.M.; Morrison, J.L.; Rattanatray, L.; Zhang, S.; Ozanne, S.E.; McMillen, I.C. The early origins of obesity and insulin resistance: Timing, programming and mechanisms. Int. J. Obes. 2016, 40, 229–238. [Google Scholar] [CrossRef]
- Bringhenti, I.; Moraes-Teixeira, J.A.; Cunha, M.R.; Ornellas, F.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Maternal obesity during the preconception and early life periods alters pancreatic development in early and adult life in male mouse offspring. PLoS ONE 2013, 8, e55711. [Google Scholar] [CrossRef]
- Su, Y.; Jiang, X.; Li, Y.; Li, F.; Cheng, Y.; Peng, Y.; Song, D.; Hong, J.; Ning, G.; Cao, Y.; et al. Maternal low protein isocaloric diet suppresses pancreatic β-cell proliferation in mouse offspring via mir-15b. Endocrinology 2016, 157, 4782–4793. [Google Scholar] [CrossRef]
- Petrik, J.; Reusens, B.; Arany, E.; Remacle, C.; Coelho, C.; Hoet, J.J.; Hill, D.J. A low protein diet alters the balance of islet cell replication and apoptosis in the fetal and neonatal rat and is associated with a reduced pancreatic expression of insulin-like growth factor-ii. Endocrinology 1999, 140, 4861–4873. [Google Scholar] [CrossRef]
- Reusens, B.; Remacle, C. Programming of the endocrine pancreas by the early nutritional environment. Int. J. Biochem. Cell Biol. 2006, 38, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, O.; Hinault, C.; Gautier, N.; Patouraux, S.; Casamento, V.; Van Obberghen, E. Maternal protein restriction leads to pancreatic failure in offspring: Role of misexpressed microrna-375. Diabetes 2014, 63, 3416. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Saisho, Y.; Inaishi, J.; Watanabe, Y.; Tsuchiya, T.; Makio, M.; Sato, M.; Kitago, M.; Yamada, T.; Itoh, H. Associations of birthweight and history of childhood obesity with beta cell mass in japanese adults. Diabetologia 2020, 63, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, L.M.; Nagao, M.; Kusinski, L.C.; Fernandez-Twinn, D.S.; Eliasson, L.; Ozanne, S.E. Exposure to maternal obesity programs sex differences in pancreatic islets of the offspring in mice. Diabetologia 2020, 63, 324–337. [Google Scholar] [CrossRef]
- Yokomizo, H.; Inoguchi, T.; Sonoda, N.; Sakaki, Y.; Maeda, Y.; Inoue, T.; Hirata, E.; Takei, R.; Ikeda, N.; Fujii, M.; et al. Maternal high-fat diet induces insulin resistance and deterioration of pancreatic beta-cell function in adult offspring with sex differences in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1163–E1175. [Google Scholar] [CrossRef]
- Zambrano, E.; Sosa-Larios, T.; Calzada, L.; Ibanez, C.A.; Mendoza-Rodriguez, C.A.; Morales, A.; Morimoto, S. Decreased basal insulin secretion from pancreatic islets of pups in a rat model of maternal obesity. J. Endocrinol. 2016, 231, 49–57. [Google Scholar] [CrossRef]
- Simmons, R.A.; Templeton, L.J.; Gertz, S.J. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 2001, 50, 2279. [Google Scholar] [CrossRef]
- Camacho, L.E.; Chen, X.; Hay, W.W., Jr.; Limesand, S.W. Enhanced insulin secretion and insulin sensitivity in young lambs with placental insufficiency-induced intrauterine growth restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R101–R109. [Google Scholar] [CrossRef] [PubMed]
- Long, N.M.; George, L.A.; Uthlaut, A.B.; Smith, D.T.; Nijland, M.J.; Nathanielsz, P.W.; Ford, S.P. Maternal obesity and increased nutrient intake before and during gestation in the ewe results in altered growth, adiposity, and glucose tolerance in adult offspring1. J. Anim. Sci. 2010, 88, 3546–3553. [Google Scholar] [CrossRef]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Investig. 2009, 119, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.R.; Gottheil, S.K.; Arany, E.J.; Hill, D.J. The effects of low protein during gestation on mouse pancreatic development and beta cell regeneration. Pediatr. Res. 2010, 68, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Cerf, M.E.; Williams, K.; Nkomo, X.I.; Muller, C.J.; Du Toit, D.F.; Louw, J.; Wolfe-Coote, S.A. Islet cell response in the neonatal rat after exposure to a high-fat diet during pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1122–R1128. [Google Scholar] [CrossRef] [PubMed]
- Graus-Nunes, F.; Dalla Corte Frantz, E.; Lannes, W.R.; da Silva Menezes, M.C.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Pregestational maternal obesity impairs endocrine pancreas in male f1 and f2 progeny. Nutrition 2015, 31, 380–387. [Google Scholar] [CrossRef]
- Yoon, K.H.; Ko, S.H.; Cho, J.H.; Lee, J.M.; Ahn, Y.B.; Song, K.H.; Yoo, S.J.; Kang, M.I.; Cha, B.Y.; Lee, K.W.; et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in korea. J. Clin. Endocrinol. Metab. 2003, 88, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.C.; Rahier, J. Pancreatic alpha cell mass in european subjects with type 2 diabetes. Diabetologia 2011, 54, 1720–1725. [Google Scholar] [CrossRef]
- Meier, J.J.; Ueberberg, S.; Korbas, S.; Schneider, S. Diminished glucagon suppression after β-cell reduction is due to impaired α-cell function rather than an expansion of α-cell mass. Endocrinol. Metab. 2011, 300, E717–E723. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Wortham, M.; Sander, M. Mechanisms of beta-cell functional adaptation to changes in workload. Diabetes Obes. Metab. 2016, 18 (Suppl. 1), 78–86. [Google Scholar] [CrossRef]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R134–R139. [Google Scholar] [CrossRef] [PubMed]
- Bringhenti, I.; Ornellas, F.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. The insulin-signaling pathway of the pancreatic islet is impaired in adult mice offspring of mothers fed a high-fat diet. Nutrition 2016, 32, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.M.; Matthews, P.A.; Argenton, M.; Christie, M.R.; McConnell, J.M.; Jansen, E.H.J.M.; Piersma, A.H.; Ozanne, S.E.; Twinn, D.F.; Remacle, C.; et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Katewa, S.D.; Palaniyappan, A.; Pandya, J.D.; Patel, M.S. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E792–E799. [Google Scholar] [CrossRef]
- Guo, S.; Dai, C.; Guo, M.; Taylor, B.; Harmon, J.S.; Sander, M.; Robertson, R.P.; Powers, A.C.; Stein, R. Inactivation of specific β cell transcription factors in type 2 diabetes. J. Clin. Investig. 2013, 123, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef]
- Maechler, P.; Wollheim, C.B. Mitochondrial function in normal and diabetic β-cells. Nature 2001, 414, 807–812. [Google Scholar] [CrossRef]
- Simmons, R.A.; Suponitsky-Kroyter, I.; Selak, M.A. Progressive accumulation of mitochondrial DNA mutations and decline in mitochondrial function lead to β-cell failure. J. Biol. Chem. 2005, 280, 28785–28791. [Google Scholar] [CrossRef]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef]
- Lei, X.G.; Vatamaniuk, M.Z. Two tales of antioxidant enzymes on beta cells and diabetes. Antioxid. Redox Signal. 2011, 14, 489–503. [Google Scholar] [CrossRef]
- Fernandez-Twinn, D.S.; Gascoin, G.; Musial, B.; Carr, S.; Duque-Guimaraes, D.; Blackmore, H.L.; Alfaradhi, M.Z.; Loche, E.; Sferruzzi-Perri, A.N.; Fowden, A.L.; et al. Exercise rescues obese mothers’ insulin sensitivity, placental hypoxia and male offspring insulin sensitivity. Sci. Rep. 2017, 7, 44650. [Google Scholar] [CrossRef]
- Theys, N.; Ahn, M.T.; Bouckenooghe, T.; Reusens, B.; Remacle, C. Maternal malnutrition programs pancreatic islet mitochondrial dysfunction in the adult offspring. J. Nutr. Biochem. 2011, 22, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Lupse, B.; Kido, Y.; Leibowitz, G.; Maedler, K. Mtorc1 signaling: A double-edged sword in diabetic β cells. Cell Metab. 2018, 27, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, D.; Kaestner, K.H. The dynamic methylome of islets in health and disease. Mol. Metab. 2019, 27s, S25–S32. [Google Scholar] [CrossRef] [PubMed]
- Dayeh, T.; Volkov, P.; Salo, S.; Hall, E.; Nilsson, E.; Olsson, A.H.; Kirkpatrick, C.L.; Wollheim, C.B.; Eliasson, L.; Ronn, T.; et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014, 10, e1004160. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Del Guerra, S.; Lupi, R.; Rönn, T.; Granhall, C.; Luthman, H.; Masiello, P.; Marchetti, P.; Groop, L.; Del Prato, S. Epigenetic regulation of ppargc1a in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 2008, 51, 615–622. [Google Scholar] [CrossRef]
- Volkov, P.; Bacos, K.; Ofori, J.K.; Esguerra, J.L.; Eliasson, L.; Ronn, T.; Ling, C. Whole-genome bisulfite sequencing of human pancreatic islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes 2017, 66, 1074–1085. [Google Scholar] [CrossRef]
- Yang, B.T.; Dayeh, T.A.; Volkov, P.A.; Kirkpatrick, C.L.; Malmgren, S.; Jing, X.; Renström, E.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Increased DNA methylation and decreased expression of pdx-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol. 2012, 26, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Sandovici, I.; Smith, N.H.; Nitert, M.D.; Ackers-Johnson, M.; Uribe-Lewis, S.; Ito, Y.; Jones, R.H.; Marquez, V.E.; Cairns, W.; Tadayyon, M.; et al. Maternal diet and aging alter the epigenetic control of a promoter–enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc. Natl. Acad. Sci. USA 2011, 108, 5449. [Google Scholar] [CrossRef]
- Eliasson, L.; Esguerra, J.L.S. Microrna networks in pancreatic islet cells: Normal function and type 2 diabetes. Diabetes 2020, 69, 804. [Google Scholar] [CrossRef] [PubMed]
- Grieco, F.A.; Sebastiani, G.; Juan-Mateu, J.; Villate, O.; Marroqui, L.; Ladrière, L.; Tugay, K.; Regazzi, R.; Bugliani, M.; Marchetti, P.; et al. Micrornas mir-23a-3p, mir-23b-3p, and mir-149-5p regulate the expression of proapoptotic bh3-only proteins dp5 and puma in human pancreatic β-cells. Diabetes 2017, 66, 100–112. [Google Scholar] [CrossRef]
- Nesca, V.; Guay, C.; Jacovetti, C.; Menoud, V.; Peyot, M.-L.; Laybutt, D.R.; Prentki, M.; Regazzi, R. Identification of particular groups of micrornas that positively or negatively impact on beta cell function in obese models of type 2 diabetes. Diabetologia 2013, 56, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Dekker Nitert, M.; Volkov, P.; Malmgren, S.; Mulder, H.; Bacos, K.; Ling, C. The effects of high glucose exposure on global gene expression and DNA methylation in human pancreatic islets. Mol. Cell. Endocrinol. 2018, 472, 57–67. [Google Scholar] [CrossRef]
- Hall, E.; Jönsson, J.; Ofori, J.K.; Volkov, P.; Perfilyev, A.; Dekker Nitert, M.; Eliasson, L.; Ling, C.; Bacos, K. Glucolipotoxicity alters insulin secretion via epigenetic changes in human islets. Diabetes 2019, 68, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Volkov, P.; Dayeh, T.; Bacos, K.; Rönn, T.; Nitert, M.D.; Ling, C. Effects of palmitate on genome-wide mrna expression and DNA methylation patterns in human pancreatic islets. BMC Med. 2014, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Roggli, E.; Britan, A.; Gattesco, S.; Lin-Marq, N.; Abderrahmani, A.; Meda, P.; Regazzi, R. Involvement of micrornas in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic β-cells. Diabetes 2010, 59, 978–986. [Google Scholar] [CrossRef]
- Esguerra, J.L.S.; Bolmeson, C.; Cilio, C.M.; Eliasson, L. Differential glucose-regulation of micrornas in pancreatic islets of non-obese type 2 diabetes model goto-kakizaki rat. PLoS ONE 2011, 6, e18613. [Google Scholar] [CrossRef]


{kind=link}
| Environmental Exposure | Pdx1 Expression | Mitochondrial Metabolism + | Oxidative Stress 🡪 | Functional Outcome | mTOR Abundance/Activity | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fetal Life | Adult Life | Adult Life | Birth | Adult Life | ||||||||
| Intrauterine growth restriction (due to maternal protein restriction or uterine artery ligation) | 🡫(26, 33) | 🡫(26, 33) | 🡫 🡫 | glucose-stimulated ATP (87) expression of mitochondrial-encoded genes (87) | 🡩 | oxidative stress (87) | 🡫 | glucose and mitochondrial-fuel stimulated insulin secretion (87) |  | mTOR protein (18) | 🡫 | mTOR protein (18) |
| Males and females: | Males only: | Males and females: | ||||||||||
| 🡫 | glucose-stimulated ATP (21) | 🡩 | reactive oxygen species (21) | 🡩 | glucose-stimulated insulin secretion (21) | |||||||
| Females only: | ||||||||||||
| 🡫 | expression of Tfam (21) expression of mitochondrial-encoded genes (21) | |||||||||||
| Maternal protein restriction without intrauterine growth restriction | 🡫(20) | 🡫 | mTOR activity (20) | 🡫 | mTOR activity (20) | |||||||
| Maternal caloric excess | Males: 🡫 (65,73,81) Females: (65) | Exposure to a high-fat, high-sugar diet from before and throughout pregnancy and lactation (64): | ||||||||||
| Females: | Males and females: | Females: | ||||||||||
| 🡩 🡩 🡩 | mitochondrial respiration expression of mitochondrial-encoded genes mitochondrial density | 🡩 | reactive oxygen species | 🡩 | glucose and mitochondrial-fuel stimulated insulin secretion | |||||||
| Males: | Males: | |||||||||||
| 🡫 | mitochondrial respiration expression of mitochondrial-encoded genes mitochondrial density | | glucose and mitochondrial-fuel stimulated insulin | |||||||||
| Exposure to a high-fat diet from conception and throughout pregnancy and lactation (65): | ||||||||||||
| Males but not females: | ||||||||||||
| 🡩 | oxidative stress | |||||||||||
| 🡩 | expression of genes involved in superoxide production | |||||||||||
| Exposure to a high-fat diet from conception and throughout pregnancy and lactation (91): | ||||||||||||
| 🡫 🡫 | glucose-stimulated ATP basal ATP content | 🡫 | glucose stimulated insulin secretion | |||||||||
| mitochondrial DNA content expression of Tfam expression of mitochondrial-encoded genes | |||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Hara, S.E.; Gembus, K.M.; Nicholas, L.M. Understanding the Long-Lasting Effects of Fetal Nutrient Restriction versus Exposure to an Obesogenic Diet on Islet-Cell Mass and Function. Metabolites 2021, 11, 514. https://doi.org/10.3390/metabo11080514
O’Hara SE, Gembus KM, Nicholas LM. Understanding the Long-Lasting Effects of Fetal Nutrient Restriction versus Exposure to an Obesogenic Diet on Islet-Cell Mass and Function. Metabolites. 2021; 11(8):514. https://doi.org/10.3390/metabo11080514
Chicago/Turabian StyleO’Hara, Stephanie E., Kelly M. Gembus, and Lisa M. Nicholas. 2021. "Understanding the Long-Lasting Effects of Fetal Nutrient Restriction versus Exposure to an Obesogenic Diet on Islet-Cell Mass and Function" Metabolites 11, no. 8: 514. https://doi.org/10.3390/metabo11080514
APA StyleO’Hara, S. E., Gembus, K. M., & Nicholas, L. M. (2021). Understanding the Long-Lasting Effects of Fetal Nutrient Restriction versus Exposure to an Obesogenic Diet on Islet-Cell Mass and Function. Metabolites, 11(8), 514. https://doi.org/10.3390/metabo11080514