
Facilitated Qualitative Determination of Insulin, Its Synthetic Analogs, and C-Peptide in Human Urine by Means of LC–HRMS



Abstract
1. Introduction
2. Results and Discussion
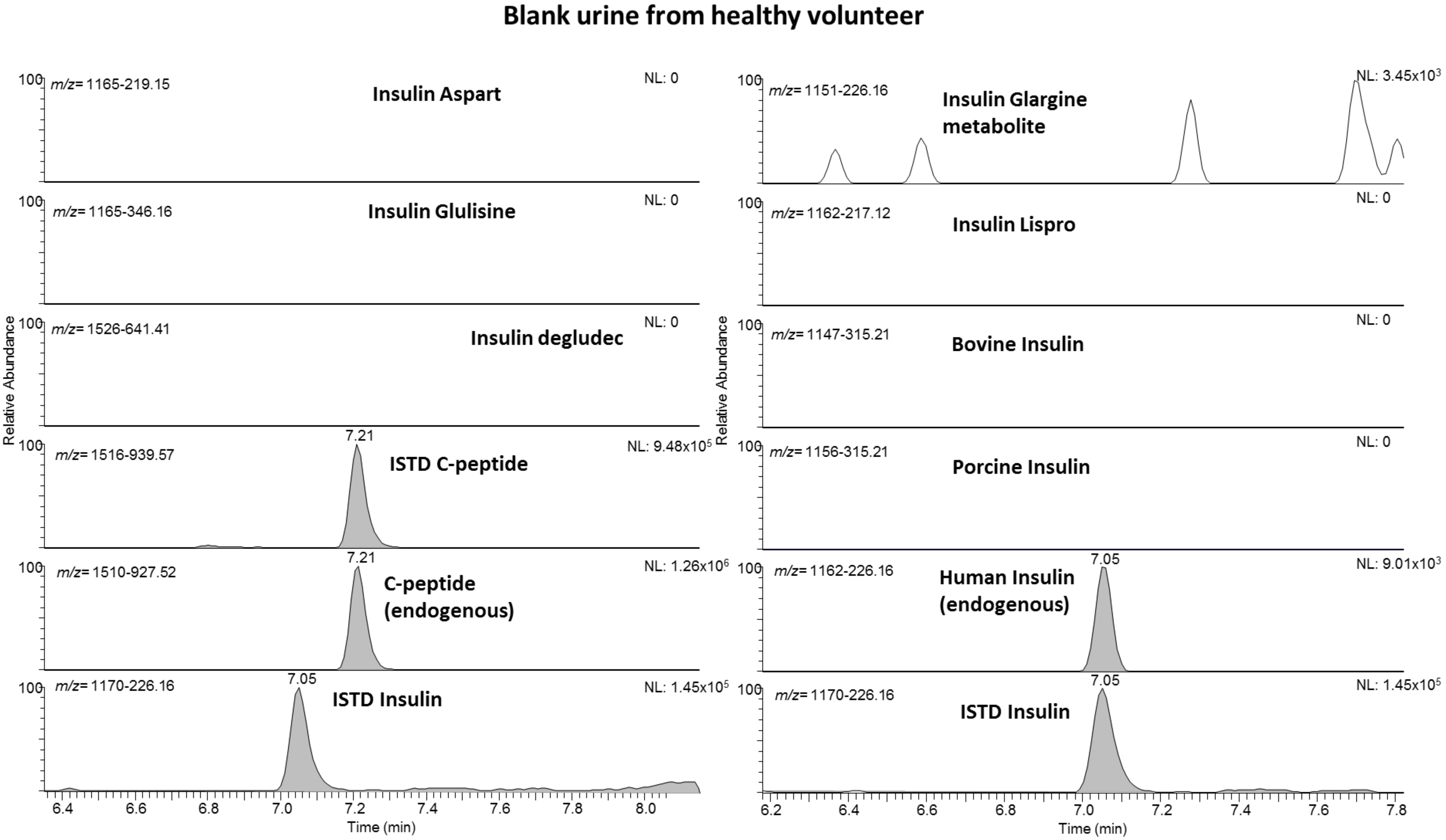
2.1. Liquid Chromatography/Mass Spectrometry
2.2. Validation
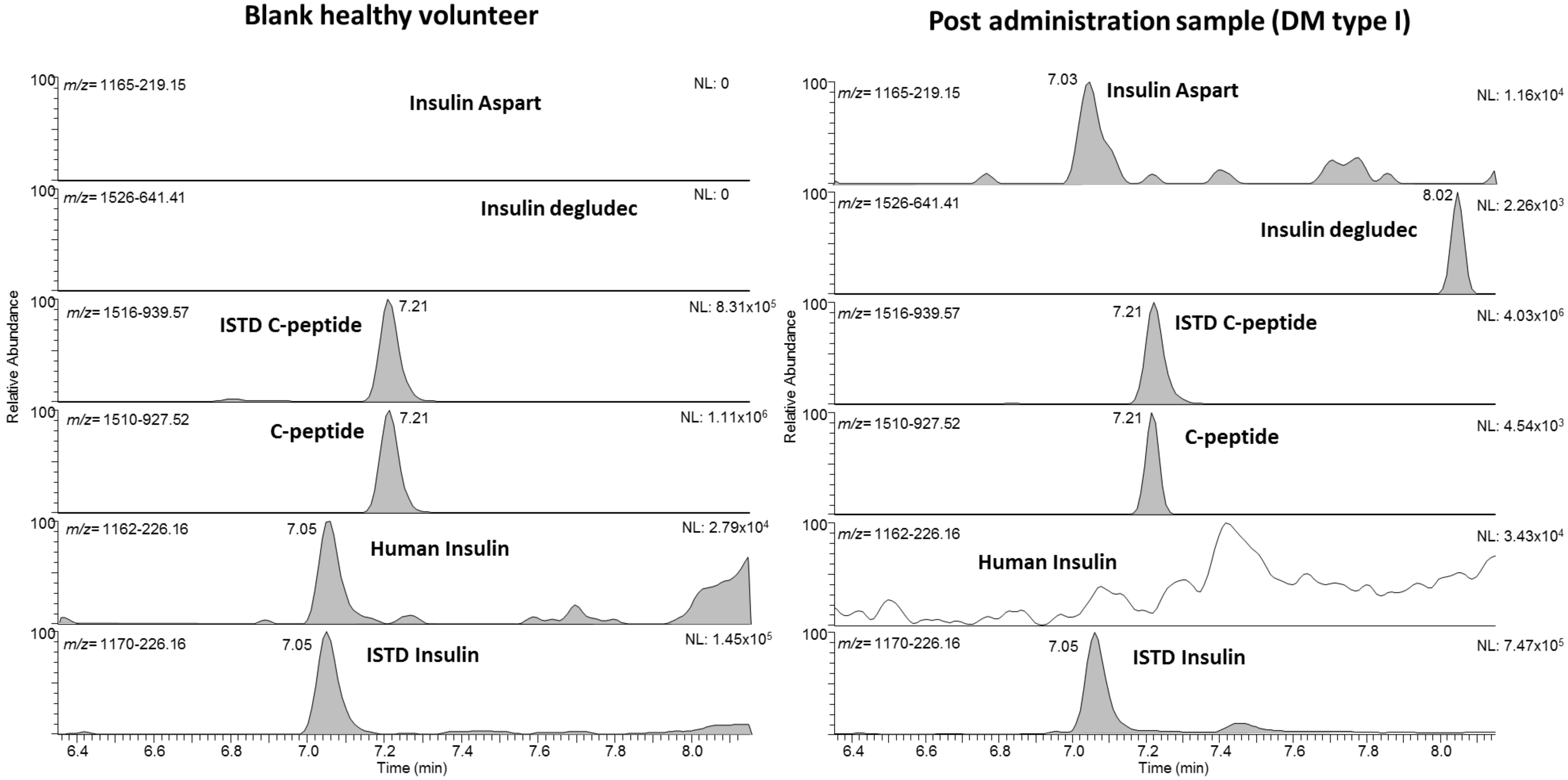
2.3. Proof of Concept
2.4. Limitations of the Assay/Potential Applications
3. Materials and Methods
3.1. Urine Samples
3.2. Sample Preparation Mixed-Cation Exchange
3.3. Liquid Chromatography
3.4. Mass Spectrometry
3.5. Validation
3.5.1. Specificity
3.5.2. Recovery
3.5.3. Limit of Detection/Limit of Identification (LOD/LOI)
3.5.4. Precision
3.5.5. Carry-Over
3.5.6. Matrix Effect
3.5.7. Robustness
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rebsomen, L.; Pitel, S.; Boubred, F.; Buffat, C.; Feuerstein, J.M.; Raccah, D.; Vague, P.; Tsimaratos, M. C-peptide replacement improves weight gain and renal function in diabetic rats. Diabetes Metab. 2006, 32, 223–228. [Google Scholar] [CrossRef]
- Lebowitz, M.R.; Blumenthal, S.A. The molar ratio of insulin to c-peptide. An aid to the diagnosis of hypoglycemia due to surreptitious (or inadvertent) insulin administration. Arch. Intern. Med. 1993, 153, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, M. Advances in the quantitation of therapeutic insulin analogues by lc–ms/ms. Bioanalysis 2013, 5, 2933–2946. [Google Scholar] [CrossRef] [PubMed]
- Cobelli, C.; Pacini, G. Insulin secretion and hepatic extraction in humans by minimal modeling of c-peptide and insulin kinetics. Diabetes 1988, 37, 223–231. [Google Scholar] [CrossRef] [PubMed]
- WADA. 2019. Available online: https://www.Wada-ama.Org/sites/default/files/resources/files/td2019mrpl_eng.Pdf (accessed on 25 March 2021).
- Judak, P.; Coppieters, G.; Lapauw, B.; Van Eenoo, P.; Deventer, K. Urinary detection of rapid-acting insulin analogs in healthy humans. Drug Test Anal. 2020, 12, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Thevis, M. Recent advances in the determination of insulins from biological fluids. In Advances in Clinical Chemistry; Makowski, G.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Mazzarino, M.; Senofonte, M.; Martinelli, F.; de la Torre, X.; Botre, F. Detection of recombinant insulins in human urine by liquid chromatography-electrospray ionization tandem mass spectrometry after immunoaffinity purification based on monolithic microcolumns. Anal. Bioanal. Chem. 2019, 411, 8153–8162. [Google Scholar] [CrossRef] [PubMed]
- Judak, P.; Van Eenoo, P.; Deventer, K. Utilizing elisa-plate based immunopurification and liquid chromatography-tandem mass spectrometry for the urinary detection of short- and long acting human insulin analogues. J. Pharm. Biomed. Anal. 2018, 153, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Schänzer, W.; Thevis, M. Immunoaffinity techniques coupled to mass spectrometry for the analysis of human peptide hormones: Advances and applications. Expert Rev. Proteom. 2017, 14, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Judak, P.; Van Eenoo, P.; Deventer, K. Adsorption effects of the doping relevant peptides insulin lispro, synachten, tb-500 and ghrp 5. Anal. Biochem. 2017, 537, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Schänzer, W.; Delahaut, P.; Thevis, M. Immunoaffinity purification of peptide hormones prior to liquid chromatography-mass spectrometry in doping controls. Methods 2012, 56, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Yang, R.; Petring, S.; Bally, L.; Thevis, M. Simplified quantification of insulin, its synthetic analogs and c-peptide in human plasma by means of lc-hrms. Drug Test. Anal. 2020, 12, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.E.; Fountain, K.J.; Smith, N.; Ashraf, L.; Karalliedde, J.; Cowan, D.; Legido-Quigley, C. Multidimensional lc-ms/ms enables simultaneous quantification of intact human insulin and five recombinant analogs in human plasma. Anal. Chem. 2014, 86, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Thevis, M.; Delahaut, P.; Bosseloir, A.; Schanzer, W. Mass spectrometric identification of degradation products of insulin and its long-acting analogues in human urine for doping control purposes. Anal. Chem. 2007, 79, 2518–2524. [Google Scholar] [CrossRef] [PubMed]
- WADA. 2021. Available online: https://www.Wada-ama.Org/en/resources/laboratories/international-standard-for-laboratories-isl (accessed on 25 March 2021).
- Wunder, C.; Kauert, G.F.; Toennes, S.W. Factors leading to the degradation/loss of insulin in postmortem blood samples. Forensic Sci. Int. 2014, 241, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Kuerzel, G.U.; Shukla, U.; Scholtz, H.E.; Pretorius, S.G.; Wessels, D.H.; Venter, C.; Potgieter, M.A.; Lang, A.M.; Koose, T.; Bernhardt, E. Biotransformation of insulin glargine after subcutaneous injection in healthy subjects. Curr. Med. Res. Opin. 2003, 19, 34–40. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Peptides Included | Amino Acid Sequence | Monoisotopic Mass [Da] | Precursor Ion [m/z] | Monitored Charge State | Product Ions | Multiple × Group | ~Ret. Time [min] |
|---|---|---|---|---|---|---|---|
| Human insulin | GIVEQCCTSICSLYQLENYCN—FVNQHLCGSHLVEALYLVCGERGFFYTPKT | 5803.6 | 1452/1162 | 4+/5+ | 226y, 219b, 143a, 345y | 2 | 7.05 |
| Insulin aspart | GIVEQCCTSICSLYQLENYCN—FVNQHLCGSHLVEALYLVCGERGFFYTDKT | 5821.6 | 1457/1166 | 4+/5+ | 226y, 219b, 248y, 464y | 1 | 7.02 |
| Insulin glulisine | GIVEQCCTSICSLYQLENYCN—FVKQHLCGSHLVEALYLVCGERGFFYTPET | 5818.6 | 1456/1166 | 4+/5+ | 227y, 346y, 199y | 1 | 7.01 |
| Insulin lispro | GIVEQCCTSICSLYQLENYCN—FVNQHLCGSHLVEALYLVCGERGFFYTKPT | 5803.6 | 1452/1162 | 4+/5+ | 217y, 230y | 2 | 7.01 |
| Insulin glargine met | GIVEQCCTSICSLYQLENYCG—FVNQHLCGSHLVEALYLVCGERGFFYTPKT | 5746.6 | 1438 /1151 | 4+/5+ | 226y, 219b | 1 | 7.09 |
| Insulin degludec | GIVEQCCTSICSLYQLENYCN—FVNQHLCGSHLVEALYLVCGERGFFYTPK-γ-L-Glu-Pal | 6099.8 | 1527 | 4+ | 641y, 244y | 2 | 8.00 |
| Porcine insulin | GIVEQCCTSICSLYQLENYCN—FVNQHLCGSHLVEALYLVCGERGFFYTPKA | 5773.6 | 1445/1156 | 4+/5+ | 226y, 315y | 3 | 7.06 |
| Bovine insulin | GIVEQCCASVCSLYQLENYCN—FVNQHLCGSHLVEALYLVCGERGFFYTPKA | 5729.6 | 1433/1147 | 4+/5+ | 226y, 315y | 2 | 6.99 |
| C-peptide | EAEDLQVGQVELGGGPGAGSLQPLALEGSLQ | 3018.5 | 1510 | 2+ | 927, 260y, 785b | 3 | 7.22 |
| labeled insulin | GIVEQCCTSICSLYQLENYCN—FVNQHL*CGSHL*VEAL*YL*VCGERGFFYTPKT | 5843.9 | 1462/1170 | 4+/5+ | 226y, 219b, 143a, 345y | 4 | 7.01 |
| labeled C-peptide | EAEDLQVGQVELGGGPGAGSLQPLAL*EGSL*Q | 3030.6 | 1516 | 2+ | 939y, 266y, 785b | 4 | 7.21 |
| Specificity | LOD | LOI | Precision | Recovery | Carry Over | Matrix Effect | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| [pg/mL] | At 50 pg/mL | At 25 pg/mL | At 10 pg/mL | At 5 pg/mL | At 100 pg/mL [%] | [%] | [%] | [%] | ||
| Human insulin | ok | 10 | - | - | - | - | 2 | 35 | <1% | - |
| Insulin aspart | ok | 10 | 6/6 | 6/6 | 4/6 | 3/6 | 9 | 26 | <1% | 80–120 |
| Insulin glulisine | ok | 10 | 6/6 | 6/6 | 4/6 | 3/6 | 8 | 26 | <2% | 80–120 |
| Insulin lispro | ok | 10 | 6/6 | 6/6 | 5/6 | 3/6 | 8 | 31 | <2% | 80–120 |
| Insulin glargine met | ok | 10 | 6/6 | 6/6 | 4/6 | 2/6 | 13 | 39 | <1% | 80–120 |
| Insulin degludec | ok | 10 | 6/6 | 6/6 | 5/6 | 4/6 | 24 | 51 | <2% | 80–120 |
| Porcine Insulin | ok | 25 | 6/6 | 4/6 | 2/6 | 0/6 | 9 | 34 | <2% | 80–120 |
| Bovine Insulin | ok | 10 | 6/6 | 6/6 | 5/6 | 4/6 | 10 | 35 | <1% | 80–120 |
| C-peptide | ok | 25 | - | - | - | - | 3 | 100 | <2% | - |
| Patient Number | Diabetes Type | Insulin Regimen | Detected Peptides |
|---|---|---|---|
| 1 | II | Tujon (glargine) | glargine metabolite, human insulin, C-peptide |
| 2 | I | Fiasp (aspart), 44 U/d | aspart |
| 3 | II | Fiasp (aspart), tiny doses | aspart, human insulin, C-peptide |
| 4 | unclear | Novorapid, 24 U/d; Lantus 24 U/d | aspart, glargine (lantus) metabolite, human insulin, C-peptide |
| 5 | unknown | Novorapid, 24 U/d; Tresiba 19 U/d | aspart, tresiba (degludec), C-peptide (traces) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, A.; Benzenberg, L.; Bally, L.; Thevis, M. Facilitated Qualitative Determination of Insulin, Its Synthetic Analogs, and C-Peptide in Human Urine by Means of LC–HRMS. Metabolites 2021, 11, 309. https://doi.org/10.3390/metabo11050309
Thomas A, Benzenberg L, Bally L, Thevis M. Facilitated Qualitative Determination of Insulin, Its Synthetic Analogs, and C-Peptide in Human Urine by Means of LC–HRMS. Metabolites. 2021; 11(5):309. https://doi.org/10.3390/metabo11050309
Chicago/Turabian StyleThomas, Andreas, Lukas Benzenberg, Lia Bally, and Mario Thevis. 2021. "Facilitated Qualitative Determination of Insulin, Its Synthetic Analogs, and C-Peptide in Human Urine by Means of LC–HRMS" Metabolites 11, no. 5: 309. https://doi.org/10.3390/metabo11050309
APA StyleThomas, A., Benzenberg, L., Bally, L., & Thevis, M. (2021). Facilitated Qualitative Determination of Insulin, Its Synthetic Analogs, and C-Peptide in Human Urine by Means of LC–HRMS. Metabolites, 11(5), 309. https://doi.org/10.3390/metabo11050309