
Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease



Abstract
1. Introduction
2. Steroid Hormones and Cognate Receptors
2.1. Steroid Hormones
2.1.1. Estrogens
2.1.2. Androgens
2.1.3. Progestogens
2.1.4. Glucocorticoids
2.1.5. Mineralocorticoids
2.1.6. Vitamin D
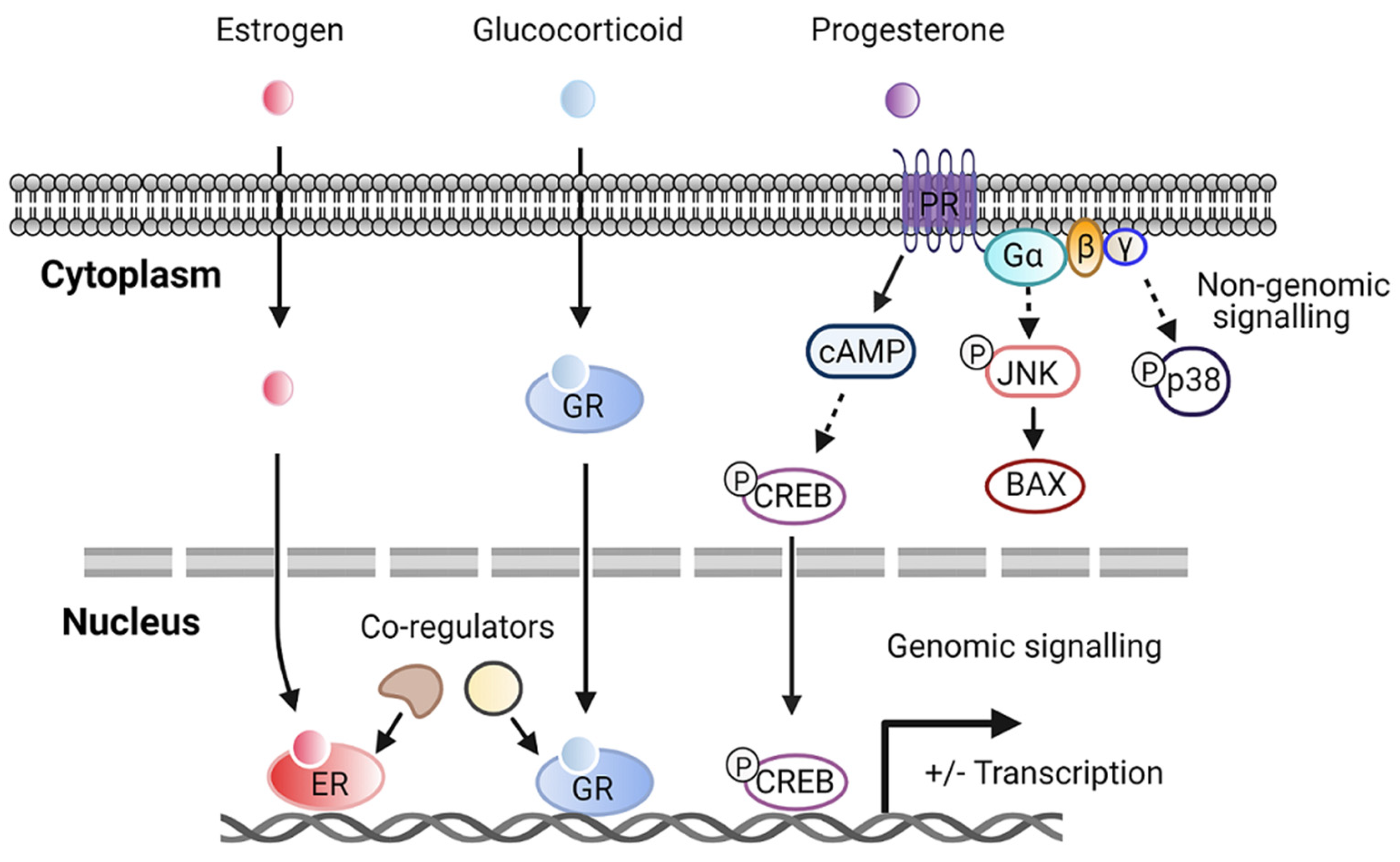
2.2. Steroid Hormone Receptors
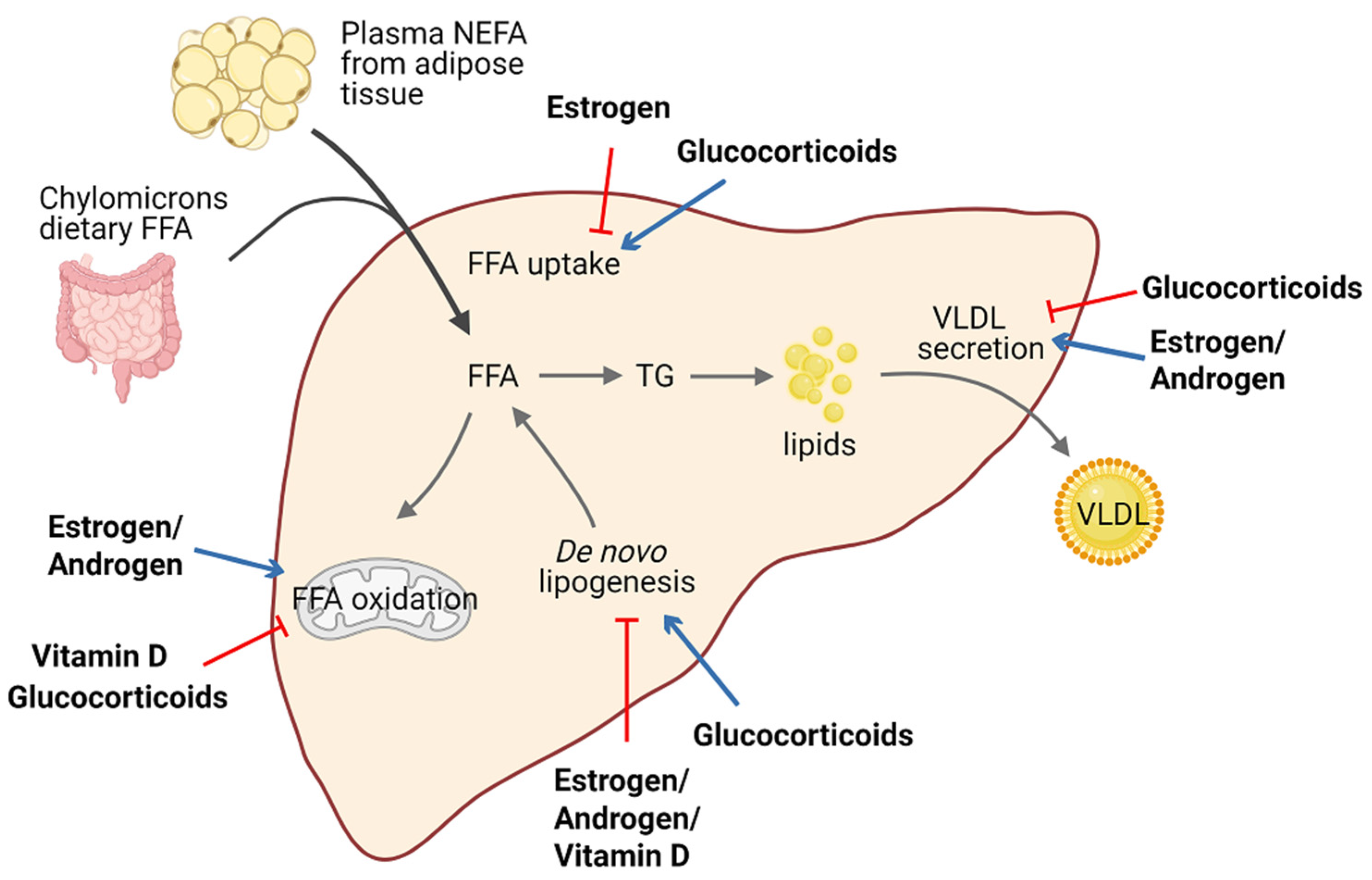
3. Role of Steroid Hormones in Hepatic Steatosis and Metabolism

{kind=link}
{kind=link}
{kind=link}
| Steroid Hormones | Model(s) Used | Major Phenotypes Examined |
|---|---|---|
| Estrogen | - Female ERα-deficient mice fed HFD for 10 weeks [71] - Male hepatic ERα-deficient mice fed HFD for 12 weeks [72] - OVX mice treated with E2 and fed HFD for 6 weeks [74] | - Liver weight, hepatic steatosis, and ALT level ↑ - Hepatic steatosis and insulin resistance ↑ - Hepatic steatosis and insulin- mediated suppression of VLDL secretion ↓ |
| Androgen | - Male hepatic AR-deficient mice fed HFD for 8 weeks [76] | - Body weight, hepatic steatosis, and insulin resistance ↑ |
| Glucocorticoid | - db/db mice treated with GC shRNA for 14 days [77] - SD rats treated with exogenous corticosterone and fed HFD for 16 days [78] | - Hepatic steatosis and genes critical for lipid storage and transport ↓ - Hepatic steatosis, uptake of FA into liver, and ALT level ↑ |
| Mineralocorticoid | - Myeloid MR-deficient ob/ob mice [79] - Aldosterone synthase-deficient mice fed HFD for 12 weeks [80] | - Hepatic steatosis, lipogenesis, and insulin resistance ↓ - HFD-feeding-induced hepatic steatosis ↓ |
| Vitamin D | - C57BL6 mice fed a high-fat/ high-sucrose diet followed by treatment with vitamin D for 15 weeks [81] - SD rats fed HFD followed by treatment with vitamin D for 16 weeks [82] | - Hepatic steatosis and hepatic de novo lipogenesis ↓ - Liver weight, hepatic steatosis, and ALT level ↓ |
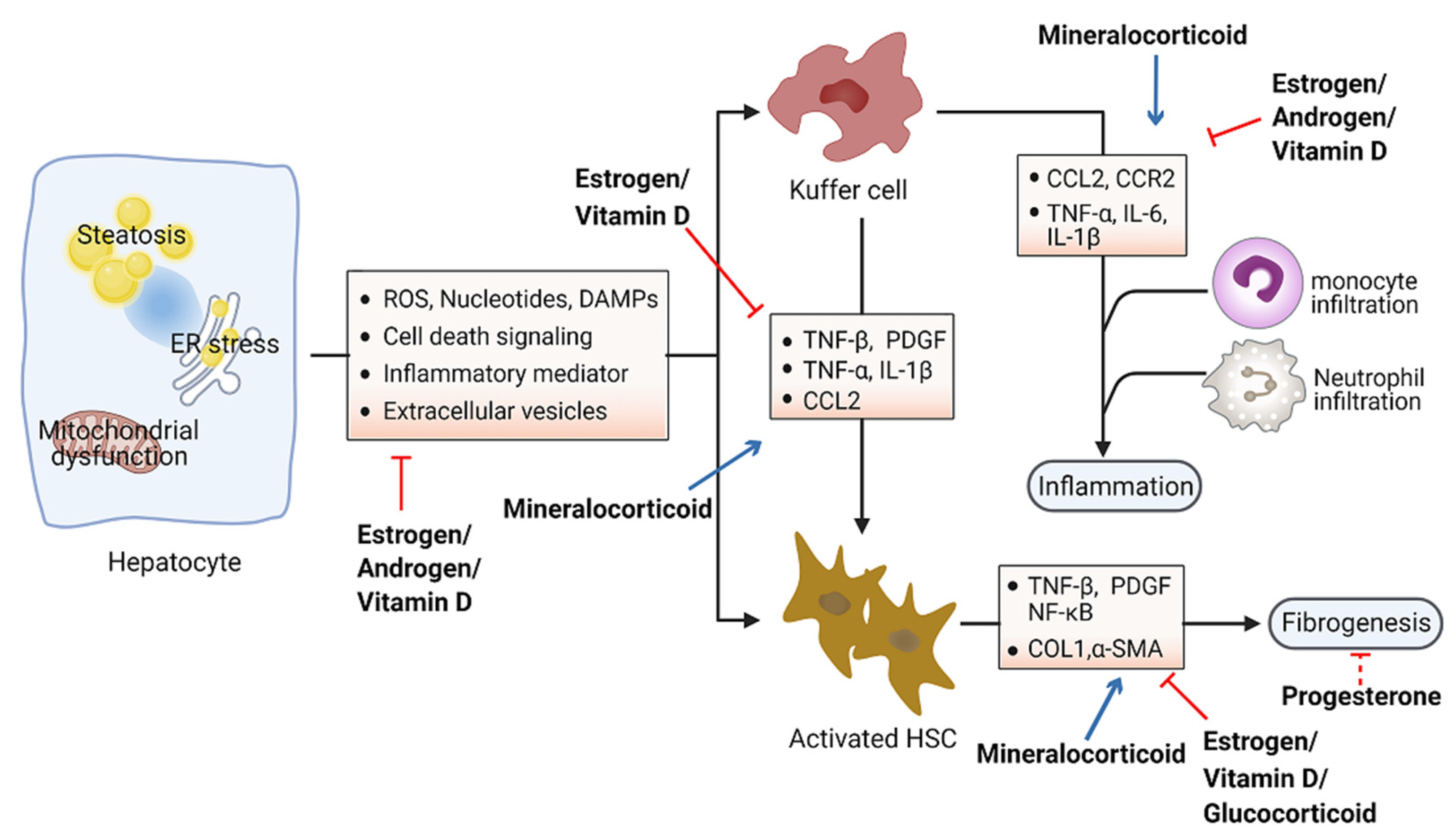
4. Role of Steroid Hormones in Hepatic Inflammation and Fibrosis
| Steroid Hormones | Model (s) Used | Major Phenotypes Examined |
|---|---|---|
| Estrogen | - OVX mice fed HFD and high-fructose water for 12 weeks [111] - OVX mice fed a high-fat and high-cholic-acid diet for 6 weeks [109] - Old female zebrafish fed a high-calorie diet for 24 weeks [112] - Orchidectomized C57/BL6 mice treated with estradiol benzoate-fed MCD for 4 weeks [110] - Male C57BL6 mice treated with β-LGND2 and fed HFD for 10 weeks [115] | - Hepatic inflammation and fibrosis, ALT level and ballooning degeneration ↑ - Liver fibrosis, inflammation, and hepatocyte ballooning degeneration ↑ - Liver fibrosis, IL-6, and TNF-β ↑ - Hepatic inflammation, MyD88, and IL-6 ↓ - Hepatic steatosis and insulin resistance ↓ |
| Androgen | - Orchidectomized male SD rats treated with dihydrotestosterone and fed HFD for 75 days [117] | - Portal inflammation, TNF-α, and IL-6 ↓ |
| Progesterone | - Hepatic fibrosis model of New Zealand male rabbits treated with progesterone for 180 days [119] | - Liver fibrosis, fat metamorphosis, and inflammatory infiltrate ↓ |
| Glucocorticoid | - Immune cell-specific GR-knockout mice treated with CCl4 and dexamethasone for 6 weeks [120] - HSC-specific GR-knockout mice treated with CCl4 and dexamethasone for 6 weeks [120] | - Inflammation and monocyte recruitment ↓ - Hepatic fibrosis and fibrotic gene expression ↓ |
| Mineralocorticoid | - C57BL6 mice fed HFFD mixed with eplerenone for 12 weeks [121] - Male C57BL6 mice fed a CDAA diet for 22 weeks with eplerenone [122] - Male SD rats treated with aldosterone for 4 weeks [123] | - Lipid accumulation, lobular inflammation, and collagen deposition ↓ - Hepatic fibrosis, steatosis, and inflammation ↓ - Hepatic fibrosis, oxidative stress, and DNA double-strand breaks ↑ |
| Vitamin D | - Vitamin D-deficient SD rats fed WD for 10 weeks [124] - CDAA diet-induced rat NASH model with phototherapy for 6 or 12 weeks [125] | - Foci of lobular inflammation and ballooning degeneration ↑ - Collagen fibrosis, insulin and leptin resistance, inflammation, and HSC activation ↓ |
5. Conclusions and Perspective
Funding
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of Nafld and Nash: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: An Emerging Menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of Disease Progression in Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2008, 28, 370–379. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.K. Extrahepatic Complications of Nonalcoholic Fatty Liver Disease. Hepatology 2014, 59, 1174–1197. [Google Scholar] [CrossRef]
- Cusi, K. Role of Obesity and Lipotoxicity in the Development of Nonalcoholic Steatohepatitis: Pathophysiology and Clinical Implications. Gastroenterology 2012, 142, 711–725. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (Nafld). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-Alcoholic Fatty Liver Disease (Nafld) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. Review: The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, M.; Liu, Q.; Xu, T.; Huang, T.; Yao, D.; Wong, C.W.; Liu, J.; Guan, M. 18beta-Glycyrrhetinic Acid Acts through Hepatocyte Nuclear Factor 4 Alpha to Modulate Lipid and Carbohydrate Metabolism. Pharmacol. Res. 2020, 157, 104840. [Google Scholar] [CrossRef]
- Guan, M.; Qu, L.; Tan, W.; Chen, L.; Wong, C.W. Hepatocyte Nuclear Factor-4 Alpha Regulates Liver Triglyceride Metabolism in Part through Secreted Phospholipase a(2) Gxiib. Hepatology 2011, 53, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Song, K.X.; Qu, S. Nonalcoholic Fatty Liver Disease: Pathogenesis and Therapeutics from a Mitochondria-Centric Perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, M.; Wang, N.; Liu, Q.; Wang, B.; Huang, T.; Tong, Y.; Ming, Y.; Wong, C.W.; Liu, J.; et al. Andrographolide Modulates Hnf4alpha Activity Imparting on Hepatic Metabolism. Mol. Cell. Endocrinol. 2020, 513, 110867. [Google Scholar] [CrossRef] [PubMed]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of Non-Alcoholic Fatty Liver Disease. QJM 2010, 103, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of Nash. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef]
- Handa, P.; Maliken, B.D.; Nelson, J.E.; Morgan-Stevenson, V.; Messner, D.J.; Dhillon, B.K.; Klintworth, H.M.; Beauchamp, M.; Yeh, M.M.; Elfers, C.T.; et al. Reduced Adiponectin Signaling Due to Weight Gain Results in Nonalcoholic Steatohepatitis through Impaired Mitochondrial Biogenesis. Hepatology 2014, 60, 133–145. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Gramlich, T.; Liu, Y.C.; Matteoni, C.; Petrelli, M.; Goldblum, J.; Rybicki, L.; McCullough, A.J. Nonalcoholic Fatty Liver Disease: Assessment of Variability in Pathologic Interpretations. Mod. Pathol. 1998, 11, 560–565. [Google Scholar]
- Beste, L.A.; Leipertz, S.L.; Green, P.K.; Dominitz, J.A.; Ross, D.; Ioannou, G.N. Trends in Burden of Cirrhosis and Hepatocellular Carcinoma by Underlying Liver Disease in Us Veterans, 2001-2013. Gastroenterology 2015, 149, 1471–1482.e5. [Google Scholar] [CrossRef]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic Reticulum Stress and Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Tarantino, G.; Finelli, C. Pathogenesis of Hepatic Steatosis: The Link between Hypercortisolism and Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2013, 19, 6735–6743. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Aloni-Grinstein, R.; Osher, E.; Rotter, V. Liver and Steroid Hormones-Can a Touch of P53 Make a Difference? Front. Endocrinol. 2019, 10, 374. [Google Scholar] [CrossRef]
- Morris, E.M.; Fletcher, J.A.; Thyfault, J.P.; Rector, R.S. The Role of Angiotensin Ii in Nonalcoholic Steatohepatitis. Mol. Cell. Endocrinol. 2013, 378, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Lacin, S.; Abdel-Wahab, R.; Uemura, M.; Hassan, M.; Rashid, A.; Duda, D.G.; Kaseb, A.O. Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma: Is There a Role for the Androgen Receptor Pathway? Onco Targets Ther. 2017, 10, 1403–1412. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef]
- Schneider, A.E.; Karpati, E.; Schuszter, K.; Toth, E.A.; Kiss, E.; Kulcsar, M.; Laszlo, G.; Matko, J. A Dynamic Network of Estrogen Receptors in Murine Lymphocytes: Fine-Tuning the Immune Response. J. Leukoc. Biol. 2014, 96, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.K.; Paramanik, V. Role of Steroid Hormone Coregulators in Health and Disease. Horm. Res. 2009, 71, 194–200. [Google Scholar] [CrossRef]
- McLachlan, J.A.; Newbold, R.R. Estrogens and Development. Environ Health Perspect 1987, 75, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, Y.; Li, R. Estrogen Synthesis and Signaling Pathways During Aging: From Periphery to Brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef]
- Lobo, R.A.; Pickar, J.H.; Stevenson, J.C.; Mack, W.J.; Hodis, H.N. Back to the Future: Hormone Replacement Therapy as Part of a Prevention Strategy for Women at the Onset of Menopause. Atherosclerosis 2016, 254, 282–290. [Google Scholar] [CrossRef]
- Trabert, B.; Wentzensen, N.; Yang, H.P.; Sherman, M.E.; Hollenbeck, A.R.; Park, Y.; Brinton, L.A. Is Estrogen Plus Progestin Menopausal Hormone Therapy Safe with Respect to Endometrial Cancer Risk? Int. J. Cancer 2013, 132, 417–426. [Google Scholar] [CrossRef]
- Pierard-Franchimont, C.; Pierard, G.E. Postmenopausal Aging of the Sebaceous Follicle: A Comparison between Women Receiving Hormone Replacement Therapy or Not. Dermatology 2002, 204, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.P.; Lyu, K.; Goldberg, E.L.; Zhang, D.; Cline, G.W.; Jurczak, M.J.; Dixit, V.D.; Petersen, K.F.; Shulman, G.I. Anti-Inflammatory Effects of Oestrogen Mediate the Sexual Dimorphic Response to Lipid-Induced Insulin Resistance. J. Physiol. 2019, 597, 3885–3903. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [PubMed]
- Shen, M.; Kumar, S.P.; Shi, H. Estradiol Regulates Insulin Signaling and Inflammation in Adipose Tissue. Horm. Mol. Biol. Clin. Investig. 2014, 17, 99–107. [Google Scholar] [CrossRef]
- Wilson, J.D.; Griffin, J.E.; Leshin, M.; George, F.W. Role of Gonadal Hormones in Development of the Sexual Phenotypes. Hum. Genet. 1981, 58, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Quigley, C.A.; De Bellis, A.; Marschke, K.B.; el-Awady, M.K.; Wilson, E.M.; French, F.S. Androgen Receptor Defects: Historical, Clinical, and Molecular Perspectives. Endocr. Rev. 1995, 16, 271–321. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Morley, J.E.; Korenman, S.G. Biological Actions of Androgens. Endocr. Rev. 1987, 8, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Y.; Zhao, J.; Wang, H.; Tan, J.; Yang, M.; Li, Y.; Deng, S.; Gao, S.; Li, H.; et al. Distinct Cardiac Energy Metabolism and Oxidative Stress Adaptations between Obese and Non-Obese Type 2 Diabetes Mellitus. Theranostics 2020, 10, 2675–2695. [Google Scholar] [CrossRef]
- Pikler, G.M.; Webster, R.A.; Spelsberg, T.C. Nuclear Binding of Progesterone in Hen Oviduct. Binding to Multiple Sites in Vitro. Biochem. J. 1976, 156, 399–408. [Google Scholar] [CrossRef]
- Oettel, M.; Mukhopadhyay, A.K. Progesterone: The Forgotten Hormone in Men? Aging Male 2004, 7, 236–257. [Google Scholar] [CrossRef] [PubMed]
- Manyonda, I.; Talaulikar, V.S.; Pirhadi, R.; Onwude, J. Progestogens Are the Problem in Hormone Replacement Therapy: Time to Reappraise Their Use. Post Reprod. Health 2020, 26, 26–31. [Google Scholar] [CrossRef]
- Adcock, I.M.; Mumby, S. Glucocorticoids. Handb. Exp. Pharmacol. 2017, 237, 171–196. [Google Scholar]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory Action of Glucocorticoids--New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Pazirandeh, A.; Xue, Y.; Prestegaard, T.; Jondal, M.; Okret, S. Effects of Altered Glucocorticoid Sensitivity in the T Cell Lineage on Thymocyte and T Cell Homeostasis. FASEB J. 2002, 16, 727–729. [Google Scholar] [CrossRef]
- Scheller, K.; Sekeris, C.E. The Effects of Steroid Hormones on the Transcription of Genes Encoding Enzymes of Oxidative Phosphorylation. Exp. Physiol. 2003, 88, 129–140. [Google Scholar] [CrossRef]
- Allan, E.H.; Chisholm, A.B.; Titheradge, M.A. The Stimulation of Hepatic Oxidative Phosphorylation Following Dexamethasone Treatment of Rats. Biochim. Biophys. Acta 1983, 725, 71–76. [Google Scholar] [CrossRef]
- Briet, M.; Schiffrin, E.L. Aldosterone: Effects on the Kidney and Cardiovascular System. Nat. Rev. Nephrol. 2010, 6, 261–273. [Google Scholar] [CrossRef]
- Pearce, D.; Bhargava, A.; Cole, T.J. Aldosterone: Its Receptor, Target Genes, and Actions. Vitam. Horm. 2003, 66, 29–76. [Google Scholar]
- Weinberger, M.H. Mineralocorticoids and Blood Pressure. Curr. Opin. Nephrol. Hypertens. 1994, 3, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J. Contribution of Aldosterone to Cardiovascular and Renal Inflammation and Fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.B.; McGraw, A.P.; Jaffe, I.Z.; Sowers, J.R. Mineralocorticoid Receptor-Mediated Vascular Insulin Resistance: An Early Contributor to Diabetes-Related Vascular Disease? Diabetes 2013, 62, 313–319. [Google Scholar] [CrossRef]
- Feraco, A.; Armani, A.; Mammi, C.; Fabbri, A.; Rosano, G.M.; Caprio, M. Role of Mineralocorticoid Receptor and Renin-Angiotensin-Aldosterone System in Adipocyte Dysfunction and Obesity. J. Steroid Biochem. Mol. Biol. 2013, 137, 99–106. [Google Scholar] [CrossRef]
- Garg, R.; Adler, G.K. Role of Mineralocorticoid Receptor in Insulin Resistance. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Calvo, M.S.; Whiting, S.J.; Barton, C.N. Vitamin D Intake: A Global Perspective of Current Status. J. Nutr. 2005, 135, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W. From Vitamin D to Hormone D: Fundamentals of the Vitamin D Endocrine System Essential for Good Health. Am. J. Clin. Nutr. 2008, 88, 491S–499S. [Google Scholar] [CrossRef]
- Alkharfy, K.M.; Al-Daghri, N.M.; Yakout, S.M.; Ahmed, M. Calcitriol Attenuates Weight-Related Systemic Inflammation and Ultrastructural Changes in the Liver in a Rodent Model. Basic Clin. Pharmacol. Toxicol. 2013, 112, 42–49. [Google Scholar] [CrossRef]
- Kumar, R.; Litwack, G. Structural and Functional Relationships of the Steroid Hormone Receptors’ N-Terminal Transactivation Domain. Steroids 2009, 74, 877–883. [Google Scholar] [CrossRef]
- Weigel, N.L. Steroid Hormone Receptors and Their Regulation by Phosphorylation. Biochem. J. 1996, 319 (Pt. 3), 657–667. [Google Scholar] [CrossRef]
- Robinson-Rechavi, M.; Escriva Garcia, H.; Laudet, V. The Nuclear Receptor Superfamily. J. Cell Sci. 2003, 116, 585–586. [Google Scholar] [CrossRef]
- Liao, R.S.; Ma, S.; Miao, L.; Li, R.; Yin, Y.; Raj, G.V. Androgen Receptor-Mediated Non-Genomic Regulation of Prostate Cancer Cell Proliferation. Transl. Androl. Urol. 2013, 2, 187–196. [Google Scholar]
- Pupo, M.; Maggiolini, M.; Musti, A.M. Gper Mediates Non-Genomic Effects of Estrogen. Methods Mol. Biol. 2016, 1366, 471–488. [Google Scholar] [PubMed]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular Mechanism of Estrogen-Estrogen Receptor Signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Scheller, K.; Seibel, P.; Sekeris, C.E. Glucocorticoid and Thyroid Hormone Receptors in Mitochondria of Animal Cells. Int. Rev. Cytol. 2003, 222, 1–61. [Google Scholar] [PubMed]
- Valadez-Cosmes, P.; Vazquez-Martinez, E.R.; Cerbon, M.; Camacho-Arroyo, I. Membrane Progesterone Receptors in Reproduction and Cancer. Mol. Cell. Endocrinol. 2016, 434, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Dressing, G.E.; Goldberg, J.E.; Charles, N.J.; Schwertfeger, K.L.; Lange, C.A. Membrane Progesterone Receptor Expression in Mammalian Tissues: A Review of Regulation and Physiological Implications. Steroids 2011, 76, 11–17. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Madak-Erdogan, Z. Estrogens and Female Liver Health. Steroids 2018, 133, 38–43. [Google Scholar] [CrossRef]
- Hart-Unger, S.; Arao, Y.; Hamilton, K.J.; Lierz, S.L.; Malarkey, D.E.; Hewitt, S.C.; Freemark, M.; Korach, K.S. Hormone Signaling and Fatty Liver in Females: Analysis of Estrogen Receptor Alpha Mutant Mice. Int. J. Obes. 2017, 41, 945–954. [Google Scholar] [CrossRef]
- Zhu, L.; Martinez, M.N.; Emfinger, C.H.; Palmisano, B.T.; Stafford, J.M. Estrogen Signaling Prevents Diet-Induced Hepatic Insulin Resistance in Male Mice with Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1188–E1197. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, Q.; Huang, T.; Tan, W.; Qu, L.; Chen, T.; Pan, H.; Chen, L.; Liu, J.; Wong, C.W.; et al. Dysfunction of Estrogen-Related Receptor Alpha-Dependent Hepatic Vldl Secretion Contributes to Sex Disparity in Nafld/Nash Development. Theranostics 2020, 10, 10874–10891. [Google Scholar] [CrossRef]
- Della Torre, S. Non-Alcoholic Fatty Liver Disease as a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence: Relevance of Estrogen Signaling. Front. Endocrinol. 2020, 11, 572490. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.T.; Pan, H.J.; Lee, C.H. Prevention of Tamoxifen-Related Nonalcoholic Fatty Liver Disease in Breast Cancer Patients. Clin. Breast Cancer 2018, 18, e677–e685. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Yu, I.C.; Wang, R.S.; Chen, Y.T.; Liu, N.C.; Altuwaijri, S.; Hsu, C.L.; Ma, W.L.; Jokinen, J.; Sparks, J.D.; et al. Increased Hepatic Steatosis and Insulin Resistance in Mice Lacking Hepatic Androgen Receptor. Hepatology 2008, 47, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Lemke, U.; Krones-Herzig, A.; Berriel Diaz, M.; Narvekar, P.; Ziegler, A.; Vegiopoulos, A.; Cato, A.C.; Bohl, S.; Klingmuller, U.; Screaton, R.A.; et al. The Glucocorticoid Receptor Controls Hepatic Dyslipidemia through Hes1. Cell Metab. 2008, 8, 212–223. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.M.; Beaudry, J.L.; Szigiato, A.A.; Trumble, S.J.; Snook, L.A.; Bonen, A.; Giacca, A.; Riddell, M.C. Consumption of a High-Fat Diet Rapidly Exacerbates the Development of Fatty Liver Disease That Occurs with Chronically Elevated Glucocorticoids. Am. J. Physiol Gastrointest. Liver Physiol. 2012, 302, G850–G863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Li, C.; Yao, G.F.; Du, L.J.; Liu, Y.; Zheng, X.J.; Yan, S.; Sun, J.Y.; Liu, Y.; Liu, M.Z.; et al. Deletion of Macrophage Mineralocorticoid Receptor Protects Hepatic Steatosis and Insulin Resistance through Eralpha/Hgf/Met Pathway. Diabetes 2017, 66, 1535–1547. [Google Scholar] [CrossRef]
- Luo, P.; Dematteo, A.; Wang, Z.; Zhu, L.; Wang, A.; Kim, H.S.; Pozzi, A.; Stafford, J.M.; Luther, J.M. Aldosterone Deficiency Prevents High-Fat-Feeding-Induced Hyperglycaemia and Adipocyte Dysfunction in Mice. Diabetologia 2013, 56, 901–910. [Google Scholar] [CrossRef]
- Marziou, A.; Philouze, C.; Couturier, C.; Astier, J.; Obert, P.; Landrier, J.F.; Riva, C. Vitamin D Supplementation Improves Adipose Tissue Inflammation and Reduces Hepatic Steatosis in Obese C57bl/6j Mice. Nutrients 2020, 12, 342. [Google Scholar] [CrossRef]
- Zhu, C.G.; Liu, Y.X.; Wang, H.; Wang, B.P.; Qu, H.Q.; Wang, B.L.; Zhu, M. Active Form of Vitamin D Ameliorates Non-Alcoholic Fatty Liver Disease by Alleviating Oxidative Stress in a High-Fat Diet Rat Model. Endocr. J. 2017, 64, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Jaruvongvanich, V.; Sanguankeo, A.; Riangwiwat, T.; Upala, S. Testosterone, Sex Hormone-Binding Globulin and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2017, 16, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, H.; Park, J.H.; Cho, B.; Kim, D.; Oh, S.W.; Lee, C.M.; Choi, H.C. A Low Level of Serum Total Testosterone Is Independently Associated with Nonalcoholic Fatty Liver Disease. BMC Gastroenterol. 2012, 12, 69. [Google Scholar] [CrossRef]
- Eguchi, Y.; Eguchi, T.; Mizuta, T.; Ide, Y.; Yasutake, T.; Iwakiri, R.; Hisatomi, A.; Ozaki, I.; Yamamoto, K.; Kitajima, Y.; et al. Visceral Fat Accumulation and Insulin Resistance Are Important Factors in Nonalcoholic Fatty Liver Disease. J. Gastroenterol. 2006, 41, 462–469. [Google Scholar] [CrossRef]
- Ma, W.L.; Lai, H.C.; Yeh, S.; Cai, X.; Chang, C. Androgen Receptor Roles in Hepatocellular Carcinoma, Fatty Liver, Cirrhosis and Hepatitis. Endocr. Relat. Cancer 2014, 21, R165–R182. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Gray, N.E.; Kuo, T.; Harris, C.A. Regulation of Triglyceride Metabolism by Glucocorticoid Receptor. Cell Biosci. 2012, 2, 19. [Google Scholar] [CrossRef]
- Sorensen, H.N.; Gautik, K.M.; Bremer, J.; Spydevold, O. Induction of the Three Peroxisomal Beta-Oxidation Enzymes Is Synergistically Regulated by Dexamethasone and Fatty Acids, and Counteracted by Insulin in Morris 7800c1 Hepatoma Cells in Culture. Eur. J. Biochem. 1992, 208, 705–711. [Google Scholar] [CrossRef]
- Andrews, R.C.; Walker, B.R. Glucocorticoids and Insulin Resistance: Old Hormones, New Targets. Clin. Sci. 1999, 96, 513–523. [Google Scholar] [CrossRef]
- Rockall, A.G.; Sohaib, S.A.; Evans, D.; Kaltsas, G.; Isidori, A.M.; Monson, J.P.; Besser, G.M.; Grossman, A.B.; Reznek, R.H. Hepatic Steatosis in Cushing’s Syndrome: A Radiological Assessment Using Computed Tomography. Eur. J. Endocrinol. 2003, 149, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Belden, Z.; Deiuliis, J.A.; Dobre, M.; Rajagopalan, S. The Role of the Mineralocorticoid Receptor in Inflammation: Focus on Kidney and Vasculature. Am. J. Nephrol. 2017, 46, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Carotti, S.; Labbadia, G.; Gentilucci, U.V.; Muda, A.O.; Angelico, F.; Silecchia, G.; Leonetti, F.; Fraioli, A.; Picardi, A.; et al. Liver Vitamin D Receptor, Cyp2r1, and Cyp27a1 Expression: Relationship with Liver Histology and Vitamin D3 Levels in Patients with Nonalcoholic Steatohepatitis or Hepatitis C Virus. Hepatology 2012, 56, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D and Metabolic Dysfunction-Associated Fatty Liver Disease (Mafld): An Update. Nutrients 2020, 12, 3302. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Pathogenesis of Nonalcoholic Steatohepatitis and Hormone-Based Therapeutic Approaches. Front. Endocrinol. 2018, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Broom, L.J.; Kogut, M.H. Inflammation: Friend or Foe for Animal Production? Poult. Sci. 2018, 97, 510–514. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Kamei, Y.; Xu, L.; Heinzel, T.; Torchia, J.; Kurokawa, R.; Gloss, B.; Lin, S.C.; Heyman, R.A.; Rose, D.W.; Glass, C.K.; et al. A Cbp Integrator Complex Mediates Transcriptional Activation and Ap-1 Inhibition by Nuclear Receptors. Cell 1996, 85, 403–414. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver Inflammation and Fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver Fibrosis: Pathophysiology, Pathogenetic Targets and Clinical Issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic Stellate Cells as Key Target in Liver Fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular Mediators of Hepatic Steatosis and Liver Injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; Network, N.C.R. Nonalcoholic Fatty Liver Disease (Nafld) Activity Score and the Histopathologic Diagnosis in Nafld: Distinct Clinicopathologic Meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Florentino, G.S.; Cotrim, H.P.; Vilar, C.P.; Florentino, A.V.; Guimaraes, G.M.; Barreto, V.S. Nonalcoholic Fatty Liver Disease in Menopausal Women. Arq. Gastroenterol. 2013, 50, 180–185. [Google Scholar] [CrossRef]
- Suzuki, A.; Abdelmalek, M.F. Nonalcoholic Fatty Liver Disease in Women. Womens Health 2009, 5, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Galmes-Pascual, B.M.; Martinez-Cignoni, M.R.; Moran-Costoya, A.; Bauza-Thorbrugge, M.; Sbert-Roig, M.; Valle, A.; Proenza, A.M.; Llado, I.; Gianotti, M. 17beta-Estradiol Ameliorates Lipotoxicity-Induced Hepatic Mitochondrial Oxidative Stress and Insulin Resistance. Free Radic. Biol. Med. 2020, 150, 148–160. [Google Scholar] [CrossRef]
- Kamada, Y.; Kiso, S.; Yoshida, Y.; Chatani, N.; Kizu, T.; Hamano, M.; Tsubakio, M.; Takemura, T.; Ezaki, H.; Hayashi, N.; et al. Estrogen Deficiency Worsens Steatohepatitis in Mice Fed High-Fat and High-Cholesterol Diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1031–G1043. [Google Scholar] [CrossRef]
- Xin, G.; Qin, S.; Wang, S.; Wang, X.; Zhang, Y.; Wang, J. Sex Hormone Affects the Severity of Non-Alcoholic Steatohepatitis through the Myd88-Dependent Il-6 Signaling Pathway. Exp. Biol. Med. 2015, 240, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Kato, M.; Yamasaki, A.; Kuwano, A.; Suzuki, H.; Kohjima, M.; Ogawa, Y. Effects of High Fructose Intake on Liver Injury Progression in High Fat Diet Induced Fatty Liver Disease in Ovariectomized Female Mice. Food Chem. Toxicol. 2018, 118, 190–197. [Google Scholar] [CrossRef]
- Turola, E.; Petta, S.; Vanni, E.; Milosa, F.; Valenti, L.; Critelli, R.; Miele, L.; Maccio, L.; Calvaruso, V.; Fracanzani, A.L.; et al. Ovarian Senescence Increases Liver Fibrosis in Humans and Zebrafish with Steatosis. Dis. Model. Mech. 2015, 8, 1037–1046. [Google Scholar] [CrossRef]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp-Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. A Longer Duration of Estrogen Deficiency Increases Fibrosis Risk among Postmenopausal Women with Nonalcoholic Fatty Liver Disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Shimizu, I.; Cui, X.; Itonaga, M.; Tamaki, K.; Fukuno, H.; Inoue, H.; Honda, H.; Ito, S. Antioxidant and Antiapoptotic Activities of Idoxifene and Estradiol in Hepatic Fibrosis in Rats. Life Sci. 2004, 74, 897–907. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Tran, Q.T.; Thiyagarajan, T.; Miller, D.D.; Bridges, D.; Narayanan, R. An Estrogen Receptor Beta-Selective Agonist Inhibits Non-Alcoholic Steatohepatitis in Preclinical Models by Regulating Bile Acid and Xenobiotic Receptors. Exp. Biol. Med. 2017, 242, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, N.V.; Wong, S.K.; Wan Hasan, W.N.; Jolly, J.J.; Nur-Farhana, M.F.; Ima-Nirwana, S.; Chin, K.Y. The Relationship between Circulating Testosterone and Inflammatory Cytokines in Men. Aging Male 2019, 22, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Wang, L.; Li, Z.; Zhang, H.; Wu, J.; Rahman, N.; Guo, Y.; Li, D.; Li, N.; et al. Differential Effects of Estrogen/Androgen on the Prevention of Nonalcoholic Fatty Liver Disease in the Male Rat. J. Lipid Res. 2013, 54, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Morita, A.; Ishigaki, Y. Gender-Difference in Diabetes Mellitus. Nihon Rinsho 2015, 73, 606–610. [Google Scholar]
- Lanari, A.; Garegnani, T.C.; Heinrichs, G.; Castresana, M.P. [Hepatic Fibrosis and Progesterone]. Acta Gastroenterol. Latinoam. 1988, 18, 161–171. [Google Scholar]
- Kim, K.H.; Lee, J.M.; Zhou, Y.; Harpavat, S.; Moore, D.D. Glucocorticoids Have Opposing Effects on Liver Fibrosis in Hepatic Stellate and Immune Cells. Mol. Endocrinol. 2016, 30, 905–916. [Google Scholar] [CrossRef]
- Wada, T.; Miyashita, Y.; Sasaki, M.; Aruga, Y.; Nakamura, Y.; Ishii, Y.; Sasahara, M.; Kanasaki, K.; Kitada, M.; Koya, D.; et al. Eplerenone Ameliorates the Phenotypes of Metabolic Syndrome with Nash in Liver-Specific Srebp-1c Tg Mice Fed High-Fat and High-Fructose Diet. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1415–E1425. [Google Scholar] [CrossRef]
- Pizarro, M.; Solis, N.; Quintero, P.; Barrera, F.; Cabrera, D.; Rojas-de Santiago, P.; Arab, J.P.; Padilla, O.; Roa, J.C.; Moshage, H.; et al. Beneficial Effects of Mineralocorticoid Receptor Blockade in Experimental Non-Alcoholic Steatohepatitis. Liver Int. 2015, 35, 2129–2138. [Google Scholar] [CrossRef]
- Queisser, N.; Happ, K.; Link, S.; Jahn, D.; Zimnol, A.; Geier, A.; Schupp, N. Aldosterone Induces Fibrosis, Oxidative Stress and DNA Damage in Livers of Male Rats Independent of Blood Pressure Changes. Toxicol. Appl. Pharmacol. 2014, 280, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D Deficiency in Obese Rats Exacerbates Nonalcoholic Fatty Liver Disease and Increases Hepatic Resistin and Toll-Like Receptor Activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Cheng, Y.F.; Lai, C.Y.; Hsu, L.W.; Chang, Y.C.; Deng, J.Y.; Huang, Y.Z.; Honda, H.; Chen, K.D.; Wang, C.C.; et al. Impact of Artificial Sunlight Therapy on the Progress of Non-Alcoholic Fatty Liver Disease in Rats. J. Hepatol. 2011, 55, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Nolly, M.B.; Caldiz, C.I.; Yeves, A.M.; Villa-Abrille, M.C.; Morgan, P.E.; Amado Mondaca, N.; Portiansky, E.L.; Chiappe de Cingolani, G.E.; Cingolani, H.E.; Ennis, I.L. The Signaling Pathway for Aldosterone-Induced Mitochondrial Production of Superoxide Anion in the Myocardium. J. Mol. Cell. Cardiol. 2014, 67, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Artunc, F.; Lang, F. Mineralocorticoid and Sgk1-Sensitive Inflammation and Tissue Fibrosis. Nephron Physiol. 2014, 128, 35–39. [Google Scholar] [CrossRef]
- Eliades, M.; Spyrou, E. Vitamin D: A New Player in Non-Alcoholic Fatty Liver Disease? World J. Gastroenterol. 2015, 21, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.B.; Chen, Y.H.; Zhang, C.; Shi, C.E.; Hu, K.F.; Zhou, J.; Xu, D.X.; Chen, X. Low Vitamin D Status Is Associated with Advanced Liver Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Endocrine 2017, 55, 582–590. [Google Scholar] [CrossRef]
- Breitkopf, K.; Godoy, P.; Ciuclan, L.; Singer, M.V.; Dooley, S. Tgf-Beta/Smad Signaling in the Injured Liver. Z. Gastroenterol. 2006, 44, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D Supplementation and Non-Alcoholic Fatty Liver Disease: Present and Future. Nutrients 2017, 9, 1015. [Google Scholar] [CrossRef]
- Beilfuss, A.; Sowa, J.P.; Sydor, S.; Beste, M.; Bechmann, L.P.; Schlattjan, M.; Syn, W.K.; Wedemeyer, I.; Mathe, Z.; Jochum, C.; et al. Vitamin D Counteracts Fibrogenic Tgf-Beta Signalling in Human Hepatic Stellate Cells Both Receptor-Dependently and Independently. Gut 2015, 64, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Kojima, T.; Ohbora, A.; Takeda, N.; Fukui, M.; Kato, T. Aging Is a Risk Factor of Nonalcoholic Fatty Liver Disease in Premenopausal Women. World J. Gastroenterol. 2012, 18, 237–243. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, M.; Hu, Z.; Shrestha, U.K. Prevalence of Nonalcoholic Fatty Liver Disease and Its Metabolic Risk Factors in Women of Different Ages and Body Mass Index. Menopause 2015, 22, 667–673. [Google Scholar] [CrossRef]
- Caballeria, L.; Pera, G.; Auladell, M.A.; Toran, P.; Munoz, L.; Miranda, D.; Aluma, A.; Casas, J.D.; Sanchez, C.; Gil, D.; et al. Prevalence and Factors Associated with the Presence of Nonalcoholic Fatty Liver Disease in an Adult Population in Spain. Eur. J. Gastroenterol. Hepatol. 2010, 22, 24–32. [Google Scholar] [CrossRef]
- Camhi, S.M.; Bray, G.A.; Bouchard, C.; Greenway, F.L.; Johnson, W.D.; Newton, R.L.; Ravussin, E.; Ryan, D.H.; Smith, S.R.; Katzmarzyk, P.T. The Relationship of Waist Circumference and Bmi to Visceral, Subcutaneous, and Total Body Fat: Sex and Race Differences. Obesity 2011, 19, 402–408. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-Alcoholic Fatty Liver Disease—A Global Public Health Perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Y.; Wang, E.; Zhang, Z.; Xiong, X.; Zhang, H.; Lu, J.; Zheng, S.; Yang, J.; Xia, X.; et al. Hepatic Estrogen Receptor Alpha Improves Hepatosteatosis through Upregulation of Small Heterodimer Partner. J. Hepatol. 2015, 63, 183–190. [Google Scholar] [CrossRef]
- Pan, J.J.; Fallon, M.B. Gender and Racial Differences in Nonalcoholic Fatty Liver Disease. World J. Hepatol. 2014, 6, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Summart, U.; Thinkhamrop, B.; Chamadol, N.; Khuntikeo, N.; Songthamwat, M.; Kim, C.S. Gender Differences in the Prevalence of Nonalcoholic Fatty Liver Disease in the Northeast of Thailand: A Population-Based Cross-Sectional Study. F1000Research 2017, 6, 1630. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.K.; Boulet, S.L.; Mehta, A.; Hotaling, J.; Eisenberg, M.L.; Honig, S.C.; Warner, L.; Kissin, D.M.; Nangia, A.K.; Ross, L.S. Trends in Testosterone Replacement Therapy Use from 2003 to 2013 among Reproductive-Age Men in the United States. J. Urol. 2017, 197, 1121–1126. [Google Scholar] [CrossRef]
- Phan, H.; Richard, A.; Lazo, M.; Nelson, W.G.; Denmeade, S.R.; Groopman, J.; Kanarek, N.; Platz, E.A.; Rohrmann, S. The Association of Sex Steroid Hormone Concentrations with Non-Alcoholic Fatty Liver Disease and Liver Enzymes in Us Men. Liver Int. 2021, 41, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, S.; Meng, Y.; Yu, Q.; Wang, Q.; Xu, H.; Yuan, H.; Li, X.; Chen, L. Effects of Vitamin D Supplementation in Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Int. J. Endocrinol. Metab. 2020, 18, e97205. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Ma, F.; Guan, M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 320. https://doi.org/10.3390/metabo11050320
Yang M, Ma F, Guan M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites. 2021; 11(5):320. https://doi.org/10.3390/metabo11050320
Chicago/Turabian StyleYang, Meng, Feng Ma, and Min Guan. 2021. "Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease" Metabolites 11, no. 5: 320. https://doi.org/10.3390/metabo11050320
APA StyleYang, M., Ma, F., & Guan, M. (2021). Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites, 11(5), 320. https://doi.org/10.3390/metabo11050320