
Improved One-Class Modeling of High-Dimensional Metabolomics Data via Eigenvalue-Shrinkage

 , , ,
, , ,
Abstract
1. Introduction
2. Results
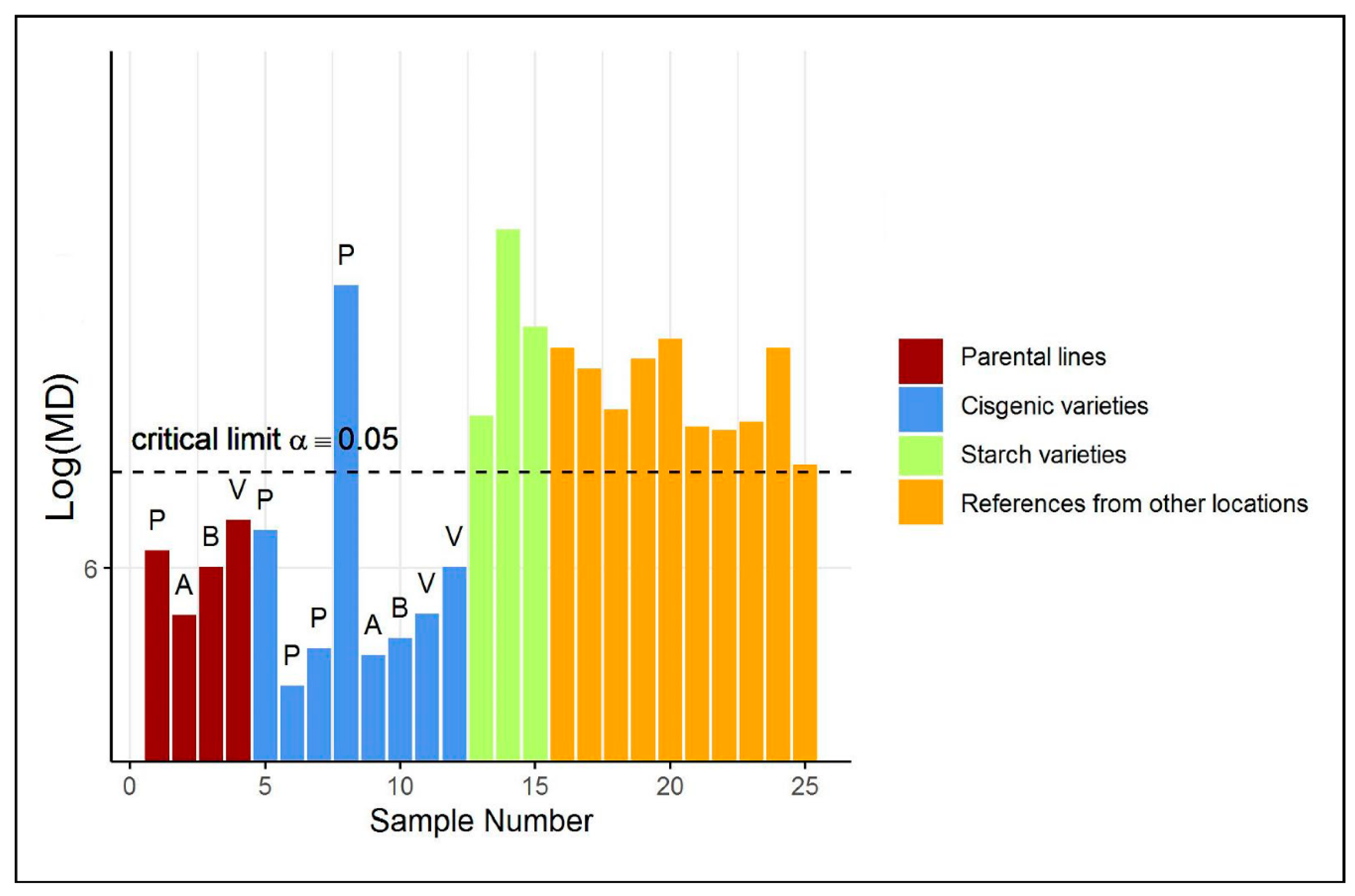
2.1. Case Study 1: Compositional Safety Assessment of Tubers from Commercial and Cisgenic Potato Varieties
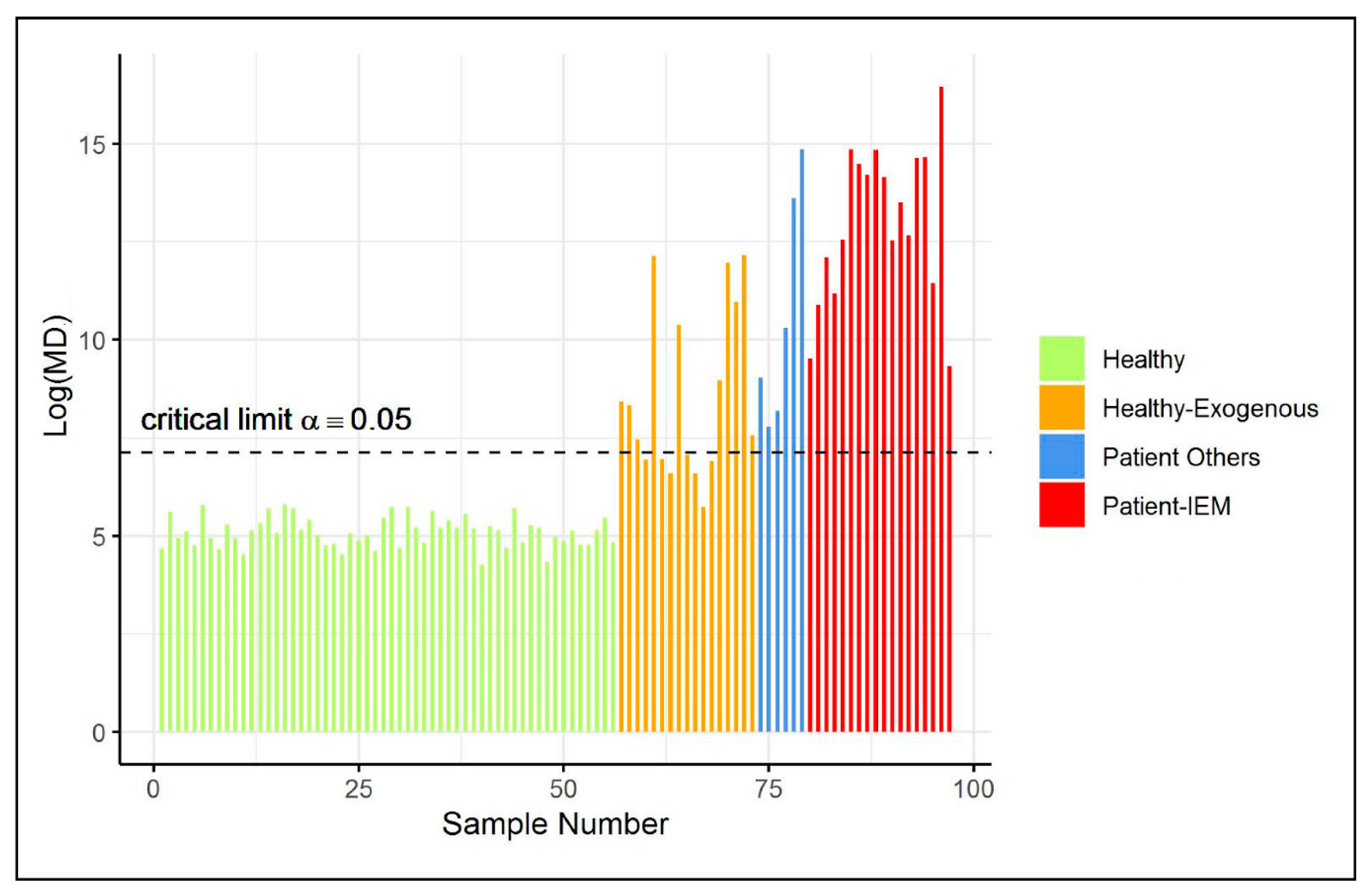
2.2. Case Study 2: Diagnosis of Inborn Error of Metabolism
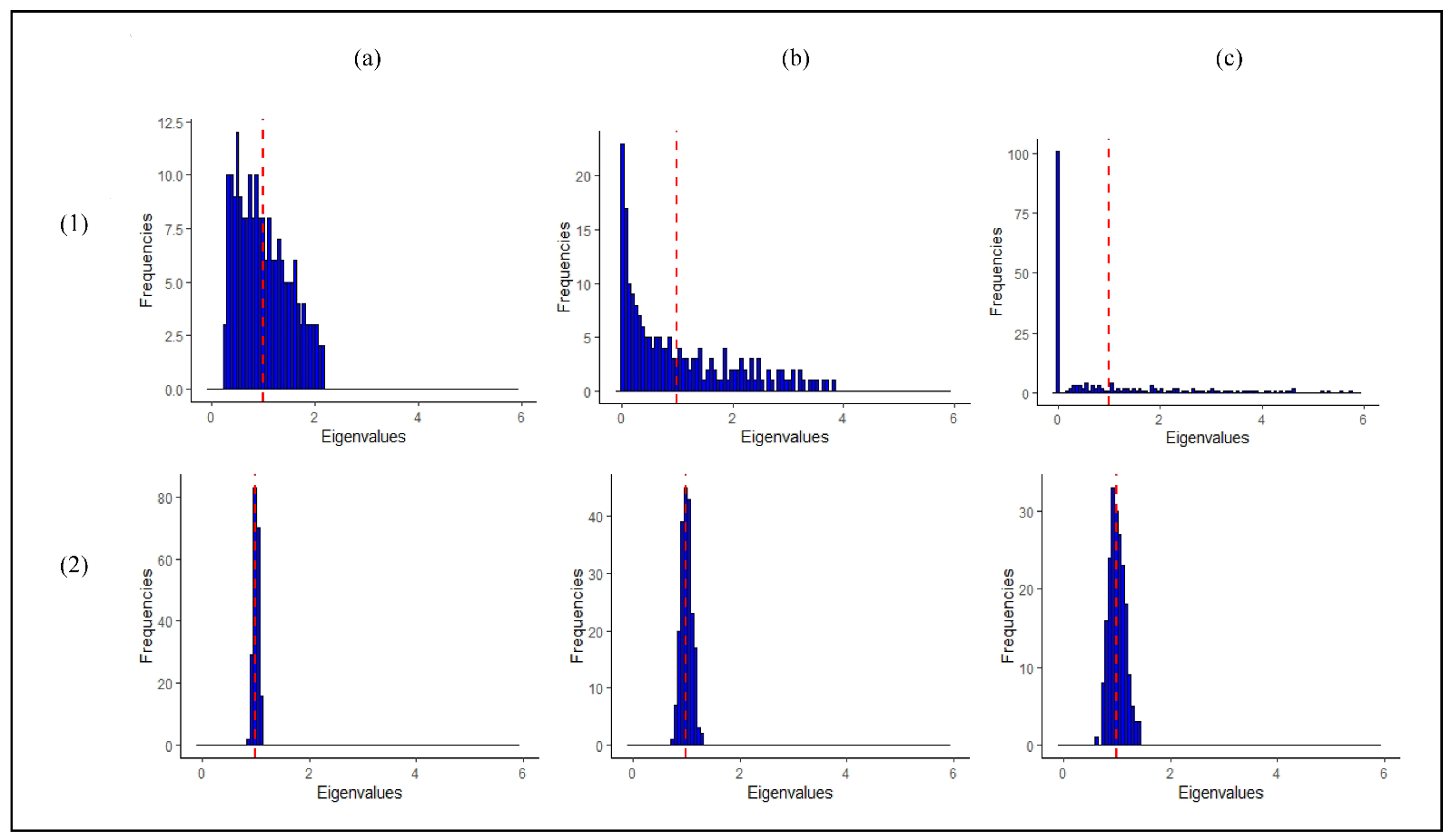
2.3. Synthetic Data Experiments
2.3.1. Assessment of the Computational Speed
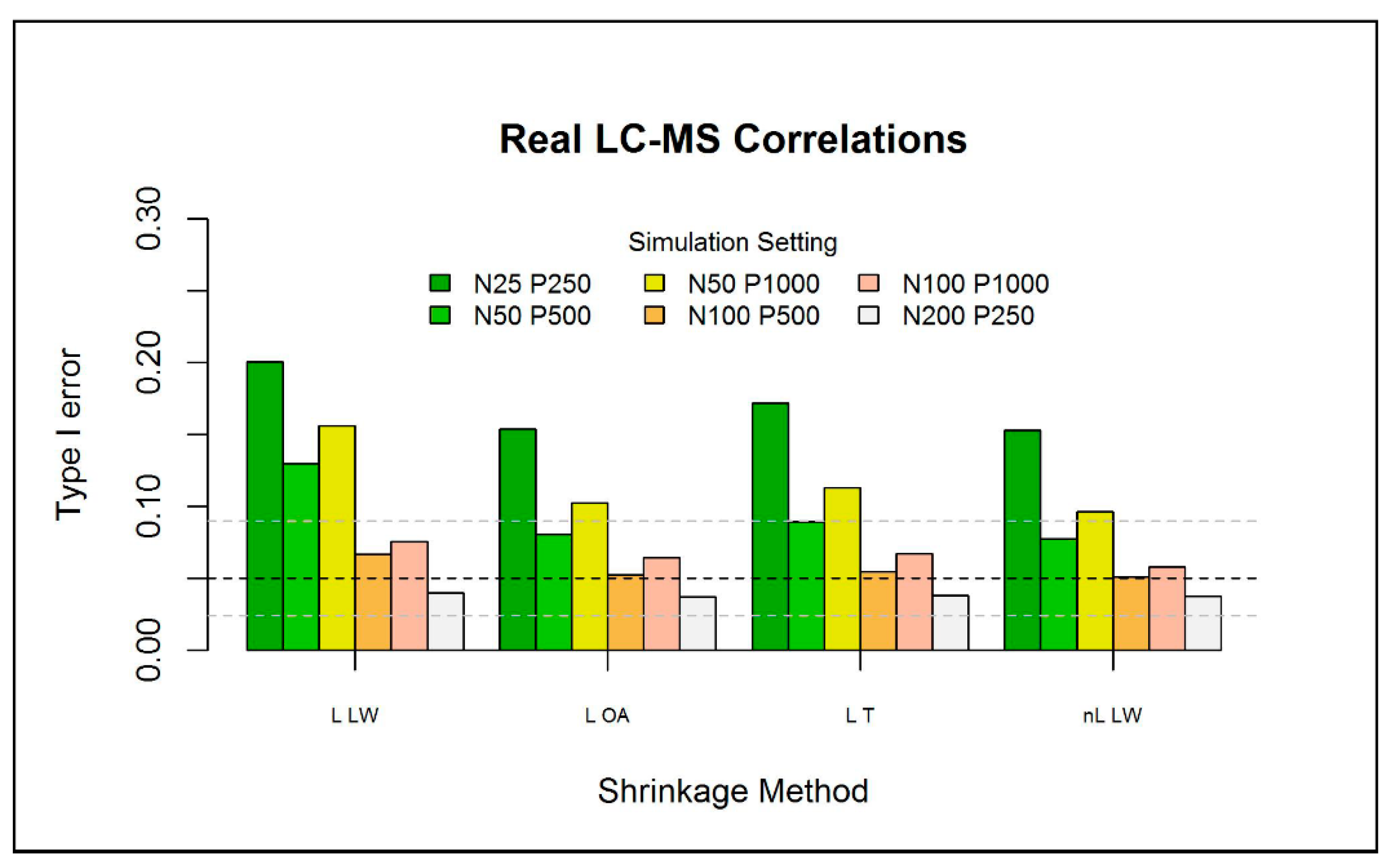
2.3.2. Type I Error of the Eigenvalue-Shrinkage Approaches
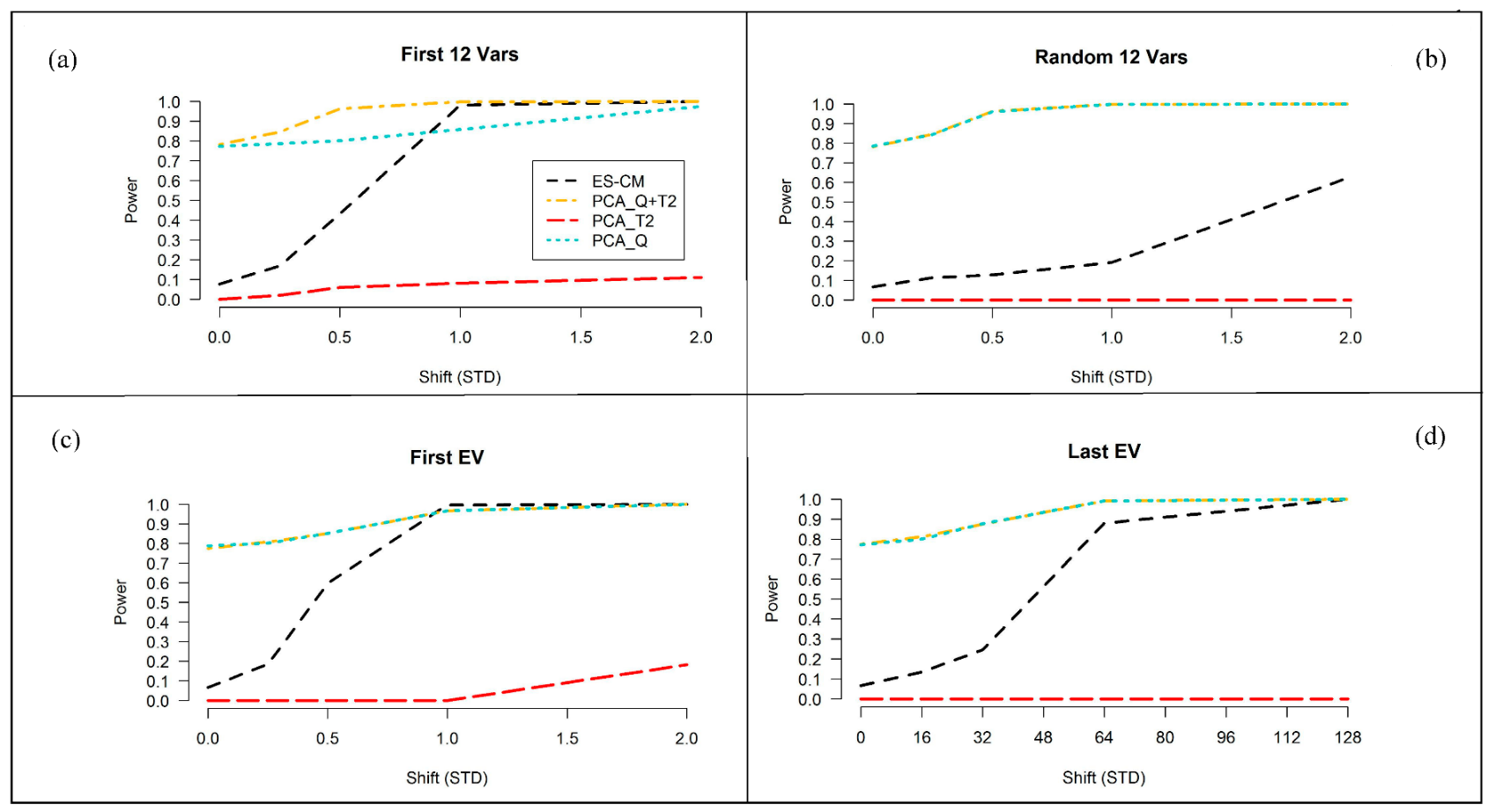
2.3.3. Improved Power of ES-CM over the Standard PCA-Based Decision Criteria
3. Discussion
4. Materials and Methods
4.1. Case Study 1: Compositional Safety Assessment of Tubers from Commercial and Cisgenic Potato Varieties
4.2. Case Study 2: Diagnosis of Inborn Error of Metabolism
4.3. Simulation Set Up
4.4. One-Class Modelling Based on
4.5. (Shrinkage) Estimators of the Covariance Matrix
4.6. One-Class Modelling Based on Shrunken
- (1)
- For each reference observation from to :
- Leave the observation out and compute the (shrinkage) covariance estimate of the set of references using “L T”.
- Compute the for the left-out observation,
- (2)
- Calculate the critical limit for ES-CM using the quantile of the scaled distribution for a predetermined significance level (i.e., 5%), given the scale factor and the degrees of freedom with and being the (sample) mean and standard deviation of the (cross-validated) values.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, S.S.; Madden, M.G. One-class classification: Taxonomy of study and review of techniques. Knowl. Eng. Rev. 2014, 29, 345–374. [Google Scholar] [CrossRef]
- Wallace, E.D.; Todd, D.A.; Harnly, J.M.; Cech, N.B.; Kellogg, J.J. Identification of adulteration in botanical samples with untargeted metabolomics. Anal. Bioanal. Chem. 2020, 412, 4273–4286. [Google Scholar] [CrossRef]
- Engel, J.; Blanchet, L.; Engelke, U.F.H.; Wevers, R.A.; Buydens, L.M.C. Towards the Disease Biomarker in an Individual Patient Using Statistical Health Monitoring. PLoS ONE 2014, 9, e92452. [Google Scholar] [CrossRef]
- Kok, E.; Van Dijk, J.; Voorhuijzen, M.; Staats, M.; Slot, M.; Lommen, A.; Venema, D.; Pla, M.; Corujo, M.; Barros, E.; et al. Omics analyses of potato plant materials using an improved one-class classification tool to identify aberrant compositional profiles in risk assessment procedures. Food Chem. 2019, 292, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Koeman, M.; Engel, J.; Jansen, J.; Buydens, L. Critical comparison of methods for fault diagnosis in metabolomics data. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Del Carratore, F.; Lussu, M.; Kowalik, M.A.; Perra, A.; Griffin, J.L.; Atzori, L.; Grosso, M. Statistical Health Monitoring Applied to a Metabolomic Study of Experimental Hepatocarcinogenesis: An Alternative Approach to Supervised Methods for the Identification of False Positives. Anal. Chem. 2016, 88, 7921–7929. [Google Scholar] [CrossRef] [PubMed]
- Van der Voet, H.; Goedhart, P.W.; Schmidt, K. Equivalence testing using existing reference data: An example with genetically modified and conventional crops in animal feeding studies. Food Chem. Toxicol. 2017, 109, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Ullah, I.; Pawley, M.D.; Smith, A.N.; Jones, B. Improving the detection of unusual observations in high-dimensional settings. Aust. N. Z. J. Stat. 2017, 59, 449–462. [Google Scholar] [CrossRef]
- Stanfill, B.A.; Nakayasu, E.S.; Bramer, L.M.; Thompson, A.M.; Ansong, C.K.; Clauss, T.R.; Gritsenko, M.A.; Monroe, M.E.; Moore, R.J.; Orton, D.J.; et al. Quality Control Analysis in Real-time (QC-ART): A Tool for Real-time Quality Control Assessment of Mass Spectrometry-based Proteomics Data. Mol. Cell. Proteom. 2018, 17, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- De Maesschalck, R.; Jouan-Rimbaud, D.; Massart, D. The Mahalanobis distance. Chemom. Intell. Lab. Syst. 2000, 50, 1–18. [Google Scholar] [CrossRef]
- Engel, J.; Buydens, L.; Blanchet, L. An overview of large-dimensional covariance and precision matrix estimators with applications in chemometrics. J. Chemom. 2017, 31, e2880. [Google Scholar] [CrossRef]
- De Maesschalck, R.; Candolfi, A.; Massart, D.; Heuerding, S. Decision criteria for soft independent modelling of class analogy applied to near infrared data. Chemom. Intell. Lab. Syst. 1999, 47, 65–77. [Google Scholar] [CrossRef]
- Ramaker, H.-J.; Van Sprang, E.N.; Westerhuis, J.A.; Smilde, A.K. The effect of the size of the training set and number of principal components on the false alarm rate in statistical process monitoring. Chemom. Intell. Lab. Syst. 2004, 73, 181–187. [Google Scholar] [CrossRef]
- Ledoit, O.; Wolf, M. A well-conditioned estimator for large-dimensional covariance matrices. J. Multivar. Anal. 2004, 88, 365–411. [Google Scholar] [CrossRef]
- Touloumis, A. Nonparametric Stein-type shrinkage covariance matrix estimators in high-dimensional settings. Comput. Stat. Data Anal. 2015, 83, 251–261. [Google Scholar] [CrossRef]
- Chen, Y.; Wiesel, A.; Eldar, Y.C.; Hero, A.O. Shrinkage Algorithms for MMSE Covariance Estimation. IEEE Trans. Signal Process. 2010, 58, 5016–5029. [Google Scholar] [CrossRef]
- Van Wieringen, W.N.; Peeters, C.F. Ridge estimation of inverse covariance matrices from high-dimensional data. Comput. Stat. Data Anal. 2016, 103, 284–303. [Google Scholar] [CrossRef]
- Ledoit, O.; Wolf, M. Nonlinear shrinkage estimation of large-dimensional covariance matrices. Ann. Stat. 2012, 40, 1024–1060. [Google Scholar] [CrossRef]
- Herman, R.A.; Price, W.D. Unintended Compositional Changes in Genetically Modified (GM) Crops: 20 Years of Research. J. Agric. Food Chem. 2013, 61, 11695–11701. [Google Scholar] [CrossRef]
- Van Dijk, J.P.; De Mello, C.S.; Voorhuijzen, M.M.; Hutten, R.C.; Arisi, A.C.M.; Jansen, J.J.; Buydens, L.M.; Van Der Voet, H.; Kok, E.J. Safety assessment of plant varieties using transcriptomics profiling and a one-class classifier. Regul. Toxicol. Pharmacol. 2014, 70, 297–303. [Google Scholar] [CrossRef]
- Jo, K.-R.; Kim, C.-J.; Kim, S.-J.; Kim, T.-Y.; Bergervoet, M.; Jongsma, M.A.; Visser, R.G.F.; Jacobsen, E.; Vossen, J.H. Development of late blight resistant potatoes by cisgene stacking. BMC Biotechnol. 2014, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Engel, J. Chemometrics on Its Way towards Personalized Health Care. Ph.D. Thesis, Radboud University Nijmegen, Nijmegen, The Netherlands, 2016. [Google Scholar]
- Kennard, R.W.; Stone, L.A. Computer Aided Design of Experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Ledoit, O.; Wolf, M. Analytical Nonlinear Shrinkage of Large-Dimensional Covariance Matrices Analytical Nonlinear Shrinkage of Large-Dimensional Covariance Matrices. 2018. Available online: http://www.econ.uzh.ch/static/wp/econwp264.pdf (accessed on 12 April 2021).
- Warton, D.I. Penalized Normal Likelihood and Ridge Regularization of Correlation and Covariance Matrices. J. Am. Stat. Assoc. 2008, 103, 340–349. [Google Scholar] [CrossRef]
- Kucheryavskiy, S. mdatools—R package for chemometrics. Chemom. Intell. Lab. Syst. 2020, 198, 103937. [Google Scholar] [CrossRef]
- Pomerantsev, A.L. Acceptance areas for multivariate classification derived by projection methods. J. Chemom. 2008, 22, 601–609. [Google Scholar] [CrossRef]
- Qin, S.J. Statistical process monitoring: Basics and beyond. J. Chemom. 2003, 17, 480–502. [Google Scholar] [CrossRef]
- Zimek, A.; Schubert, E.; Kriegel, H.-P. A survey on unsupervised outlier detection in high-dimensional numerical data. Stat. Anal. Data Min. ASA Data Sci. J. 2012, 5, 363–387. [Google Scholar] [CrossRef]
- Kuismin, M.O.; Kemppainen, J.T.; Sillanpää, M.J. Precision Matrix Estimation with ROPE. J. Comput. Graph. Stat. 2017, 26, 682–694. [Google Scholar] [CrossRef][Green Version]
- Kuismin, M.O.; Sillanpää, M.J. Estimation of covariance and precision matrix, network structure, and a view toward systems biology. Wiley Interdiscip. Rev. Comput. Stat. 2017, 9, e1415. [Google Scholar] [CrossRef]
- Witten, D.M.; Friedman, J.H.; Simon, N. New Insights and Faster Computations for the Graphical Lasso. J. Comput. Graph. Stat. 2011, 20, 892–900. [Google Scholar] [CrossRef]
- Hubert, M.; Debruyne, M.; Rousseeuw, P.J. Minimum covariance determinant and extensions. Wiley Interdiscip. Rev. Comput. Stat. 2017, 10, 1–11. [Google Scholar] [CrossRef]
- Rousseeuw, P.J.; Hubert, M. Anomaly detection by robust statistics. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2017, 8, 1236. [Google Scholar] [CrossRef]
- Cabana, E.; Lillo, R.E.; Laniado, H. Multivariate outlier detection based on a robust Mahalanobis distance with shrinkage estimators. Stat. Pap. 2019, 1–27. [Google Scholar] [CrossRef]
- Gnanadesikan, R.; Kettenring, J.R. Robust Estimates, Residuals, and Outlier Detection with Multiresponse Data. Biometrics 1972, 28, 81. [Google Scholar] [CrossRef]
- Öllerer, V.; Croux, C. Robust High-Dimensional Precision Matrix Estimation. In Modern Nonparametric, Robust and Multivariate Methods; Springer: Cham, Switzerland, 2015; pp. 325–350. [Google Scholar] [CrossRef]
- Agostinelli, C.; Leung, A.; Yohai, V.J.; Zamar, R.H. Robust estimation of multivariate location and scatter in the presence of cellwise and casewise contamination. TEST 2015, 24, 441–461. [Google Scholar] [CrossRef]
- Tarr, G.; Müller, S.; Weber, N. Robust estimation of precision matrices under cellwise contamination. Comput. Stat. Data Anal. 2016, 93, 404–420. [Google Scholar] [CrossRef]
- Loh, P.-L.; Tan, X.L. High-dimensional robust precision matrix estimation: Cellwise corruption under ϵ-contamination. Electron. J. Stat. 2018, 12, 1429–1467. [Google Scholar] [CrossRef]
- Avagyan, V.; Mei, X. Precision matrix estimation under data contamination with an application to minimum variance portfolio selection. Commun. Stat. Simul. Comput. 2019, 1–20. [Google Scholar] [CrossRef]
- De Vos, R.C.H.; Moco, S.; Lommen, A.; Keurentjes, J.J.B.; Bino, R.J.; Hall, R.D. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2007, 2, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Lommen, A. MetAlign: Interface-Driven, Versatile Metabolomics Tool for Hyphenated Full-Scan Mass Spectrometry Data Preprocessing. Anal. Chem. 2009, 81, 3079–3086. [Google Scholar] [CrossRef]
- Tikunov, Y.M.; Laptenok, S.; Hall, R.D.; Bovy, A.; De Vos, R.C.H. MSClust: A tool for unsupervised mass spectra extraction of chromatography-mass spectrometry ion-wise aligned data. Metabolomics 2011, 8, 714–718. [Google Scholar] [CrossRef]
- Stekhoven, D.J.; Bühlmann, P. MissForest—Non-parametric missing value imputation for mixed-type data. Bioinformatics 2011, 28, 112–118. [Google Scholar] [CrossRef]
- Dunn, W.B.; Lin, W.; Broadhurst, D.; Begley, P.; Brown, M.; Zelena, E.; Vaughan, A.A.; Halsall, A.; Harding, N.; Knowles, J.D.; et al. Molecular phenotyping of a UK population: Defining the human serum metabolome. Metabolomics 2015, 11, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Camp, T. The incredible shrinking pipeline. ACM Sigcse Bull. 2002, 34, 129–134. [Google Scholar] [CrossRef]
- Fisher, T.J.; Sun, X. Improved Stein-type shrinkage estimators for the high-dimensional multivariate normal covariance matrix. Comput. Stat. Data Anal. 2011, 55, 1909–1918. [Google Scholar] [CrossRef]
- Ledoit, O.; Wolf, M. Improved estimation of the covariance matrix of stock returns with an application to portfolio selection. J. Empir. Financ. 2003, 10, 603–621. [Google Scholar] [CrossRef]
- Schäfer, J.; Strimmer, K. A Shrinkage Approach to Large-Scale Covariance Matrix Estimation and Implications for Functional Genomics. Stat. Appl. Genet. Mol. Biol. 2005, 4, 32. [Google Scholar] [CrossRef]
- Theiler, J. The incredible shrinking covariance estimator. In Proceedings of the Automatic Target Recognition XXII, Baltimore, MD, USA, 23–24 April 2012; Volume 8391. [Google Scholar] [CrossRef]
- Ledoit, O.; Wolf, M. Spectrum estimation: A unified framework for covariance matrix estimation and PCA in large dimensions. J. Multivar. Anal. 2015, 139, 360–384. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L LW | L OA | L T | nL LW | nL vW | |
|---|---|---|---|---|---|
| Time [s] | 1.19 | 1.00 | 0.79 | 189.00 | 490.00 |
| Type I error | 0.14 | 0.097 | 0.087 | 0.079 | 0.081 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brini, A.; Avagyan, V.; de Vos, R.C.H.; Vossen, J.H.; van den Heuvel, E.R.; Engel, J. Improved One-Class Modeling of High-Dimensional Metabolomics Data via Eigenvalue-Shrinkage. Metabolites 2021, 11, 237. https://doi.org/10.3390/metabo11040237
Brini A, Avagyan V, de Vos RCH, Vossen JH, van den Heuvel ER, Engel J. Improved One-Class Modeling of High-Dimensional Metabolomics Data via Eigenvalue-Shrinkage. Metabolites. 2021; 11(4):237. https://doi.org/10.3390/metabo11040237
Chicago/Turabian StyleBrini, Alberto, Vahe Avagyan, Ric C. H. de Vos, Jack H. Vossen, Edwin R. van den Heuvel, and Jasper Engel. 2021. "Improved One-Class Modeling of High-Dimensional Metabolomics Data via Eigenvalue-Shrinkage" Metabolites 11, no. 4: 237. https://doi.org/10.3390/metabo11040237
APA StyleBrini, A., Avagyan, V., de Vos, R. C. H., Vossen, J. H., van den Heuvel, E. R., & Engel, J. (2021). Improved One-Class Modeling of High-Dimensional Metabolomics Data via Eigenvalue-Shrinkage. Metabolites, 11(4), 237. https://doi.org/10.3390/metabo11040237