
Butyrate Prevents TGF-β1-Induced Alveolar Myofibroblast Differentiation and Modulates Energy Metabolism

 , , ,
, , ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
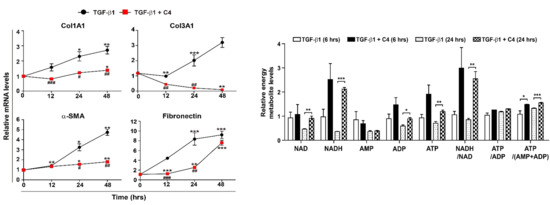
2.1. TGF-β1 Treatment Increases Expression of Fibrotic Markers and Decreases Energy Metabolism in Pulmonary Fibroblasts
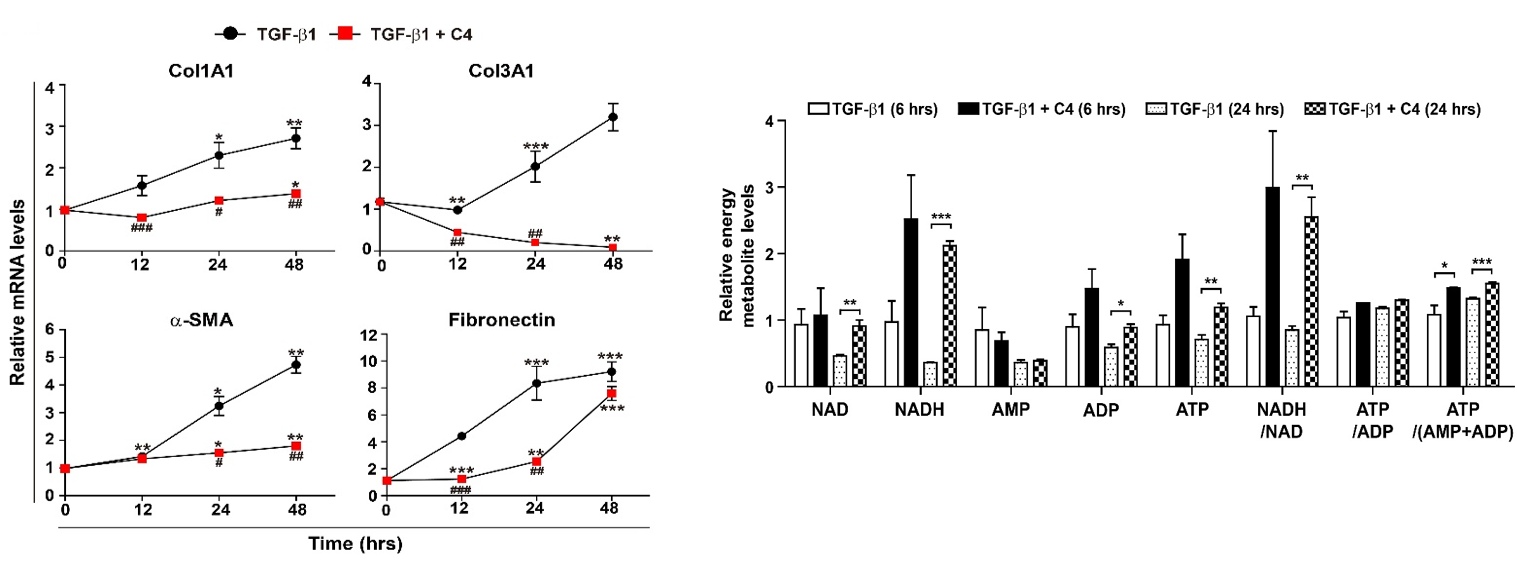
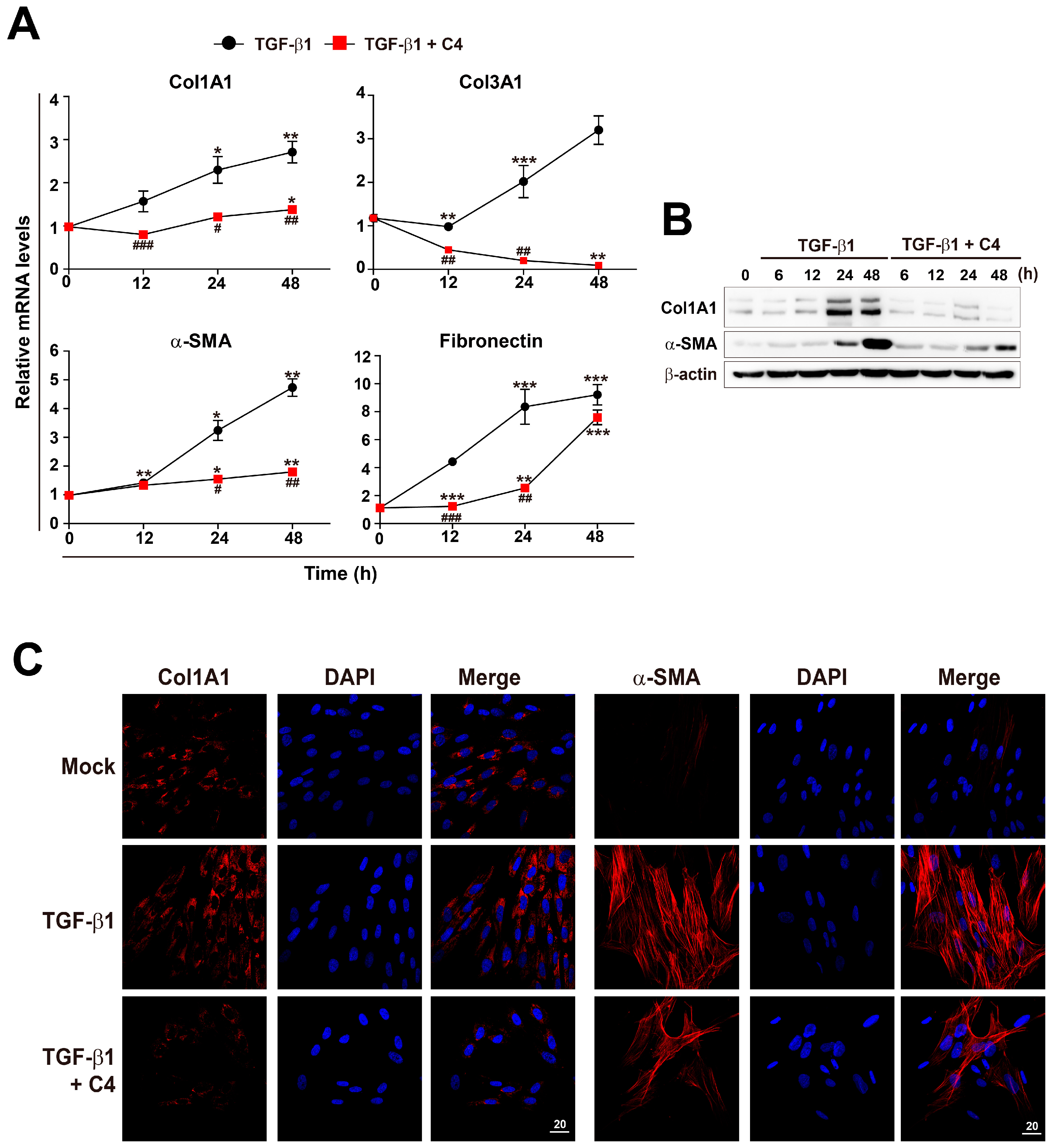
2.2. Effect of SCFAs on Fibrotic Marker Expression of TGF-β1-Treated Pulmonary Fibroblasts
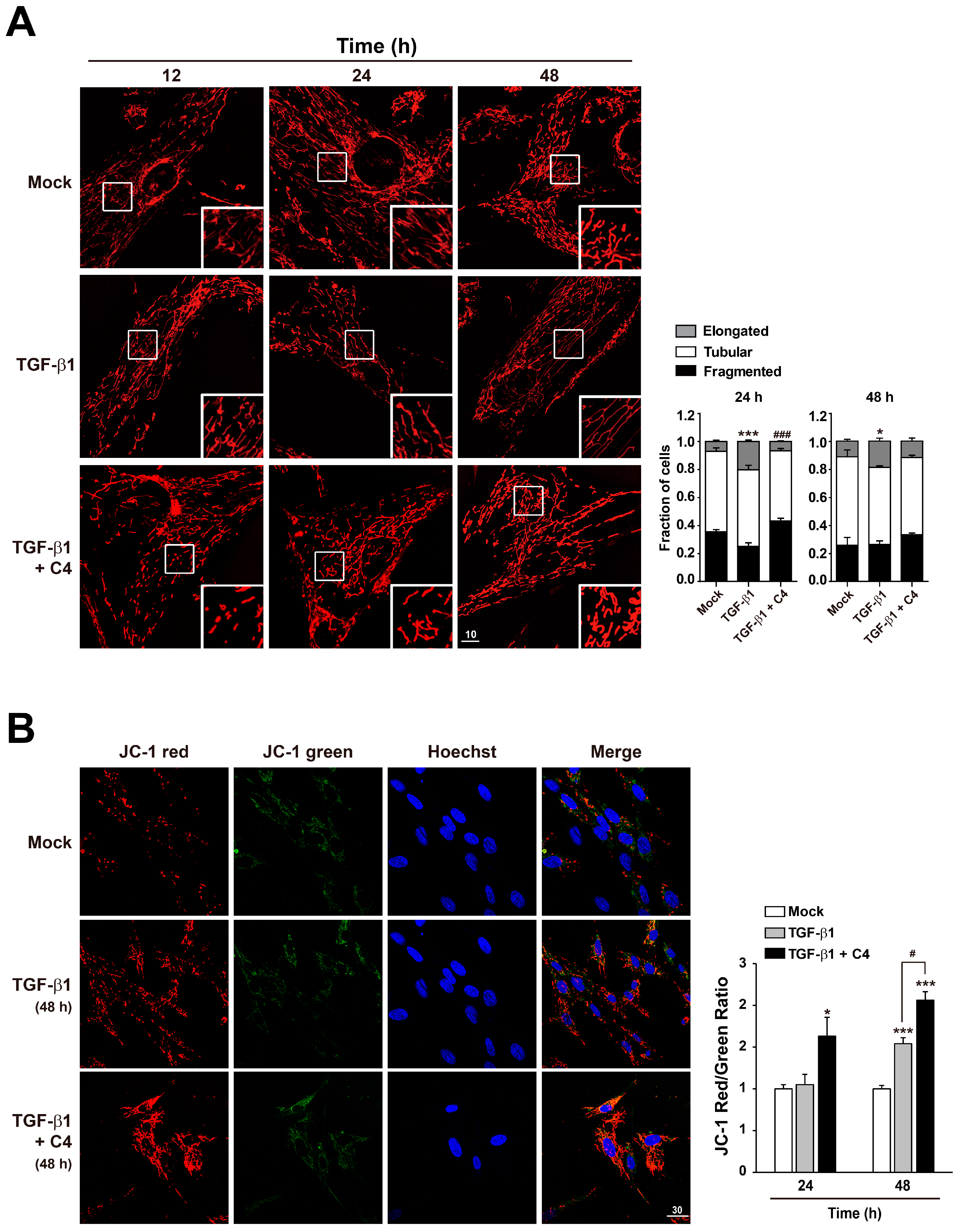
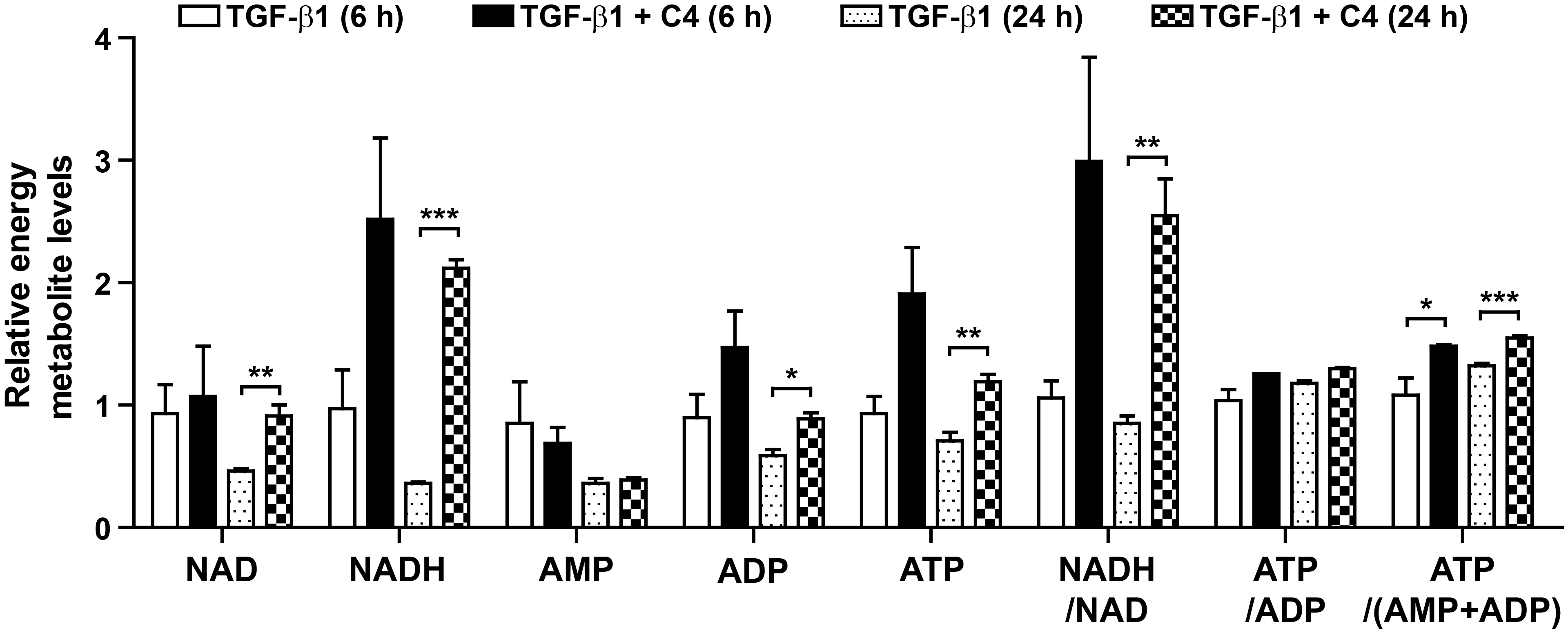
2.3. Butyrate Enhances Mitochondrial Function in Pulmonary Fibroblasts Whose Differentiation Is Surpressed
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Cell Culture and Treatments
3.3. Western Blot Analysis and Antibodies
3.4. RNA Isolation and Quantitative Polymerase Chain Reaction (qPCR)
3.5. Microscopy and Image Analysis
3.6. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef]
- Sheppard, D. Transforming growth factor beta: A central modulator of pulmonary and airway inflammation and fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Phan, S.H. Fibroblast phenotypes in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2003, 29, S87–S92. [Google Scholar] [PubMed]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef]
- Man, W.H.; de Steenhuijsen Piters, W.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef]
- Spagnolo, P.; Molyneaux, P.L.; Bernardinello, N.; Cocconcelli, E.; Biondini, D.; Fracasso, F.; Tine, M.; Saetta, M.; Maher, T.M.; Balestro, E. The Role of the Lung’s Microbiome in the Pathogenesis and Progression of Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2019, 20, 5618. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Willis-Owen, S.A.G.; Cox, M.J.; James, P.; Cowman, S.; Loebinger, M.; Blanchard, A.; Edwards, L.M.; Stock, C.; Daccord, C.; et al. Host-Microbial Interactions in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Cox, M.J.; Wells, A.U.; Kim, H.C.; Ji, W.; Cookson, W.O.; Moffatt, M.F.; Kim, D.S.; Maher, T.M. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Ashley, S.L.; Gurczynski, S.J.; Xia, M.; Wilke, C.; Falkowski, N.R.; Norman, K.C.; Arnold, K.B.; Huffnagle, G.B.; Salisbury, M.L.; et al. Lung Microbiota Contribute to Pulmonary Inflammation and Disease Progression in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Han, M.K.; Zhou, Y.; Murray, S.; Tayob, N.; Noth, I.; Lama, V.N.; Moore, B.B.; White, E.S.; Flaherty, K.R.; Huffnagle, G.B.; et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: An analysis of the COMET study. Lancet Respir. Med. 2014, 2, 548–556. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, S.F.; Espindola, M.S.; Vij, R.; Oldham, J.M.; Huffnagle, G.B.; Erb-Downward, J.R.; Flaherty, K.R.; Moore, B.B.; White, E.S.; et al. Microbes Are Associated with Host Innate Immune Response in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 208–219. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Segal, L.N.; Clemente, J.C.; Li, Y.; Ruan, C.; Cao, J.; Danckers, M.; Morris, A.; Tapyrik, S.; Wu, B.G.; Diaz, P.; et al. Anaerobic Bacterial Fermentation Products Increase Tuberculosis Risk in Antiretroviral-Drug-Treated HIV Patients. Cell Host Microbe 2017, 21, 530–537.e534. [Google Scholar] [CrossRef]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Mortensen, P.B.; Clausen, M.R. Short-chain fatty acids in the human colon: Relation to gastrointestinal health and disease. Scand. J. Gastroenterol. Suppl. 1996, 216, 132–148. [Google Scholar] [CrossRef]
- Ghorbani, P.; Santhakumar, P.; Hu, Q.; Djiadeu, P.; Wolever, T.M.; Palaniyar, N.; Grasemann, H. Short-chain fatty acids affect cystic fibrosis airway inflammation and bacterial growth. Eur. Respir. J. 2015, 46, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Niccum, D.; Dunitz, J.M.; Hunter, R.C. Evidence and Role for Bacterial Mucin Degradation in Cystic Fibrosis Airway Disease. PLoS Pathog. 2016, 12, e1005846. [Google Scholar] [CrossRef] [PubMed]
- Sowah, S.A.; Hirche, F.; Milanese, A.; Johnson, T.S.; Grafetstatter, M.; Schubel, R.; Kirsten, R.; Ulrich, C.M.; Kaaks, R.; Zeller, G.; et al. Changes in Plasma Short-Chain Fatty Acid Levels after Dietary Weight Loss Among Overweight and Obese Adults over 50 Weeks. Nutrients 2020, 12, 452. [Google Scholar] [CrossRef]
- Lewis, G.; Wang, B.; Shafiei Jahani, P.; Hurrell, B.P.; Banie, H.; Aleman Muench, G.R.; Maazi, H.; Helou, D.G.; Howard, E.; Galle-Treger, L.; et al. Dietary Fiber-Induced Microbial Short Chain Fatty Acids Suppress ILC2-Dependent Airway Inflammation. Front. Immunol. 2019, 10, 2051. [Google Scholar] [CrossRef]
- Tahara, Y.; Yamazaki, M.; Sukigara, H.; Motohashi, H.; Sasaki, H.; Miyakawa, H.; Haraguchi, A.; Ikeda, Y.; Fukuda, S.; Shibata, S. Gut Microbiota-Derived Short Chain Fatty Acids Induce Circadian Clock Entrainment in Mouse Peripheral Tissue. Sci. Rep. 2018, 8, 1395. [Google Scholar] [CrossRef]
- Byrne, C.S.; Chambers, E.S.; Morrison, D.J.; Frost, G. The role of short chain fatty acids in appetite regulation and energy homeostasis. Int. J. Obes. 2015, 39, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef]
- Correa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef]
- Mirkovic, B.; Murray, M.A.; Lavelle, G.M.; Molloy, K.; Azim, A.A.; Gunaratnam, C.; Healy, F.; Slattery, D.; McNally, P.; Hatch, J.; et al. The Role of Short-Chain Fatty Acids, Produced by Anaerobic Bacteria, in the Cystic Fibrosis Airway. Am. J. Respir. Crit. Care Med. 2015, 192, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef]
- Tanjore, H.; Blackwell, T.S.; Lawson, W.E. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L721–L729. [Google Scholar] [CrossRef]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Barja, G. Mitochondrial oxygen radical generation and leak: Sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr. 1999, 31, 347–366. [Google Scholar] [CrossRef]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Shan, B.; Lasky, J.A. TGF-beta: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6. [Google Scholar] [CrossRef]
- Khalil, N.; Greenberg, A.H. The role of TGF-beta in pulmonary fibrosis. Ciba Found. Symp. 1991, 157, 194–207; discussion 207–211. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Lee, S.B.; Lee, J.M.; Kim, H.M.; Hong, J.Y.; Lee, W.J.; Choi, C.W.; Shin, H.K.; Kim, D.J.; Koh, E.S.; et al. Metabolic Profiling Regarding Pathogenesis of Idiopathic Pulmonary Fibrosis. J. Proteome Res. 2016, 15, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Choudhury, M.; Kang, J.H.; Schaefbauer, K.J.; Jung, M.Y.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-beta. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Rutting, S.; Xenaki, D.; Malouf, M.; Horvat, J.C.; Wood, L.G.; Hansbro, P.M.; Oliver, B.G. Short-chain fatty acids increase TNFalpha-induced inflammation in primary human lung mesenchymal cells through the activation of p38 MAPK. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L157–L174. [Google Scholar] [CrossRef] [PubMed]
- Theiler, A.; Barnthaler, T.; Platzer, W.; Richtig, G.; Peinhaupt, M.; Rittchen, S.; Kargl, J.; Ulven, T.; Marsh, L.M.; Marsche, G.; et al. Butyrate ameliorates allergic airway inflammation by limiting eosinophil trafficking and survival. J. Allergy Clin. Immunol. 2019, 144, 764–776. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Yoon, D.S.; Lim, I.K.; Yoon, S.H.; Chung, H.Y.; Rojo, M.; Malka, F.; Jou, M.J.; Martinou, J.C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.W.; Yoon, J.; Yim, N.; Choi, K.; Choi, C. Downregulation of OPA3 is responsible for transforming growth factor-beta-induced mitochondrial elongation and F-actin rearrangement in retinal pigment epithelial ARPE-19 cells. PLoS ONE 2013, 8, e63495. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.B. Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 1988, 4, 155–181. [Google Scholar] [CrossRef]
- Salvioli, S.; Ardizzoni, A.; Franceschi, C.; Cossarizza, A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: Implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997, 411, 77–82. [Google Scholar] [CrossRef]
- Levine, E.; Hwa, T. Stochastic fluctuations in metabolic pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 9224–9229. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Muller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M.; et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Choi, W.G.; Lee, P.J.; Chung, S.W.; Kim, B.S.; Chung, H.T.; Cho, S.; Kim, J.H.; Kang, B.H.; Kim, H.; et al. The novel resveratrol derivative 3,5-diethoxy-3′,4′-dihydroxy-trans-stilbene induces mitochondrial ROS-mediated ER stress and cell death in human hepatoma cells in vitro. Acta Pharmacol. Sin. 2017, 38, 1486–1500. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.Y.; Nam, S.; Kim, M.J.; Kim, S.J.; Back, S.H.; Yoo, H.J. Butyrate Prevents TGF-β1-Induced Alveolar Myofibroblast Differentiation and Modulates Energy Metabolism. Metabolites 2021, 11, 258. https://doi.org/10.3390/metabo11050258
Lee HY, Nam S, Kim MJ, Kim SJ, Back SH, Yoo HJ. Butyrate Prevents TGF-β1-Induced Alveolar Myofibroblast Differentiation and Modulates Energy Metabolism. Metabolites. 2021; 11(5):258. https://doi.org/10.3390/metabo11050258
Chicago/Turabian StyleLee, Hyo Yeong, Somi Nam, Mi Jeong Kim, Su Jung Kim, Sung Hoon Back, and Hyun Ju Yoo. 2021. "Butyrate Prevents TGF-β1-Induced Alveolar Myofibroblast Differentiation and Modulates Energy Metabolism" Metabolites 11, no. 5: 258. https://doi.org/10.3390/metabo11050258
APA StyleLee, H. Y., Nam, S., Kim, M. J., Kim, S. J., Back, S. H., & Yoo, H. J. (2021). Butyrate Prevents TGF-β1-Induced Alveolar Myofibroblast Differentiation and Modulates Energy Metabolism. Metabolites, 11(5), 258. https://doi.org/10.3390/metabo11050258