
CRISPR Gene Editing in Lipid Disorders and Atherosclerosis: Mechanisms and Opportunities


 ,
,  ,
, 


Abstract
:1. Introduction
2. CRISPR Gene Editing
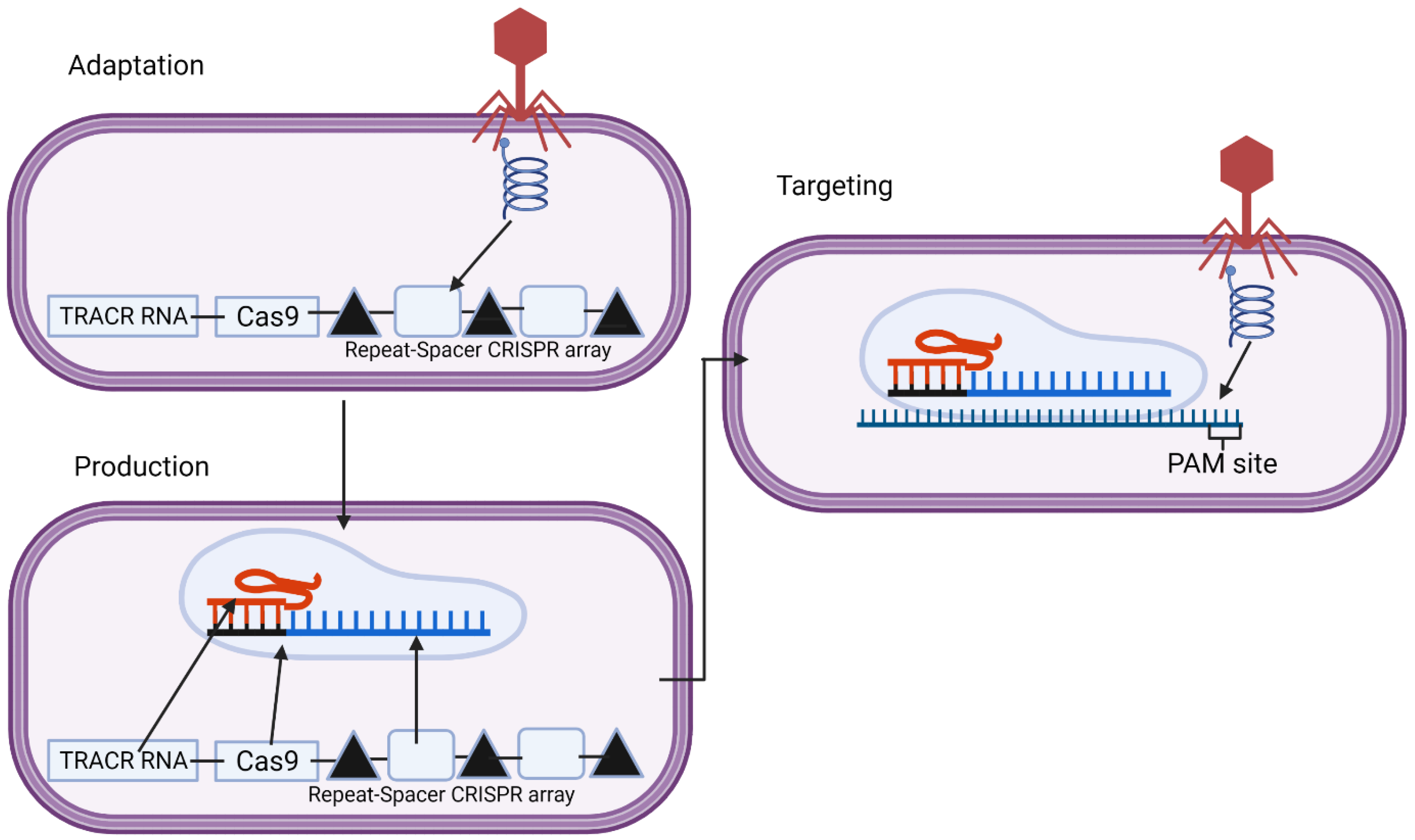
2.1. CRISPR in Bacterial Adaptive Immunity
2.2. CRISPR as a Potential Technology for Gene Therapy
2.3. Problems and Issues with CRISPR in Genetic Engineering
3. CRISPR Gene Editing of PCSK9
3.1. Specific Mechanisms
3.2. Experimental Evidence of Proof-of-Concept
3.3. Other Potential Uses of CRISPR in Atherosclerosis and Cardiovascular Disease
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef] [Green Version]
- Cybulska, B.; Kłosiewicz-Latoszek, L.; Penson, P.E.; Nabavi, S.M.; Lavie, C.J.; Banach, M. How much should LDL cholesterol be lowered in secondary prevention? Clinical efficacy and safety in the era of PCSK9 inhibitors. Prog. Cardiovasc. Dis. 2020, 67, 65–74. [Google Scholar] [CrossRef]
- Penson, P.E.; Pirro, M.; Banach, M. LDL-C: Lower is better for longer—even at low risk. BMC Med. 2020, 18, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Penson, P.E.; Vrablik, M.; Bunc, M.; Dyrbus, K.; Fedacko, J.; Gaita, D.; Gierlotka, M.; Jarai, Z.; Magda, S.L.; et al. Optimal use of lipid-lowering therapy after acute coronary syndromes: A Position Paper endorsed by the International Lipid Expert Panel (ILEP). Pharmacol. Res. 2021, 166, 105499. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Penson, P.E. Lipid-lowering therapies: Better together. Atherosclerosis 2021, 320, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Serban, M.-C.; Colantonio, L.; Manthripragada, A.D.; Monda, K.L.; Bittner, V.A.; Banach, M.; Chen, L.; Huang, L.; Dent, R.; Kent, S.T.; et al. Statin Intolerance and Risk of Coronary Heart Events and All-Cause Mortality Following Myocardial Infarction. J. Am. Coll. Cardiol. 2017, 69, 1386–1395. [Google Scholar] [CrossRef]
- Bytyçi, I.; Penson, P.E.; Mikhailidis, D.P.; Wong, N.D.; Hernandez, A.V.; Sahebkar, A.; Thomson, P.; Mazidi, M.; Rysz, J.; Pella, D.; et al. The prevalence of statin intolerance worldwide: A systematic review and meta-analysis with 4,143,517 patients. In Proceedings of the European Society of Cardiology Congress, Online, 27–30 August 2021. [Google Scholar]
- Banach, M.; Patti, A.M.; Giglio, R.V.; Cicero, A.F.; Atanasov, A.; Bajraktari, G.; Bruckert, E.; Descamps, O.; Djuric, D.M.; Ezhov, M.; et al. The role of nutraceuticals in statin intolerant patients. J. Am. Coll. Cardiol. 2018, 72, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Rizzo, M.; Toth, P.P.; Farnier, M.; Davidson, M.H.; Al-Rasadi, K.; Aronow, W.S.; Athyros, V.; Djuric, D.M.; Ezhov, M.V.; et al. Statin intolerance—An attempt at a unified definition. Position paper from an International Lipid Expert Panel. Expert Opin. Drug Saf. 2015, 14, 935–955. [Google Scholar] [CrossRef] [PubMed]
- Penson, P.; Toth, P.; Mikhailidis, D.; Ezhov, M.; Fras, Z.; Mitchenko, O.; Pella, D.; Sahebkar, A.; Rysz, J.; Reiner, Z.; et al. P705Step by step diagnosis and management of statin intolerance: Position paper from an international lipid expert panel. Eur. Hear. J. 2019, 40, 312. [Google Scholar] [CrossRef]
- Penson, P.E.; Mancini, G.B.J.; Toth, P.P.; Martin, S.S.; Watts, G.F.; Sahebkar, A.; Mikhailidis, D.P.; Banach, M.; on behalf of Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) Group & International Lipid Expert Panel (ILEP). Introducing the ‘drucebo’ effect in statin therapy: A systematic review of studies comparing reported rates of statin-associated muscle symptoms, under blinded and open-label conditions. J. Cachex-Sarcopenia Muscle 2018, 9, 1023–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; Penson, P.E. Drucebo effect—The challenge we should all definitely face! Arch. Med Sci. 2021, 17, 542–543. [Google Scholar] [CrossRef] [PubMed]
- Penson, P.; Banach, M. Nocebo/drucebo effect in statin-intolerant patients: An attempt at recommendations. Eur. Hear. J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bytyci, I.; Bajraktari, G.; Sahebkar, A.; Penson, P.; Rysz, J.; Banach, M. The prevalence of statin intolerance worldwide: A systematic review and meta-analysis with 4,143,517 patients. Eur. Heart J. 2021, 42, 2943. [Google Scholar] [CrossRef]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Banach, M.; Penson, P. What have we learned about lipids and cardiovascular risk from PCSK9 inhibitor outcome trials: ODYSSEY and FOURIER? Cardiovasc. Res. 2019, 115, e26–e31. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.; et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. New Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyrbuś, K.; Gąsior, M.; Penson, P.; Ray, K.K.; Banach, M. Inclisiran—New hope in the management of lipid disorders? J. Clin. Lipidol. 2020, 14, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.S.; Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Friedman, A.; et al. Pooled patient-level analysis of inclisiran trials in patients with familial hypercholesterolemia or atherosclerosis. J. Am. Coll. Cardiol. 2021, 77, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Henney, N.C.; Banach, M.; Penson, P.E. RNA Silencing in the management of dyslipidemias. Curr. Atheroscler. Rep. 2021, 23, 69. [Google Scholar] [CrossRef]
- Momtazi, A.A.; Jaafari, M.R.; Badiee, A.; Banach, M.; Sahebkar, A. Therapeutic effect of nanoliposomal PCSK9 vaccine in a mouse model of atherosclerosis. BMC Med. 2019, 17, 223. [Google Scholar] [CrossRef]
- Sahebkar, A.; Momtazi-Borojeni, A.A.; Banach, M. PCSK9 vaccine: So near, yet so far! Eur. Hear. J. 2021, 42, 4007–4010. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Jaafari, M.R.; Afshar, M.; Banach, M.; Sahebkar, A. PCSK9 immunization using nanoliposomes: Preventive efficacy against hypercholesterolemia and atherosclerosis. Arch. Med Sci. 2021, 17, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Rizzo, M.; Obradovic, M.; Montalto, G.; Rysz, J.; Mikhailidis, D.P.; Isenovic, E.R. PCSK9 Inhibition—A novel mechanism to treat lipid disorders? Curr. Pharm. Des. 2013, 19, 3869–3877. [Google Scholar] [CrossRef]
- Banerjee, Y.; Stoian, A.P.; Cicero, A.F.G.; Fogacci, F.; Nikolic, D.; Sachinidis, A.; Rizvi, A.A.; Janez, A.; Rizzo, M. Inclisiran: A small interfering RNA strategy targeting PCSK9 to treat hypercholesterolemia. Expert Opin. Drug Saf. 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, Y.; Santos, R.D.; Al-Rasadi, K.; Rizzo, M. Targeting PCSK9 for therapeutic gains: Have we addressed all the concerns? Atherosclerosis 2016, 248, 62–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abifadel, M.; Rabès, J.-P.; Devillers, M.; Munnich, A.; Erlich, D.; Junien, C.; Varret, M.; Boileau, C. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum. Mutat. 2009, 30, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.H.; Peloso, G.M.; Auer, P.L.; Crosslin, D.R.; Stitziel, N.; Lange, A.L.; Lu, Y.; Tang, Z.Z.; Zhang, H. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med. 2014, 371, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.P.; Pazdernik, N.J.; McGehee, M.R. Genome defense. In Molecular Biology, 3rd ed.; Clark, D.P., Pazdernik, N.J., McGehee, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 622–653. [Google Scholar]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The biology of CRISPR-Cas: Backward and forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Dhingra, Y.; Sashital, D.G. The Cas4-Cas1-Cas2 complex mediates precise prespacer processing during CRISPR adaptation. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Shiimori, M.; Garrett, S.C.; Graveley, B.R.; Terns, M.P. Cas4 nucleases define the PAM, length, and orientation of DNA fragments integrated at CRISPR Loci. Mol. Cell 2018, 70, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Mir, A.; Edraki, A.; Lee, J.; Sontheimer, E.J. Type II-C CRISPR-Cas9 biology, mechanism, and application. ACS Chem. Biol. 2017, 13, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Koonin, E.V. Annotation and classification of crispr-cas systems. Methods Mol. Biol. 2015, 1311, 47–75. [Google Scholar] [CrossRef] [Green Version]
- Gleditzsch, D.; Pausch, P.; Esparza, H.M.; Özcan, A.; Guo, X.; Bange, G.; Randau, L. PAM identification by CRISPR-Cas effector complexes: Diversified mechanisms and structures. RNA Biol. 2018, 16, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Rath, D.; Amlinger, L.; Rath, A.; Lundgren, M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015, 117, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Karginov, F.V.; Hannon, G.J. The CRISPR system: Small RNA-guided defense in bacteria and archaea. Mol. Cell 2010, 37, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainy, J.; Garrett, S.; Graveley, B.R.; Terns, M.P. CRISPR repeat sequences and relative spacing specify DNA integration by Pyrococcus furiosus Cas1 and Cas2. Nucleic Acids Res. 2019, 47, 7518–7531. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Chang, J.T.; Fineran, P.C. Function and regulation of clustered regularly interspaced short palindromic repeats (CRISPR) / CRISPR associated (Cas) systems. Viruses 2012, 4, 2291–2311. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Rutkauskas, M.; Krivoy, A.; Szczelkun, M.; Rouillon, C.; Seidel, R. Single-Molecule Insight Into Target Recognition by CRISPR–Cas Complexes. Methods Enzymol. 2017, 582, 239–273. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.; Zhang, Y.; Yuan, G.; De, K.; Chen, J.-G.; Muchero, W.; Tuskan, G.A.; Qi, Y.; Yang, X. Construct design for CRISPR/Cas-based genome editing in plants. Trends Plant Sci. 2021, 26, 1133–1152. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Zhao, X.; Zhang, Q.; Li, W.; Zu, Y. Comparison of various nuclear localization signal-fused Cas9 proteins and Cas9 mRNA for genome editing in zebrafish. G3 Genes Genomes Genet. 2018, 8, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [Green Version]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9, 691. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Hou, Y.; Zhang, P.; Zhang, Z.; Xu, Y.; Zhang, L.; Niu, L.; Yang, Y.; Liang, D.; Yi, F.; et al. Profiling single-guide RNA specificity reveals a mismatch sensitive core sequence. Sci. Rep. 2017, 7, 40638. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.Q.; Joung, J.K. Defining and improving the genome-wide specificities of CRISPR–Cas9 nucleases. Nat. Rev. Genet. 2016, 17, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E.; Ledford, H. Pioneers of revolutionary CRISPR gene editing win chemistry Nobel. Nature 2020, 586, 346–347. [Google Scholar] [CrossRef]
- Dai, W.-J.; Zhu, L.-Y.; Yan, Z.-Y.; Xu, Y.; Wang, Q.-L.; Lu, X.-J. CRISPR-Cas9 for in vivo gene therapy: Promise and hurdles. Mol. Ther. Nucleic Acids 2016, 5, e349. [Google Scholar] [CrossRef] [Green Version]
- Behr, M.; Zhou, J.; Xu, B.; Zhang, H. In vivo delivery of CRISPR-Cas9 therapeutics: Progress and challenges. Acta Pharm. Sin. B 2021, 11, 2150–2171. [Google Scholar] [CrossRef]
- Wilbie, D.; Walther, J.; Mastrobattista, E. Delivery aspects of CRISPR/Cas for in vivo genome editing. Accounts Chem. Res. 2019, 52, 1555–1564. [Google Scholar] [CrossRef] [Green Version]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.L.; Ruan, M.Z.C.; Mahajan, V.B.; Tsang, S.H. Viral Delivery Systems for CRISPR. Viruses 2019, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledford, H. CRISPR treatment inserted directly into the body for first time. Nature 2020, 579, 185. [Google Scholar] [CrossRef] [Green Version]
- Janik, E.; Niemcewicz, M.; Ceremuga, M.; Krzowski, L.; Saluk-Bijak, J.; Bijak, M. Various Aspects of a Gene Editing System—CRISPR–Cas9. Int. J. Mol. Sci. 2020, 21, 9604. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Zhou, Y.; Sun, Q.; Tang, B. Novel epigenetic techniques provided by the CRISPR/Cas9 System. Stem Cells Int. 2018, 2018, 7834175. [Google Scholar] [CrossRef]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR gene therapy: Applications, limitations, and implications for the future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef]
- Wang, L.; Breton, C.; Warzecha, C.C.; Bell, P.; Yan, H.; He, Z.; White, J.; Zhu, Y.; Li, M.; Buza, E.L.; et al. Long-term stable reduction of low-density lipoprotein in nonhuman primates following in vivo genome editing of PCSK9. Mol. Ther. 2021, 29, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Xu, Y.; Dong, Z.; Zhou, Z.; Cong, N.; Gao, M.; Huang, W.; Wang, Y.; Liu, G.; Xian, X. Inactivation of ApoC3 by CRISPR/Cas9 protects against atherosclerosis in hamsters. Circ. Res. 2020, 127, 1456–1458. [Google Scholar] [CrossRef]
- Banach, M.; Penson, P.E. Statins and LDL-C in secondary prevention—So much progress, so far to go. JAMA Netw. Open 2020, 3, e2025675. [Google Scholar] [CrossRef]
- Cybulska, B.; Kłosiewicz-Latoszek, L.; Penson, P.E.; Banach, M. What do we know about the role of lipoprotein(a) in atherogenesis 57 years after its discovery? Prog. Cardiovasc. Dis. 2020, 63, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Dyrbuś, K.; Gąsior, M.; Desperak, P.; Trzeciak, P.; Nowak, J.; Penson, P.E.; Osadnik, T.; Banach, M. Risk-factors associated with extremely high cardiovascular risk of mid- and long-term mortality following myocardial infarction: Analysis of the hyperlipidaemia therapy in tertiary cardiological center (TERCET) registry. Atherosclerosis 2021, 333, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Dyrbuś, K.; Gąsior, M.; E Penson, P.; Banach, M. Extreme cardiovascular risk—do we need a new risk category? Eur. Hear. J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Waghulde, H.; Cheng, X.; Galla, S.; Mell, B.; Cai, J.; Pruett-Miller, S.M.; Vazquez, G.; Patterson, A.; Kumar, M.V.; Joe, B. Attenuation of microbiotal dysbiosis and hypertension in a CRISPR/Cas9 gene ablation rat model of GPER1. Hypertension 2018, 72, 1125–1132. [Google Scholar] [CrossRef]
- Limpitikul, W.B.; Dick, I.; Tester, D.J.; Boczek, N.J.; Limphong, P.; Yang, W.; Choi, M.H.; Babich, J.; DiSilvestre, D.; Kanter, R.J.; et al. A precision medicine approach to the rescue of function on malignant calmodulinopathic long-QT syndrome. Circ. Res. 2017, 120, 39–48. [Google Scholar] [CrossRef]
- Xie, C.; Zhang, Y.-P.; Song, L.; Luo, J.; Qi, W.; Hu, J.; Lu, D.; Yang, Z.; Zhang, J.; Xiao, J.; et al. Genome editing with CRISPR/Cas9 in postnatal mice corrects PRKAG2 cardiac syndrome. Cell Res. 2016, 26, 1099–1111. [Google Scholar] [CrossRef] [Green Version]
- Furgurson, M.; Lagor, W.R. CRISPR. Curr. Opin. Lipidol. 2019, 30, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Y.; Xing, R.; Cui, X.; Xiao, Y.; Xie, L.; You, P.; Wang, T.; Zeng, L.; Peng, W.; et al. Hyperlipidemia induces typical atherosclerosis development in Ldlr and Apoe deficient rats. Atherosclerosis 2018, 271, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hua, Z.; Xiao, H.; Cheng, Y.; Xu, K.; Gao, Q.; Xia, Y.; Liu, Y.; Zhang, X.; Zheng, X.; et al. CRISPR/Cas9-mediated ApoE-/- and LDLR-/- double gene knockout in pigs elevates serum LDL-C and TC levels. Oncotarget 2017, 8, 37751–37760. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| CRISPR System | Identification | Formation and Parts | DNA Binding | Action Upon Binding DNA | Ref. |
|---|---|---|---|---|---|
| Natural | A target site in the bacteriophage genome is identified through a Cas1–Cas2 complex which binds the invading viral DNA and takes a region that it then integrates into the CRISPR array, ensuring a PAM sequence adjacent to this site is present before choosing this spacer. Different subtypes of type II systems require other proteins, such as II-A needs Cas9 and Csn2 and II-B needs Cas4. | Upon future bacteriophage infection, the CRISPR array is transcribed, transcribing the spacers from past infections. These then complex with Cas9 and the tracrRNA, forming the CRISPR complex. | The correct spacer for the current infection and Cas9 will then bind the viral DNA at the site. The spacer is specific and complementary to Cas9, recognizing the PAM sequence, as is required for binding. | The Cas9 protein, specifically the RuvC and HnH domains, cleave the double-stranded DNA genome of the bacteriophage, stopping the infection. | [33,36,38] |
| Engineered | A disease is identified through the phenotype shown with the disease before studying the genome to find an appropriate target gene and a target site within this gene with an adjacent PAM sequence which a guide RNA can be designed against for targeting. | The tracrRNA and spacer are merged to form a guide RNA. This is then put in a vector with the Cas9 gene, which is delivered to cells and the guide RNA and Cas9 are expressed, forming the CRISPR complex. | The guide RNA is designed to be complementary to the target DNA, which is causing a disease, so it will bind at the target site with Cas9, which recognizes the adjacent PAM sequence, allowing binding so that Cas9 can then cleave the DNA. | The RuvC and HnH domains of Cas9 cleave double-stranded DNA, which is then repaired either through NHEJ or HDR. HDR can be used with an exogenous template to insert a sequence into the genome and correct mutations. | [38,46,48,50] |
| Transcriptional editors | The aberrant phenotype shown is used to identify the disease before analyzing the genome to find the gene responsible, and a guide RNA is then designed against a site within this gene with an adjacent PAM sequence to change the gene expression through epigenetic modification. | A guide RNA is put in a vector with s-gene coding for the deactivated Cas9 fused to a transcriptional activator or repressor. This is then expressed in the cell, forming the complex. | The guide RNA and Cas9 bind the DNA through Cas9, recognizing the PAM sequence allowing the guide RNA to bind the region it is targeted against. Here Cas9 binds, not for cleavage, as it is deactivated, but for the attached transcriptional control genes to function. | Upon binding the DNA, the transcriptional control gene fused to Cas9 will perform its function by either introducing or removing an epigenetic modification such as a methyl group, which will increase or decrease the gene expression, depending on the modification made. | [38,46,60] |
| Base editors | The phenotype shown is used to identify the disease before analyzing the genome to find the mutation responsible, and a guide RNA is designed against the mutated site. An adjacent PAM sequence is still required for binding, so some mutated sites may not be possible to target if there is no PAM sequence present. These can only correct transition mutations, not transversion mutations or indels. | Base editors contain a guide RNA with a partially inactivated Cas9 (Cas9 nickase) to cleave one strand. They also have a base editor—either a cytosine or adenosine deaminase fused tthis Cas9 nickase. This is all included in a vector which is then expressed in the cell where the different elements will come together to form the complex. | The guide RNA binds as it does in engineered systems with Cas9 nickase recognizing the PAM site and the guide RNA binding to its target region. Cas9 nickase will only nick one strand whilst the deaminase associated with it acts on the other strand. | Either cytosine or adenine is deaminated depending on which deaminase is being used, to go to uracil or guanine, respectively. Cas9 nickase will nick the other strand, which are then repaired to match the changed base. | [38,46,60] |
| Prime editors | Identification is carried out in the same way as for base editors, including ensuring the mutated site has an adjacent PAM sequence, but when the guide RNA sequence is made, a correct sequence to be copied in, in place of the mutated sequence, is included. This method means both transition and transversion mutations can be corrected. Indels can be repaired using this method through the inclusion or removal of a base/bases in the prime editing guide RNA that has/have been inserted or deleted in the mutated genome. | Prime editors contain Cas9 nickase, which will cleave one strand fused to reverse transcriptase and a prime editing guide RNA made up of a normal guide RNA, a reverse transcriptase primer binding site, and a sequence to be copied in. This is all put in a vector which is expressed in cells forming the CRISPR complex. | Cas9 nickase recognizes the PAM sequence with the prime editing guide RNA binding the target sequence. Reverse transcriptase binds with Cas9 as it is fused to it. | Firstly, Cas9 nickase nicks one strand. The prime editing guide RNA used in prime editors is longer, containing a sequence that is copied into the genome by reverse transcriptase to correct a mutation, with the original mutated strand replaced by this new edited strand. The other strand is then cleaved and repaired to match the edited strand. | [38,46,60,62] |
| Parameter | Model | Number of Experimental Animals | Effect |
|---|---|---|---|
| Splice site editing | Human hepatocytes in vitro | NA | >60% splice site editing achieved at splice site |
| PCSK9 expression | Human hepatocytes in vitro | NA | PCSK9 expression reduced by 55% |
| Splice site editing | Mouse in vivo | 4–5 | A 70% liver base editing of the splice site |
| Splice site editing | Cynomolgus monkeys in vivo | 2–3 | A 63% base-editing frequency of the PCSK9 splice-site after two weeks |
| PCSK9 expression | Cynomolgus monkeys in vivo | 2–3 | A 81% reduction in blood PCSK9 |
| LDL-C | Cynomolgus monkeys in vivo | 2–3 | A 65% reduction in blood LDL-C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, H.E.; Rizzo, M.; Fras, Z.; Jug, B.; Banach, M.; Penson, P.E. CRISPR Gene Editing in Lipid Disorders and Atherosclerosis: Mechanisms and Opportunities. Metabolites 2021, 11, 857. https://doi.org/10.3390/metabo11120857
Walker HE, Rizzo M, Fras Z, Jug B, Banach M, Penson PE. CRISPR Gene Editing in Lipid Disorders and Atherosclerosis: Mechanisms and Opportunities. Metabolites. 2021; 11(12):857. https://doi.org/10.3390/metabo11120857
Chicago/Turabian StyleWalker, Harry E., Manfredi Rizzo, Zlatko Fras, Borut Jug, Maciej Banach, and Peter E. Penson. 2021. "CRISPR Gene Editing in Lipid Disorders and Atherosclerosis: Mechanisms and Opportunities" Metabolites 11, no. 12: 857. https://doi.org/10.3390/metabo11120857
APA StyleWalker, H. E., Rizzo, M., Fras, Z., Jug, B., Banach, M., & Penson, P. E. (2021). CRISPR Gene Editing in Lipid Disorders and Atherosclerosis: Mechanisms and Opportunities. Metabolites, 11(12), 857. https://doi.org/10.3390/metabo11120857