
β-Cell Death in Diabetes: Past Discoveries, Present Understanding, and Potential Future Advances

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Understanding β-Cell Death in the Pathogenesis of Diabetes Mellitus
2.1. Early Observations of Insulin Deficiency as a Driver of Hyperglycemia in Diabetes
2.2. Observations of β-Cell Loss in Human Diabetes
2.2.1. β-Cell Loss in T1D
2.2.2. β-Cell Loss in T2D
2.3. Loss of β-Cell Function in Human Diabetes
2.3.1. β-Cell Dysfunction in T1D
2.3.2. β-Cell Dysfunction in T2D
2.4. Observations of β-Cell Death in Human Diabetes
2.4.1. Evidence for β-Cell Death in Human T1D
2.4.2. Evidence for β-Cell Death in Human T2D
2.4.3. Considerations for Quantifying β-Cell Death in Human Diabetes
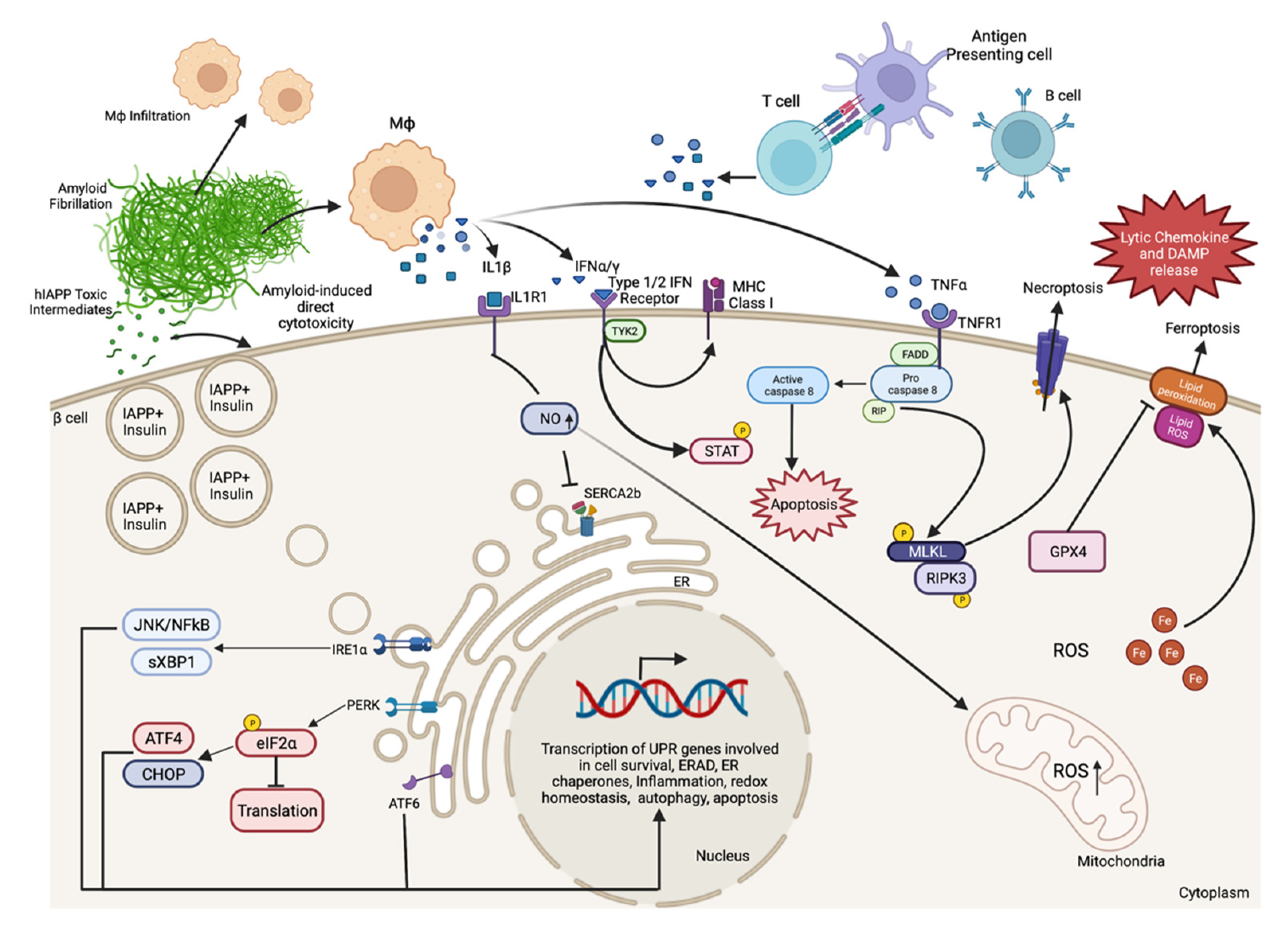
3. Advances in Understanding the Mechanisms of β-Cell Death in Diabetes
3.1. Autoimmune-Associated β-Cell Death in T1D
3.1.1. The Immune System in T1D-Associated β-Cell Death
3.1.2. The β-Cell in T1D-Associated β-Cell Death
3.2. Islet Amyloid-Induced β-Cell Death in T2D
3.3. Mechanisms of Proinflammatory Cytokine-Induced β-Cell Death
3.3.1. IL-1β Signaling in β-Cell Death
3.3.2. TNFα Signaling in β-Cell Death
3.3.3. IFNγ and IFNα Signaling in β-Cell Death
3.4. Endoplasmic Reticulum Stress-Induced β-Cell Death
3.4.1. ER Stress-Induced β-Cell Death in Models of T2D
3.4.2. ER Stress-Induced β-Cell Death in Models of T1D
3.5. Oxidative-Stress-Induced β-Cell Death
3.6. Glucotoxicity and Lipotoxicity in β-Cell Death
4. Potential Future Advances in Understanding β-Cell Death in Diabetes
4.1. Evidence for β-Cell Apoptosis, Necrosis, and Regulated Necrosis
4.1.1. β-Cell Apoptosis
4.1.2. β-Cell Necrosis
4.1.3. β-Cell Regulated Necrosis
4.2. Differentiating between Forms of β-Cell Death
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Beard, H.A.; Al Ghatrif, M.; Samper-Ternent, R.; Gerst, K.; Markides, K.S. Trends in Diabetes Prevalence and Diabetes-Related Complications in Older Mexican Americans From 1993–1994 to 2004. Diabetes Care 2009, 32, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Introduction: Standards of Medical Care in Diabetes. Diabetes Care 2020, 44, S1–S2. [Google Scholar] [CrossRef]
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef]
- Klöppel, G.; Löhr, M.; Habich, K.; Oberholzer, M.; Heitz, P.U. Islet Pathology and the Pathogenesis of Type 1 and Type 2 Diabetes mellitus Revisited. Surv. Synth. Pathol. Res. 1985, 4, 110–125. [Google Scholar] [CrossRef]
- Cnop, M.; Welsh, N.; Jonas, J.-C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. S2), S97–S107. [Google Scholar] [CrossRef]
- American Diabetes Association. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes. Diabetes Care 2021, 44 (Suppl. S1), S15–S33. [Google Scholar] [CrossRef]
- Frank, L.L. Diabetes mellitus in the texts of old Hindu medicine (Charaka, Susruta, Vagbhata). Am. J. Gastroenterol. 1957, 27, 76–95. [Google Scholar]
- Mering, J.V.; Minkowski, O. Diabetes mellitus nach Pankreasexstirpation. Arch. Exp. Pathol. Pharmakol. 1890, 26, 371–387. [Google Scholar] [CrossRef]
- Langerhans, P.H. Contributions to the Microscopic Anatomy of the Pancreas. The Johns Hopkins University Press. Available online: https://www.jstor.org/stable/44438182 (accessed on 20 August 2021).
- Opie, E.L. The relation of diabetes mellitus to lesions of the pancreas. Hyaline degeneration of the islands of Langerhans. J. Exp. Med. 1901, 5, 527–540. [Google Scholar] [CrossRef]
- Lane, M.A. The cytological characters of the areas of Langerhans. Am. J. Anat. 1907, 7, 409–422. [Google Scholar] [CrossRef]
- Banting, F.G.; Best, C. The Internal Secretion of the Pancreas. Am. J. Physiol. 1963, 59, 42–60. [Google Scholar] [CrossRef]
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A.A.; Macleod, J.J.R.; Noble, E.C. The effect produced on diabetes by extractions of pancreas. Trans. Amer Physicians 1922, 37, 337–347. [Google Scholar]
- Wellington, A. Leonard Thompson ‘ever remembered’: The first person to receive insulin. J. Med. Biogr. 2020, 1–3. [Google Scholar] [CrossRef]
- Bliss, M. The Discovery of Insulin: The Twenty-Fifth Anniversary Edition; University of Toronto Press: Toronto, ON, Canada, 1982; ISBN 978-0-8020-8344-9. Available online: https://www.jstor.org/stable/10.3138/j.ctt1wn0sjc (accessed on 26 August 2021).
- Wrenshall, G.A.; Bogoch, A.; Ritchie, R.C. Extractable Insulin of Pancreas: Correlation with Pathological and Clinical Findings in Diabetic and Nondiabetic Cases. Diabetes 1952, 1, 87–107. [Google Scholar] [CrossRef]
- MacLean, N.; Ogilvie, R.F. Quantitative Estimation of the Pancreatic Islet Tissue in Diabetic Subjects. Diabetes 1955, 4, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Von Engelhardt, D. Matthew Dobson (1735–1784). Clinical Investigator of Diabetes Mellitus. In Diabetes Its Medical and Cultural History: Outlines—Texts—Bibliography; von Engelhardt, D., Ed.; Springer: Berlin/Heidelberg, Germany, 1989; pp. 235–237. ISBN 978-3-642-48364-6. [Google Scholar] [CrossRef]
- Steiner, H. Insulitis beim perakuten Diabetes des Kindes. Klin. Wochenschr. 1968, 46, 417–421. [Google Scholar] [CrossRef]
- Maccuish, A.; Irvine, W.; Barnes, E.; Duncan, L. Antibodies to pancreatic islet cells in insulin-dependent diabetics with coexistent autoimmune disease. Lancet 1974, 304, 1529–1531. [Google Scholar] [CrossRef]
- Perley, M.J.; Kipnis, D.M. Plasma Insulin Responses to Oral and Intravenous Glucose: Studies in Normal and Diabetic Subjects*. J. Clin. Investig. 1967, 46, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, R.C.; Leif, G.; Kraemer, N.; Ferrannini, E.; Del Prato, S.; DeFronzo, R.A. Obesity and insulin resistance in humans: A dose-response study. Metabolism 1990, 39, 452–459. [Google Scholar] [CrossRef]
- MacLean, N.; Ogilvie, R.F. Observations on the Pancreatic Islet Tissue of Young Diabetic Subjects. Diabetes 1959, 8, 83–91. [Google Scholar] [CrossRef]
- Doniach, I.; Morgan, A.G. Islets of langerhans in juvenile diabetes mellitus. Clin. Endocrinol. 1973, 2, 233–248. [Google Scholar] [CrossRef]
- Stefan, Y.; Orci, L.; Malaisse-Lagae, F.; Perrelet, A.; Patel, Y.; Unger, R.H. Quantitation of Endocrine Cell Content in the Pancreas of Nondiabetic and Diabetic Humans. Diabetes 1982, 31, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Drenck, C.R.; Oberholzer, M.; Heitz, P.U. Morphometric evidence for a striking B-cell reduction at the clinical onset of type 1 diabetes. Virchows Arch. A 1984, 403, 441–452. [Google Scholar] [CrossRef]
- Johnson, K.H.; Stevens, J.B. Light and Electron Microscopic Studies of Islet Amyloid in Diabetic Cats. Diabetes 1973, 22, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.-C. Pancreatic β-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 32–42. [Google Scholar] [CrossRef]
- MacCallum, W.T. Hypertrophy of the islands of Langerhans in diabetes mellitus. Am. J. Med. Sci. 1907, 133, 432. [Google Scholar] [CrossRef]
- Ogilvie, R.F. The islands of langerhans in 19 cases of obesity. J. Pathol. Bacteriol. 1933, 37, 473–481. [Google Scholar] [CrossRef]
- Prentki, M. Islet cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef]
- Guiot, Y.; Sempoux, C.; Moulin, P.; Rahier, J. No decrease of the beta-cell mass in type 2 diabetic patients. Diabetes 2001, 50, S188. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, C.M.; Panzer, J.K.; Drotar, D.M.; Enos, S.J.; Kipke, N.; Chen, C.; Bozsak, R.; Schöniger, E.; Ehehalt, F.; Distler, M.; et al. Dysfunction of Persisting β Cells Is a Key Feature of Early Type 2 Diabetes Pathogenesis. Cell Rep. 2020, 31, 107469. [Google Scholar] [CrossRef]
- Saito, K.; Yaginuma, N.; Takahashi, T. Differential volumetry of A, B and D cells in the pancreatic islets of diabetic and nondiabetic subjects. Tohoku J. Exp. Med. 1979, 129, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, H.; Mizukami, H.; Yagihashi, N.; Wada, R.; Hanyu, C. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 2002, 45, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Jurgens, C.A.; Toukatly, M.N.; Fligner, C.L.; Udayasankar, J.; Subramanian, S.L.; Zraika, S.; Aston-Mourney, K.; Carr, D.B.; Westermark, P.; Westermark, G.T.; et al. β-Cell Loss and β-Cell Apoptosis in Human Type 2 Diabetes Are Related to Islet Amyloid Deposition. Am. J. Pathol. 2011, 178, 2632–2640. [Google Scholar] [CrossRef]
- Wigger, L.; Barovic, M.; Brunner, A.-D.; Marzetta, F.; Schöniger, E.; Mehl, F.; Kipke, N.; Friedland, D.; Burdet, F.; Kessler, C.; et al. Multi-omics profiling of living human pancreatic islet donors reveals heterogeneous beta cell trajectories towards type 2 diabetes. Nat. Metab. 2021, 3, 1017–1031. [Google Scholar] [CrossRef]
- Redondo, M.J.; Hagopian, W.A.; Oram, R.; Steck, A.K.; Vehik, K.; Weedon, M.; Balasubramanyam, A.; Dabelea, D. The clinical consequences of heterogeneity within and between different diabetes types. Diabetologia 2020, 63, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Olehnik, S.K.; Fowler, J.L.; Avramovich, G.; Hara, M. Quantitative analysis of intra- and inter-individual variability of human beta-cell mass. Sci. Rep. 2017, 7, 16398. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, E.; Prasad, R.B.; Groop, L. Subtypes of Type 2 Diabetes Determined From Clinical Parameters. Diabetes 2020, 69, 2086–2093. [Google Scholar] [CrossRef]
- Vendrame, F.; Zappaterreno, A.; Dotta, F. Markers of beta cell function in type 1 diabetes mellitus. Minerva Med. 2004, 95, 79–84. [Google Scholar]
- Cerasi, E.; Luft, R. The plasma insulin response to glucose infusion in healthy subjects and in diabetes mellitus. Eur. J. Endocrinol. 1967, 55, 278–304. [Google Scholar] [CrossRef]
- Barker, A.; Lauria, A.; Schloot, N.; Hosszufalusi, N.; Ludvigsson, J.; Mathieu, C.; Mauricio, D.; Nordwall, M.; Van der Schueren, B.; Mandrup-Poulsen, T.; et al. Age-dependent decline of β-cell function in type 1 diabetes after diagnosis: A multi-centre longitudinal study. Diabetes Obes. Metab. 2013, 16, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.G.; Keenan, H.A.; Shah, H.S.; Frodsham, S.G.; Pober, D.; He, Z.; Wolfson, E.A.; D’Eon, S.; Tinsley, L.J.; Bonner-Weir, S.; et al. Residual β cell function and monogenic variants in long-duration type 1 diabetes patients. J. Clin. Investig. 2019, 129, 3252–3263. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.; Sussel, L.; Accili, D. Pancreatic β Cell Dedifferentiation as a Mechanism of Diabetic β Cell Failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Rui, J.; Deng, S.; Arazi, A.; Perdigoto, A.L.; Liu, Z.; Herold, K.C. β Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab. 2017, 25, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Karam, J.H.; Grodsky, G.M.; Forsham, P.H.; McWilliams, N.B. Excessive Insulin Response to Glucose in Obese Subjects as Measured by Immunochemical Assay. Diabetes 1963, 12, 197–204. [Google Scholar] [CrossRef]
- Mitrakou, A.; Kelley, D.; Mokan, M.; Veneman, T.; Pangburn, T.; Reilly, J.; Gerich, J. Role of Reduced Suppression of Glucose Production and Diminished Early Insulin Release in Impaired Glucose Tolerance. N. Engl. J. Med. 1992, 326, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Bergman, R.N.; Pacini, G.; Porte, D. Pathogenesis of Age-Related Glucose Intolerance in Man: Insulin Resistance and Decreased β-Cell Function*. J. Clin. Endocrinol. Metab. 1985, 60, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, K.M.; Prigeon, R.L.; Faulenbach, M.V.; Tong, J.; Carr, D.B.; Boyko, E.J.; Leonetti, D.L.; McNeely, M.J.; Fujimoto, W.Y.; Kahn, S.E. Oral Disposition Index Predicts the Development of Future Diabetes Above and Beyond Fasting and 2-h Glucose Levels. Diabetes Care 2008, 32, 335–341. [Google Scholar] [CrossRef]
- Bottazzo, G.F. Death of a Beta Cell: Homicide or Suicide? Diabet. Med. J. Br. Diabet. Assoc. 1986, 3, 119–130. [Google Scholar] [CrossRef]
- Kahn, S.E. The Importance of β-Cell Failure in the Development and Progression of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2001, 86, 4047–4058. [Google Scholar] [CrossRef] [PubMed]
- Gorczyca, W.; Bruno, S.; Darzynkiewicz, R.; Gong, J. DNA strand breaks occurring during apoptosis—Their early insitu detection by the terminal deoxynucleotidyl transferase and nick translation assays and prevention by serine protease inhibitors. Int. J. Oncol. 1992, 1, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- Furuya, T.; Kamada, T.; Murakami, T.; Kurose, A.; Sasaki, K. Laser scanning cytometry allows detection of cell death with morphological features of apoptosis in cells stained with PI. Cytometry 1997, 29, 173–177. [Google Scholar] [CrossRef]
- Meier, J.J.; Bhushan, A.; Butler, A.E.; Rizza, R.A.; Butler, P.C. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: Indirect evidence for islet regeneration? Diabetologia 2005, 48, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J. Type 2 Diabetes-a Matter of ß-Cell Life and Death? Science 2005, 307, 380–384. [Google Scholar] [CrossRef]
- Butler, A.E.; Galasso, R.; Meier, J.J.; Basu, R.; Rizza, R.A.; Butler, P.C. Modestly increased beta cell apoptosis but no increased beta cell replication in recent-onset type 1 diabetic patients who died of diabetic ketoacidosis. Diabetologia 2007, 50, 2323–2331. [Google Scholar] [CrossRef]
- Akirav, E.M.; Lebastchi, J.; Galvan, E.M.; Henegariu, O.; Akirav, M.; Ablamunits, V.; Lizardi, P.M.; Herold, K.C. Detection of cell death in diabetes using differentially methylated circulating DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 19018–19023. [Google Scholar] [CrossRef]
- Fisher, M.M.; Chumbiauca, C.N.P.; Mather, K.J.; Mirmira, R.G.; Tersey, S.A. Detection of Islet β-Cell Death in Vivo by Multiplex PCR Analysis of Differentially Methylated DNA. Endocrinology 2013, 154, 3476–3481. [Google Scholar] [CrossRef]
- Husseiny, M.I.; Kaye, A.; Zebadua, E.; Kandeel, F.; Ferreri, K. Tissue-Specific Methylation of Human Insulin Gene and PCR Assay for Monitoring Beta Cell Death. PLoS ONE 2014, 9, e94591. [Google Scholar] [CrossRef]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [PubMed]
- Neyman, A.; Nelson, J.; Tersey, S.A.; Mirmira, R.G.; Evans-Molina, C.; Sims, E.K. Persistent elevations in circulating INS DNA among subjects with longstanding type 1 diabetes. Diabetes Obes. Metab. 2018, 21, 95–102. [Google Scholar] [CrossRef]
- O’Brien, B.A.; Harmon, B.V.; Cameron, D.P.; Allan, D.J. Apoptosis Is the Mode of -Cell Death Responsible for the Development of IDDM in the Nonobese Diabetic (NOD) Mouse. Diabetes 1997, 46, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Augstein, P.; Elefanty, A.; Allison, J.; Harrison, L.C. Apoptosis and beta-cell destruction in pancreatic islets of NOD mice with spontaneous and cyclophosphamide-accelerated diabetes. Diabetologia 1998, 41, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Lally, F.J.; Ratcliff, H.; Bone, A.J. Apoptosis and disease progression in the spontaneously diabetic BB/S rat. Diabetologia 2001, 44, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Gross, D.J.; Cerasi, E.; Kaiser, N. Hyperglycemia-induced beta-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes 1999, 48, 738–744. [Google Scholar] [CrossRef]
- Pick, A.; Clark, J.; Kubstrup, C.; Levisetti, M.; Pugh, W.; Bonner-Weir, S.; Polonsky, K.S. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 1998, 47, 358–364. [Google Scholar] [CrossRef]
- Farilla, L.; Hui, H.; Bertolotto, C.; Kang, E.; Bulotta, A.; Di Mario, U.; Perfetti, R. Glucagon-Like Peptide-1 Promotes Islet Cell Growth and Inhibits Apoptosis in Zucker Diabetic Rats. Endocrinology 2002, 143, 4397–4408. [Google Scholar] [CrossRef]
- Donath, M.Y.; Halban, P.A. Decreased beta-cell mass in diabetes: Significance, mechanisms and therapeutic implications. Diabetologia 2004, 47, 581–589. [Google Scholar] [CrossRef]
- Iwahashi, H.; Itoh, N.; Yamagata, K.; Imagawa, A.; Nakajima, H.; Tomita, K.; Moriwaki, M.; Waguri, M.; Yamamoto, K.; Miyagawa, J.; et al. Molecular mechanisms of pancreatic beta-cell destruction in autoimmune diabetes: Potential targets for preventive therapy. Cytokines Cell. Mol. Ther. 1998, 4, 45–51. [Google Scholar]
- Hull, R.L.; Westermark, G.T.; Westermark, P.; Kahn, S.E. Islet Amyloid: A Critical Entity in the Pathogenesis of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 3629–3643. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Oxidative stress and beta-cell dysfunction. Pflüg. Arch. Eur. J. Physiol. 2010, 460, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Lytrivi, M.; Castell, A.-L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2019, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C. Glucolipotoxicity, β-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2019, 69, 273–278. [Google Scholar] [CrossRef]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.; Atkinson, M.A.; Roep, B.O.; Von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef]
- Babon, J.A.B.; DeNicola, M.E.; Blodgett, D.M.; Crèvecoeur, I.; Buttrick, T.S.; Maehr, R.; Bottino, R.; Naji, A.; Kaddis, J.; Elyaman, W.; et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat. Med. 2016, 22, 1482–1487. [Google Scholar] [CrossRef]
- Bottazzo, G.; Florin-Christensen, A.; Doniach, D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974, 304, 1279–1283. [Google Scholar] [CrossRef]
- Smeets, S.; De Paep, D.L.; Stangé, G.; Verhaeghen, K.; Van der Auwera, B.; Keymeulen, B.; Weets, I.; Ling, Z.; Veld, P.I.; Gorus, F. Insulitis in the pancreas of non-diabetic organ donors under age 25 years with multiple circulating autoantibodies against islet cell antigens. Virchows Arch. 2021, 479, 295–304. [Google Scholar] [CrossRef]
- Junker, K.; Egeberg, J.; Kromann, H.; Nerup, J. An autopsy study of the islets of langerhans in acute-onset juvenile diabetes mellitus. Acta Pathol. Microbiol. Scand. Sect. A Pathol. 1977, 85A, 699–706. [Google Scholar] [CrossRef]
- Goel, A.; Chiu, H.; Felton, J.; Palmer, J.P.; Brooks-Worrell, B. T-Cell Responses to Islet Antigens Improves Detection of Autoimmune Diabetes and Identifies Patients With More Severe β-Cell Lesions in Phenotypic Type 2 Diabetes. Diabetes 2007, 56, 2110–2115. [Google Scholar] [CrossRef][Green Version]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A.; et al. Anti-CD3 Monoclonal Antibody in New-Onset Type 1 Diabetes Mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Gitelman, S.E.; Ehlers, M.R.; Gottlieb, P.A.; Greenbaum, C.J.; Hagopian, W.; Boyle, K.D.; Keyes-Elstein, L.; Aggarwal, S.; Phippard, D.; et al. Teplizumab (Anti-CD3 mAb) Treatment Preserves C-Peptide Responses in Patients With New-Onset Type 1 Diabetes in a Randomized Controlled Trial: Metabolic and Immunologic Features at Baseline Identify a Subgroup of Responders. Diabetes 2013, 62, 3766–3774. [Google Scholar] [CrossRef] [PubMed]
- Demeester, S.; Keymeulen, B.; Kaufman, L.; Van Dalem, A.; Balti, E.V.; Van De Velde, U.; Goubert, P.; Verhaeghen, K.; Davidson, H.W.; Wenzlau, J.M.; et al. Preexisting Insulin Autoantibodies Predict Efficacy of Otelixizumab in Preserving Residual β-Cell Function in Recent-Onset Type 1 Diabetes. Diabetes Care 2015, 38, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef]
- Miyazaki, A.; Hanafusa, T.; Yamada, K.; Miyagawa, J.; Fujino-Kurihara, H.; Nakajima, H.; Nonaka, K.; Tarui, S. Predominance of T lymphocytes in pancreatic islets and spleen of pre-diabetic non-obese diabetic (NOD) mice: A longitudinal study. Clin. Exp. Immunol. 1985, 60, 622–630. [Google Scholar]
- Christianson, S.W.; Shultz, L.D.; Leiter, E.H. Adoptive Transfer of Diabetes Into Immunodeficient NOD-scid/scid Mice: Relative Contributions of CD4+ and CD8+ T-Cells From Diabetic Versus Prediabetic NOD.NON-Thy-1a Donors. Diabetes 1993, 42, 44–55. [Google Scholar] [CrossRef]
- Posselt, A.M.; Barker, C.F.; Friedman, A.L.; Naji, A. Prevention of Autoimmune Diabetes in the BB Rat by Intrathymic Islet Transplantation at Birth. Science 1992, 256, 1321–1324. [Google Scholar] [CrossRef]
- Calderon, B.; Sacks, D.B. Islet Autoantibodies and Type 1 Diabetes: Does the Evidence Support Screening? Clin. Chem. 2014, 60, 438–440. [Google Scholar] [CrossRef]
- Mullen, Y. Development of the Nonobese Diabetic Mouse and Contribution of Animal Models for Understanding Type 1 Diabetes. Pancreas 2017, 46, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Orban, T.; Sosenko, J.M.; Cuthbertson, D.; Krischer, J.P.; Skyler, J.S.; Jackson, R.; Yu, L.; Palmer, J.P.; Schatz, D.; Eisenbarth, G.; et al. Pancreatic Islet Autoantibodies as Predictors of Type 1 Diabetes in the Diabetes Prevention Trial-Type. Diabetes Care 2009, 32, 2269–2274. [Google Scholar] [CrossRef] [PubMed]
- Pescovitz, M.D.; Greenbaum, C.J.; Bundy, B.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; Moran, A.; Raskin, P.; et al. B-Lymphocyte Depletion with Rituximab and—Cell Function: Two-Year Results. Diabetes Care 2013, 37, 453–459. [Google Scholar] [CrossRef]
- Mariño, E.; Silveira, P.A.; Stolp, J.; Grey, S.T. B cell-directed therapies in type 1 diabetes. Trends Immunol. 2011, 32, 287–294. [Google Scholar] [CrossRef]
- Zhang, Y.; O’Brien, B.; Trudeau, J.; Tan, R.; Santamaria, P.; Dutz, J.P. In Situ β Cell Death Promotes Priming of Diabetogenic CD8 T Lymphocytes. J. Immunol. 2002, 168, 1466–1472. [Google Scholar] [CrossRef]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet -Cell Endoplasmic Reticulum Stress Precedes the Onset of Type 1 Diabetes in the Nonobese Diabetic Mouse Model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the Unfolded Protein Response in Pancreatic β Cells Protects Mice Against Type 1 Diabetes. Sci. Transl. Med. 2013, 5, 211ra156. [Google Scholar] [CrossRef]
- Delong, T.; Wiles, T.A.; Baker, R.L.; Bradley, B.; Barbour, G.; Reisdorph, R.; Armstrong, M.; Powell, R.L.; Reisdorph, N.; Kumar, N.; et al. Pathogenic CD4 T Cells in Type 1 Diabetes Recognize Epitopes Formed by Peptide Fusion. Science 2016, 351, 711–714. Available online: https://www.science.org/doi/abs/10.1126/science.aad2791 (accessed on 31 August 2021). [CrossRef]
- Eizirik, D.L.; Colli, M.L.; Ortis, F. The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 2009, 5, 219–226. [Google Scholar] [CrossRef]
- Oram, R.A.; Sims, E.K.; Evans-Molina, C. Beta cells in type 1 diabetes: Mass and function; sleeping or dead? Diabetologia 2019, 62, 567–577. [Google Scholar] [CrossRef]
- Sims, E.K.; Mirmira, R.G.; Evans-Molina, C. The role of beta-cell dysfunction in early type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 2020, 17, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Westermark, G.T.; Krogvold, L.; Dahl-Jørgensen, K.; Ludvigsson, J. Islet amyloid in recent-onset type 1 diabetes—The DiViD study. Upsala J. Med. Sci. 2017, 122, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.F.; Ludvigsson, J.; Carlsson, A.; Casas, R.; Forsander, G.; Ivarsson, S.A.; Kockum, I.; Lernmark, Å.; Marcus, C.; Lindblad, B.; et al. High Plasma Levels of Islet Amyloid Polypeptide in Young with New-Onset of Type 1 Diabetes Mellitus. PLoS ONE 2014, 9, e93053. [Google Scholar] [CrossRef]
- Delong, T.; Baker, R.L.; Reisdorph, N.; Powell, R.L.; Armstrong, M.; Barbour, G.; Bradley, B.; Haskins, K. Islet Amyloid Polypeptide Is a Target Antigen for Diabetogenic CD4+ T Cells. Diabetes 2011, 60, 2325–2330. [Google Scholar] [CrossRef]
- Westermark, G.T.; Westermark, P.; Berne, C.; Korsgren, O.; Nordic Network for Clinical Islet Transplantation. Widespread Amyloid Deposition in Transplanted Human Pancreatic Islets. N. Engl. J. Med. 2008, 359, 977–979. [Google Scholar] [CrossRef]
- Westermark, G.T.; Davalli, A.M.; Secchi, A.; Folli, F.; Kin, T.; Toso, C.; Shapiro, A.M.J.; Korsgren, O.; Tufveson, G.; Andersson, A.; et al. Further Evidence for Amyloid Deposition in Clinical Pancreatic Islet Grafts. Transplantation 2012, 93, 219–223. [Google Scholar] [CrossRef]
- Westermark, P.; Wernstedt, C.; Wilander, E.; Hayden, D.W.; O’Brien, T.D.; Johnson, K.H. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3881–3885. [Google Scholar] [CrossRef]
- Kahn, S.E.; D’Alessio, D.A.; Schwartz, M.W.; Fujimoto, W.Y.; Ensinck, J.W.; Taborsky, G.J.; Porte, D. Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes 1990, 39, 634–638. [Google Scholar] [CrossRef]
- Scherbaum, W.A. The role of amylin in the physiology of glycemic control. Exp. Clin. Endocrinol. Diabetes 1998, 106, 97–102. [Google Scholar] [CrossRef]
- Rushing, P.A.; Hagan, M.M.; Seeley, R.J.; Lutz, T.A.; Woods, S.C. Amylin: A Novel Action in the Brain to Reduce Body Weight*. Endocrinology 2000, 141, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760. [Google Scholar] [CrossRef]
- Lopes, D.H.; Colin, C.; Degaki, T.L.; de Sousa, A.C.V.; Vieira, M.N.; Sebollela, A.; Martinez, A.M.B.; Bloch, C.; Ferreira, S.T.; Sogayar, M.C. Amyloidogenicity and Cytotoxicity of Recombinant Mature Human Islet Amyloid Polypeptide (rhIAPP). J. Biol. Chem. 2004, 279, 42803–42810. [Google Scholar] [CrossRef]
- Tenidis, K.; Waldner, M.; Bernhagen, J.; Fischle, W.; Bergmann, M.; Weber, M.; Merkle, M.-L.; Voelter, W.; Brunner, H.; Kapurniotu, A. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J. Mol. Biol. 2000, 295, 1055–1071. [Google Scholar] [CrossRef]
- Meier, J.J.; Kayed, R.; Lin, C.-Y.; Gurlo, T.; Haataja, L.; Jayasinghe, S.; Langen, R.; Glabe, C.G.; Butler, P.C. Inhibition of human IAPP fibril formation does not prevent β-cell death: Evidence for distinct actions of oligomers and fibrils of human IAPP. Am. J. Physiol. Metab. 2006, 291, E1317–E1324. [Google Scholar] [CrossRef]
- Bram, Y.; Frydman-Marom, A.; Yanai, I.; Gilead, S.; Shaltiel-Karyo, R.; Amdursky, N.; Gazit, E. Apoptosis induced by islet amyloid polypeptide soluble oligomers is neutralized by diabetes-associated specific antibodies. Sci. Rep. 2014, 4, 4267. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-J.; Armour, S.; Way, J.; Chen, G.; Watson, C.; Irving, P.; Cobb, J.; Kadwell, S.; Beaumont, K.; Rimele, T.; et al. Expression Cloning and Receptor Pharmacology of Human Calcitonin Receptors from MCF-7 Cells and Their Relationship to Amylin Receptors. Mol. Pharmacol. 1997, 52, 1164–1175. [Google Scholar] [CrossRef]
- Lee, S.; Hay, D.; Pioszak, A.A. Calcitonin and Amylin Receptor Peptide Interaction Mechanisms: Insights into peptide-binding modes and allosteric modulation of the calcitonin receptor by receptor activity-modifying proteins. J. Biol. Chem. 2016, 291, 8686–8700. [Google Scholar] [CrossRef]
- Abedini, A.; Cao, P.; Plesner, A.; Zhang, J.; He, M.; Derk, J.; Patil, S.A.; Rosario, R.; Lonier, J.; Song, F.; et al. RAGE binds preamyloid IAPP intermediates and mediates pancreatic β cell proteotoxicity. J. Clin. Investig. 2018, 128, 682–698. [Google Scholar] [CrossRef]
- Bower, R.L.; Yule, L.; Rees, T.A.; Deganutti, G.; Hendrikse, E.R.; Harris, P.W.R.; Kowalczyk, R.; Ridgway, Z.; Wong, A.G.; Swierkula, K.; et al. Molecular Signature for Receptor Engagement in the Metabolic Peptide Hormone Amylin. ACS Pharmacol. Transl. Sci. 2018, 1, 32–49. [Google Scholar] [CrossRef]
- Mirzabekov, T.A.; Lin, M.-C.; Kagan, B.L. Pore Formation by the Cytotoxic Islet Amyloid Peptide Amylin. J. Biol. Chem. 1996, 271, 1988–1992. [Google Scholar] [CrossRef]
- Janson, J.; Ashley, R.H.; Harrison, D.; McIntyre, S.; Butler, P. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 1999, 48, 491–498. [Google Scholar] [CrossRef]
- Cao, P.; Abedini, A.; Wang, H.; Tu, L.-H.; Zhang, X.; Schmidt, A.M.; Raleigh, D.P. Islet amyloid polypeptide toxicity and membrane interactions. Proc. Natl. Acad. Sci. USA 2013, 110, 19279–19284. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Kumar, S.; Rhoades, E.; Miranker, A.D. Conformational switching within dynamic oligomers underpins toxic gain-of-function by diabetes-associated amyloid. Nat. Commun. 2018, 9, 1312. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Westwell-Roper, C.Y.; Chehroudi, C.A.; Denroche, H.; Courtade, J.A.; Ehses, J.; Verchere, C.B. IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia 2014, 58, 575–585. [Google Scholar] [CrossRef]
- Schlamadinger, D.E.; Miranker, A.D. Fiber-Dependent and -Independent Toxicity of Islet Amyloid Polypeptide. Biophys. J. 2014, 107, 2559–2566. [Google Scholar] [CrossRef]
- Hopping, G.; Kellock, J.; Barnwal, R.P.; Law, P.; Bryers, J.; Varani, G.; Caughey, B.; Daggett, V. Designed α-sheet peptides inhibit amyloid formation by targeting toxic oligomers. eLife 2014, 3, e01681. [Google Scholar] [CrossRef]
- Verchere, C.B.; D’Alessio, D.A.; Palmiter, R.D.; Weir, G.C.; Bonner-Weir, S.; Baskin, D.G.; Kahn, S.E. Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proc. Natl. Acad. Sci. USA 1996, 93, 3492–3496. [Google Scholar] [CrossRef]
- Janson, J.; Soeller, W.C.; Roche, P.C.; Nelson, R.T.; Torchia, A.J.; Kreutter, D.K.; Butler, P.C. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc. Natl. Acad. Sci. USA 1996, 93, 7283–7288. [Google Scholar] [CrossRef]
- Soeller, W.C.; Janson, J.; Hart, S.E.; Parker, J.C.; Carty, M.D.; Stevenson, R.W.; Kreutter, D.K.; Butler, P.C. Islet amyloid-associated diabetes in obese A(vy)/a mice expressing human islet amyloid polypeptide. Diabetes 1998, 47, 743–750. [Google Scholar] [CrossRef]
- Butler, A.E.; Jang, J.; Gurlo, T.; Carty, M.D.; Soeller, W.C.; Butler, P.C. Diabetes due to a progressive defect in beta-cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): A new model for type 2 diabetes. Diabetes 2004, 53, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, S.; Verchere, C.B.; Terauchi, Y.; Kadowaki, T.; Kahn, S.E. beta-cell glucokinase deficiency and hyperglycemia are associated with reduced islet amyloid deposition in a mouse model of type 2 diabetes. Diabetes 2000, 49, 2056–2062. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Utzschneider, K.M.; Tong, J.; Gerchman, F.; Kahn, S.E. Glucose- and time-dependence of islet amyloid formation in vitro. Biochem. Biophys. Res. Commun. 2007, 354, 234–239. [Google Scholar] [CrossRef]
- Westwell-Roper, C.; Dai, D.L.; Soukhatcheva, G.; Potter, K.J.; Van Rooijen, N.; Ehses, J.A.; Verchere, C.B. IL-1 Blockade Attenuates Islet Amyloid Polypeptide-Induced Proinflammatory Cytokine Release and Pancreatic Islet Graft Dysfunction. J. Immunol. 2011, 187, 2755–2765. [Google Scholar] [CrossRef] [PubMed]
- Westwell-Roper, C.Y.; Ehses, J.A.; Verchere, C.B. Resident Macrophages Mediate Islet Amyloid Polypeptide–Induced Islet IL-1β Production and β-Cell Dysfunction. Diabetes 2013, 63, 1698–1711. [Google Scholar] [CrossRef] [PubMed]
- Meier, D.T.; Morcos, M.; Samarasekera, T.; Zraika, S.; Hull, R.; Kahn, S.E. Islet amyloid formation is an important determinant for inducing islet inflammation in high-fat-fed human IAPP transgenic mice. Diabetologia 2014, 57, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Templin, A.T.; Mellati, M.; Meier, D.T.; Esser, N.; Hogan, M.F.; Castillo, J.J.; Akter, R.; Raleigh, D.P.; Zraika, S.; Hull, R.L.; et al. Low concentration IL-1β promotes islet amyloid formation by increasing hIAPP release from humanised mouse islets in vitro. Diabetologia 2020, 63, 2385–2395. [Google Scholar] [CrossRef]
- Westwell-Roper, C.; Denroche, H.; Ehses, J.; Verchere, C.B. Differential Activation of Innate Immune Pathways by Distinct Islet Amyloid Polypeptide (IAPP) Aggregates. J. Biol. Chem. 2016, 291, 8908–8917. [Google Scholar] [CrossRef]
- Huang, C.-J.; Lin, C.-Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High Expression Rates of Human Islet Amyloid Polypeptide Induce Endoplasmic Reticulum Stress–Mediated β-Cell Apoptosis, a Characteristic of Humans With Type 2 but Not Type 1 Diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Gurlo, T.; Rivera, J.F.; Butler, A.E.; Cory, M.; Hoang, J.; Costes, S.; Butler, P. CHOP Contributes to, But Is Not the Only Mediator of, IAPP Induced β-Cell Apoptosis. Mol. Endocrinol. 2016, 30, 446–454. [Google Scholar] [CrossRef]
- Meng, F.; Abedini, A.; Plesner, A.; Middleton, C.T.; Potter, K.J.; Zanni, M.T.; Verchere, C.B.; Raleigh, D.P. The Sulfated Triphenyl Methane Derivative Acid Fuchsin Is a Potent Inhibitor of Amyloid Formation by Human Islet Amyloid Polypeptide and Protects against the Toxic Effects of Amyloid Formation. J. Mol. Biol. 2010, 400, 555–566. [Google Scholar] [CrossRef]
- Ren, B.; Liu, Y.; Zhang, Y.; Cai, Y.; Gong, X.; Chang, Y.; Xu, L.; Zheng, J. Genistein: A Dual Inhibitor of Both Amyloid β and Human Islet Amylin Peptides. ACS Chem. Neurosci. 2018, 9, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, M.S.; Ryazanov, S.; Leonov, A.; Nicolai, J.; Praest, P.; Giese, A.; Winter, R.; Khemtemourian, L.; Griesinger, C.; Killian, J.A. The small molecule inhibitor anle145c thermodynamically traps human islet amyloid peptide in the form of non-cytotoxic oligomers. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Oskarsson, M.E.; Hermansson, E.; Wang, Y.; Welsh, N.; Presto, J.; Johansson, J.; Westermark, G.T. BRICHOS domain of Bri2 inhibits islet amyloid polypeptide (IAPP) fibril formation and toxicity in human beta cells. Proc. Natl. Acad. Sci. USA 2018, 115, E2752–E2761. [Google Scholar] [CrossRef] [PubMed]
- Armiento, V.; Hille, K.; Naltsas, D.; Lin, J.S.; Barron, A.E.; Kapurniotu, A. The Human Host-Defense Peptide Cathelicidin LL-37 is a Nanomolar Inhibitor of Amyloid Self-Assembly of Islet Amyloid Polypeptide (IAPP). Angew. Chem. Int. Ed. 2020, 59, 12837–12841. [Google Scholar] [CrossRef]
- Röder, C.; Kupreichyk, T.; Gremer, L.; Schäfer, L.U.; Pothula, K.R.; Ravelli, R.B.G.; Willbold, D.; Hoyer, W.; Schröder, G.F. Cryo-EM structure of islet amyloid polypeptide fibrils reveals similarities with amyloid-β fibrils. Nat. Struct. Mol. Biol. 2020, 27, 660–667. [Google Scholar] [CrossRef]
- Roesti, E.S.; Boyle, C.N.; Zeman, D.T.; Sande-Melon, M.; Storni, F.; Cabral-Miranda, G.; Knuth, A.; Lutz, T.A.; Vogel, M.; Bachmann, M.F. Vaccination Against Amyloidogenic Aggregates in Pancreatic Islets Prevents Development of Type 2 Diabetes Mellitus. Vaccines 2020, 8, 116. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Gurlo, T.; Kayed, R.; Butler, A.E.; Haataja, L.; Glabe, C.G.; Butler, P.C. Toxic Human Islet Amyloid Polypeptide (h-IAPP) Oligomers Are Intracellular, and Vaccination to Induce Anti-Toxic Oligomer Antibodies Does Not Prevent h-IAPP-Induced -Cell Apoptosis in h-IAPP Transgenic Mice. Diabetes 2007, 56, 1324–1332. [Google Scholar] [CrossRef]
- Palmer, J.P.; Helqvist, S.; Spinas, G.A.; Mølvig, J.; Mandrup-Poulsen, T.; Andersen, H.U.; Nerup, J. Interaction of -Cell Activity and IL-1 Concentration and Exposure Time in Isolated Rat Islets of Langerhans. Diabetes 1989, 38, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Dror, E.; Dalmas, E.; Zeman-Meier, D.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.-M.; Rasschaert, J.; Tonnesen, M.; Van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef]
- Corbett, J.A.; Wang, J.L.; Misko, T.P.; Zhao, W.; Hickey, W.F.; Mcdaniel, M.L. Nitric Oxide Mediates IL-1β-Induced Islet Dysfunction and Destruction: Prevention by Dexamethasone. Autoimmunity 1993, 15, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Chambers, K.T.; Unverferth, J.A.; Weber, S.M.; Wek, R.C.; Urano, F.; Corbett, J.A. The Role of Nitric Oxide and the Unfolded Protein Response in Cytokine-Induced -Cell Death. Diabetes 2007, 57, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.J.; Chambers, K.T.; Meares, G.P.; Corbett, J.A. Nitric oxides mediates a shift from early necrosis to late apoptosis in cytokine-treated β-cells that is associated with irreversible DNA damage. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1187–E1196. [Google Scholar] [CrossRef]
- D’Hertog, W.; Overbergh, L.; Lage, K.; Ferreira, G.B.; Maris, M.; Gysemans, C.; Flamez, D.; Cardozo, A.K.; Bergh, G.V.D.; Schoofs, L.; et al. Proteomics Analysis of Cytokine-induced Dysfunction and Death in Insulin-producing INS-1E Cells: New insights into the pathways involved. Mol. Cell. Proteom. 2007, 6, 2180–2199. [Google Scholar] [CrossRef]
- Ehses, J.; Böni-Schnetzler, M.; Faulenbach, M.; Donath, M.Y. Macrophages, cytokines and β-cell death in Type 2 diabetes. Biochem. Soc. Trans. 2008, 36, 340–342. [Google Scholar] [CrossRef]
- Collier, J.J.; Burke, S.J.; Eisenhauer, M.E.; Lu, D.; Sapp, R.C.; Frydman, C.J.; Campagna, S. Pancreatic β-Cell Death in Response to Pro-Inflammatory Cytokines Is Distinct from Genuine Apoptosis. PLoS ONE 2011, 6, e22485. [Google Scholar] [CrossRef]
- Demine, S.; Schiavo, A.A.; Marín-Cañas, S.; Marchetti, P.; Cnop, M.; Eizirik, D.L. Pro-inflammatory cytokines induce cell death, inflammatory responses, and endoplasmic reticulum stress in human iPSC-derived beta cells. Stem Cell Res. Ther. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Corbett, J.A.; Wang, J.L.; Sweetland, M.A.; Jr, J.R.L.; McDaniel, M.L. Interleukin 1 Beta Induces the Formation of Nitric Oxide by Beta-Cells Purified from Rodent Islets of Langerhans. Evidence for the Beta-Cell as a Source and Site of Action of Nitric Oxide. Available online: https://www.jci.org/articles/view/116129/scanned-page/2384 (accessed on 27 September 2021).
- Ammendrup, A.; Maillard, A.; Nielsen, K.; Andersen, N.A.; Serup, P.; Madsen, O.D.; Mandrup-Poulsen, T.; Bonny, C. The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 2000, 49, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, H.; Zhao, B.; Fei, H. IL-1β caused pancreatic β-cells apoptosis is mediated in part by endoplasmic reticulum stress via the induction of endoplasmic reticulum Ca2+ release through the c-Jun N-terminal kinase pathway. Mol. Cell. Biochem. 2009, 324, 183–190. [Google Scholar] [CrossRef]
- Schwarznau, A.; Hanson, M.S.; Sperger, J.M.; Schram, B.R.; Danobeitia, J.S.; Greenwood, K.K.; Vijayan, A.; Fernandez, L.A. IL-1β receptor blockade protects islets against pro-inflammatory cytokine induced necrosis and apoptosis. J. Cell. Physiol. 2009, 220, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Dupraz, P.; Cottet, S.; Hamburger, F.; Dolci, W.; Felley-Bosco, E.; Thorens, B. Dominant Negative MyD88 Proteins Inhibit Interleukin-1β/Interferon-γ-mediated Induction of Nuclear Factor κB-dependent Nitrite Production and Apoptosis in β Cells. J. Biol. Chem. 2000, 275, 37672–37678. [Google Scholar] [CrossRef] [PubMed]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Lohmann-Matthes, M.L.; Gifford, G.E. Cell-associated tumor necrosis factor (TNF) as a killing mechanism of activated cytotoxic macrophages. J. Immunol. 1987, 138, 957–962. [Google Scholar]
- Ishizuka, N.; Yagui, K.; Tokuyama, Y.; Yamada, K.; Suzuki, Y.; Miyazaki, J.-I.; Hashimoto, N.; Makino, H.; Saito, Y.; Kanatsuka, A. Tumor necrosis factor alpha signaling pathway and apoptosis in pancreatic β cells. Metabolism 1999, 48, 1485–1492. [Google Scholar] [CrossRef]
- Green, E.; Eynon, E.E.; Flavell, R.A. Local Expression of TNFα in Neonatal NOD Mice Promotes Diabetes by Enhancing Presentation of Islet Antigens. Immunity 1998, 9, 733–743. [Google Scholar] [CrossRef]
- Kägi, D.; Ho, A.; Odermatt, B.; Zakarian, A.; Ohashi, P.S.; Mak, T.W. TNF receptor 1-dependent beta cell toxicity as an effector pathway in autoimmune diabetes. J. Immunol. Baltim. Md. 1950 1999, 162, 4598–4605. [Google Scholar]
- Stephens, L.A.; Thomas, H.E.; Ming, L.; Darwiche, M.G.R.; Volodin, L.; Kay, T.W.H. Tumor Necrosis Factor-α-Activated Cell Death Pathways in NIT-1 Insulinoma Cells and Primary Pancreatic β Cells*. Endocrinology 1999, 140, 3219–3227. [Google Scholar] [CrossRef]
- Quattrin, T.; Haller, M.J.; Steck, A.K.; Felner, E.I.; Li, Y.; Xia, Y.; Leu, J.H.; Zoka, R.; Hedrick, J.A.; Rigby, M.R.; et al. Golimumab and Beta-Cell Function in Youth with New-Onset Type 1 Diabetes. N. Engl. J. Med. 2020, 383, 2007–2017. [Google Scholar] [CrossRef]
- Campbell, I.L.; Wong, G.H.W.; Schrader, J.W.; Harrison, L.C. Interferon- Enhances the Expression of the Major Histocompatibility Class I Antigens on Mouse Pancreatic Beta Cells. Diabetes 1985, 34, 1205–1209. [Google Scholar] [CrossRef]
- Campbell, I.L.; Oxbrow, L.; Harrison, L.C. Interferon-γ: Pleiotropic effects on a rat pancreatic beta cell line. Mol. Cell. Endocrinol. 1987, 52, 161–167. [Google Scholar] [CrossRef]
- Thomas, H.E.; Parker, J.L.; Schreiber, R.D.; Kay, T.W. IFN-gamma action on pancreatic beta cells causes class I MHC upregulation but not diabetes. J. Clin. Investig. 1998, 102, 1249–1257. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ferreira, R.C.; Guo, H.; Coulson, R.M.; Smyth, D.J.; Pekalski, M.L.; Burren, O.S.; Cutler, A.J.; Doecke, J.D.; Flint, S.; McKinney, E.F.; et al. A Type I Interferon Transcriptional Signature Precedes Autoimmunity in Children Genetically at Risk for Type 1 Diabetes. Diabetes 2014, 63, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; dos Santos, R.S.; De Beeck, A.O.; De Brachène, A.C.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-α mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Takahashi, H.; Mine, K.; Higashimoto, K.; Inoue, K.; Kojima, M.; Kuroki, S.; Eguchi, T.; Ono, Y.; Inuzuka, S.; et al. TYK2 Promoter Variant Is Associated with Impaired Insulin Secretion and Lower Insulin Resistance in Japanese Type 2 Diabetes Patients. Genes 2021, 12, 400. [Google Scholar] [CrossRef]
- Izumi, K.; Mine, K.; Inoue, Y.; Teshima, M.; Ogawa, S.; Kai, Y.; Kurafuji, T.; Hirakawa, K.; Miyakawa, D.; Ikeda, H.; et al. Reduced Tyk2 gene expression in β-cells due to natural mutation determines susceptibility to virus-induced diabetes. Nat. Commun. 2015, 6, 6748. [Google Scholar] [CrossRef]
- Marroqui, L.; Dos Santos, R.S.; Fløyel, T.; Grieco, F.A.; Santin, I.; de Beeck, A.O.; Marselli, L.; Marchetti, P.; Pociot, F.; Eizirik, D.L. TYK2, a Candidate Gene for Type 1 Diabetes, Modulates Apoptosis and the Innate Immune Response in Human Pancreatic β-Cells. Diabetes 2015, 64, 3808–3817. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The Role for Endoplasmic Reticulum Stress in Diabetes Mellitus. Endocr. Rev. 2007, 29, 42–61. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. Delineation of a Negative Feedback Regulatory Loop That Controls Protein Translation during Endoplasmic Reticulum Stress. J. Biol. Chem. 2003, 278, 34864–34873. [Google Scholar] [CrossRef]
- Back, S.H.; Scheuner, D.; Han, J.; Song, B.; Ribick, M.; Wang, J.; Gildersleeve, R.D.; Pennathur, S.; Kaufman, R.J. Translation Attenuation through eIF2α Phosphorylation Prevents Oxidative Stress and Maintains the Differentiated State in β Cells. Cell Metab. 2009, 10, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Ron, D. Proteotoxicity in the endoplasmic reticulum: Lessons from the Akita diabetic mouse. J. Clin. Investig. 2002, 109, 443–445. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Mahadevan, J.; Kanekura, K.; Hara, M.; Lu, S.; Urano, F. Calcium Efflux From the Endoplasmic Reticulum Leads to β-Cell Death. Endocrinology 2014, 155, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.; Tong, X.; Taleb, S.; Bone, R.N.; Iida, H.; Lee, C.-C.; Sohn, P.; Gilon, P.; Roe, M.W.; Evans-Molina, C. Impaired Store-Operated Calcium Entry and STIM1 Loss Lead to Reduced Insulin Secretion and Increased Endoplasmic Reticulum Stress in the Diabetic β-Cell. Diabetes 2018, 67, 2293–2304. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Tang, C.; Koulajian, K.; Schuiki, I.; Zhang, L.; Desai, T.; Ivovic, A.; Wang, P.; Robson-Doucette, C.; Wheeler, M.B.; Minassian, B.; et al. Glucose-induced beta cell dysfunction in vivo in rats: Link between oxidative stress and endoplasmic reticulum stress. Diabetologia 2012, 55, 1366–1379. [Google Scholar] [CrossRef]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef]
- O’Sullivan-Murphy, B.; Urano, F. ER Stress as a Trigger for -Cell Dysfunction and Autoimmunity in Type 1 Diabetes. Diabetes 2012, 61, 780–781. [Google Scholar] [CrossRef]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic Reticulum Stress Sensing in the Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef] [PubMed]
- Laybutt, D.R.; Preston, A.M.; Åkerfeldt, M.C.; Kench, J.; Busch, A.K.; Biankin, A.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Bugliani, M.; Lupi, R.; Marselli, L.; Masini, M.; Boggi, U.; Filipponi, F.; Weir, G.C.; Eizirik, D.L.; Cnop, M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007, 50, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.-A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of Endoplasmic Reticulum Stress in Insulin Resistance and Diabetes. J. Biol. Chem. 2005, 280, 847–851. [Google Scholar] [CrossRef]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Marciniak, S.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress–mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- Abreu, D.; Asada, R.; Revilla, J.M.P.; Lavagnino, Z.; Kries, K.; Piston, D.W.; Urano, F. Wolfram syndrome 1 gene regulates pathways maintaining beta-cell health and survival. Lab. Investig. J. Tech. Methods Pathol. 2020, 100, 849–862. [Google Scholar] [CrossRef]
- Yang, C.; Diiorio, P.; Jurczyk, A.; O’Sullivan-Murphy, B.; Urano, F.; Bortell, R. Pathological endoplasmic reticulum stress mediated by the IRE1 pathway contributes to pre-insulitic beta cell apoptosis in a virus-induced rat model of type 1 diabetes. Diabetologia 2013, 56, 2638–2646. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, Y.-S.; Harenda, Q.; Pietrzak, S.; Oktay, H.Z.; Schreiber, S.; Liao, Y.; Sonthalia, S.; Ciecko, A.E.; Chen, Y.-G.; et al. Beta Cell Dedifferentiation Induced by IRE1α Deletion Prevents Type 1 Diabetes. Cell Metab. 2020, 31, 822–836.e5. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Villalta, S.A.; Feldman, H.C.; Register, A.C.; Rosenthal, W.; Hoffmann-Petersen, I.T.; Mehdizadeh, M.; Ghosh, R.; Wang, L.; Colon-Negron, K.; et al. Targeting ABL-IRE1α Signaling Spares ER-Stressed Pancreatic β Cells to Reverse Autoimmune Diabetes. Cell Metab. 2017, 25, 883–897.e8. [Google Scholar] [CrossRef]
- Delépine, M.; Nicolino, M.; Barrett, T.; Golamaully, M.; Lathrop, G.M.; Julier, C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 2000, 25, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.I.; Abreu, D.; McGill, J.B.; Urano, F. Monogenic and syndromic diabetes due to endoplasmic reticulum stress. J. Diabetes Its Complicat. 2020, 35, 107618. [Google Scholar] [CrossRef]
- Lu, S.; Kanekura, K.; Hara, T.; Mahadevan, J.; Spears, L.D.; Oslowski, C.M.; Martinez, R.; Yamazaki-Inoue, M.; Toyoda, M.; Neilson, A.; et al. A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, E5292–E5301. [Google Scholar] [CrossRef]
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef]
- Kaneto, H.; Fujii, J.; Seo, H.G.; Suzuki, K.; Matsuko, T.-A.; Masahiro, N.; Tatsumi, H.; Yamasaki, Y.; Kamada, T.; Taniguchi, N. Apoptotic Cell Death Triggered by Nitric Oxide in Pancreatic—Cells. Diabetes 1995, 44, 733–738. [Google Scholar] [CrossRef]
- Hong, K.; Xu, G.; Grayson, T.B.; Shalev, A. Cytokines Regulate β-Cell Thioredoxin-interacting Protein (TXNIP) via Distinct Mechanisms and Pathways. J. Biol. Chem. 2016, 291, 8428–8439. [Google Scholar] [CrossRef]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin Interacting Protein: Redox Dependent and Independent Regulatory Mechanisms. Antioxid. Redox Signal. 2012, 16, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Cha-Molstad, H.; Saxena, G.; Chen, J.; Shalev, A. Glucose-stimulated Expression of Txnip Is Mediated by Carbohydrate Response Element-binding Protein, p300, and Histone H4 Acetylation in Pancreatic Beta Cells. J. Biol. Chem. 2009, 284, 16898–16905. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hui, S.T.; Couto, F.M.; Mungrue, I.; Davis, D.B.; Attie, A.D.; Lusis, A.J.; Davis, R.A.; Shalev, A. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. 2008, 22, 3581–3594. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fontes, G.; Saxena, G.; Poitout, V.; Shalev, A. Lack of TXNIP Protects Against Mitochondria-Mediated Apoptosis but Not Against Fatty Acid–Induced ER Stress–Mediated β-Cell Death. Diabetes 2009, 59, 440–447. [Google Scholar] [CrossRef]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free. Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Pullen, T.; Rutter, G.A. When less is more: The forbidden fruits of gene repression in the adult β-cell. Diabetes Obes. Metab. 2012, 15, 503–512. [Google Scholar] [CrossRef]
- Malaisse, W.J.; Malaisse-Lagae, F.; Sener, A.; Pipeleers, D.G. Determinants of the selective toxicity of alloxan to the pancreatic B cell. Proc. Natl. Acad. Sci. USA 1982, 79, 927–930. [Google Scholar] [CrossRef]
- Xu, J.; Long, Y.-S.; Gozal, D.; Epstein, P.N. β-cell death and proliferation after intermittent hypoxia: Role of oxidative stress. Free. Radic. Biol. Med. 2008, 46, 783–790. [Google Scholar] [CrossRef]
- Grankvist, K.; Marklund, S.; Täljedal, I.-B. Superoxide dismutase is a prophylactic against alloxan diabetes. Nature 1981, 294, 158–160. [Google Scholar] [CrossRef]
- Grankvist, K.; Marklund, S.L.; Täljedal, I.B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem. J. 1981, 199, 393–398. [Google Scholar] [CrossRef]
- Johansson, L.H.; Borg, L.A.H. A spectrophotometric method for determination of catalase activity in small tissue samples. Anal. Biochem. 1988, 174, 331–336. [Google Scholar] [CrossRef]
- Tersey, S.A.; Maier, B.; Nishiki, Y.; Maganti, A.V.; Nadler, J.L.; Mirmira, R.G. 12-Lipoxygenase Promotes Obesity-Induced Oxidative Stress in Pancreatic Islets. Mol. Cell. Biol. 2014, 34, 3735–3745. [Google Scholar] [CrossRef]
- Hernandez-Perez, M.; Chopra, G.; Fine, J.; Conteh, A.M.; Anderson, R.M.; Linnemann, A.K.; Benjamin, C.; Nelson, J.B.; Benninger, K.S.; Nadler, J.L.; et al. Inhibition of 12/15-Lipoxygenase Protects Against β-Cell Oxidative Stress and Glycemic Deterioration in Mouse Models of Type 1 Diabetes. Diabetes 2017, 66, 2875–2887. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Corkey, B.E. Are the beta-Cell Signaling Molecules Malonyl-CoA and Cystolic Long-Chain Acyl-CoA Implicated in Multiple Tissue Defects of Obesity and NIDDM? Diabetes 1996, 45, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Zhang, H.J.; Pyzdrowski, K.L.; Walseth, T.F. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J. Clin. Investig. 1992, 90, 320–325. [Google Scholar] [CrossRef]
- Olson, L.K.; Redmon, J.B.; Towle, H.C.; Robertson, R.P. Chronic exposure of HIT cells to high glucose concentrations paradoxically decreases insulin gene transcription and alters binding of insulin gene regulatory protein. J. Clin. Investig. 1993, 92, 514–519. [Google Scholar] [CrossRef]
- Gleason, C.E.; Gonzalez, M.; Harmon, J.S.; Robertson, R.P. Determinants of glucose toxicity and its reversibility in the pancreatic islet β-cell line, HIT-T. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E997–E1002. [Google Scholar] [CrossRef] [PubMed]
- Harmon, J.S.; Stein, R.; Robertson, R.P. Oxidative Stress-mediated, Post-translational Loss of MafA Protein as a Contributing Mechanism to Loss of Insulin Gene Expression in Glucotoxic Beta Cells. J. Biol. Chem. 2005, 280, 11107–11113. [Google Scholar] [CrossRef]
- Boland, B.B.; Brown, C.; Boland, M.L.; Cann, J.; Sulikowski, M.; Hansen, G.; Grønlund, R.V.; King, W.; Rondinone, C.; Trevaskis, J.; et al. Pancreatic β-Cell Rest Replenishes Insulin Secretory Capacity and Attenuates Diabetes in an Extreme Model of Obese Type 2 Diabetes. Diabetes 2018, 68, 131–140. [Google Scholar] [CrossRef]
- Elks, M.L. Chronic perifusion of rat islets with palmitate suppresses glucose-stimulated insulin release. Endocrinology 1993, 133, 208–214. [Google Scholar] [CrossRef]
- Zhou, Y.P.; Grill, V.E. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J. Clin. Investig. 1994, 93, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Mittelman, S.D.; Lamarche, B.; Bergman, R.N.; Giacca, A.; Lewis, G.F. Acute enhancement of insulin secretion by FFA in humans is lost with prolonged FFA elevation. Am. J. Physiol. Endocrinol. Metab. 1999, 276, E1055–E1066. [Google Scholar] [CrossRef] [PubMed]
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patané, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged Exposure to Free Fatty Acids Has Cytostatic and Pro-Apoptotic Effects on Human Pancreatic Islets: Evidence that -Cell Death Is Caspase Mediated, Partially Dependent on Ceramide Pathway, and Bcl-2 Regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef]
- Cnop, M.; Hannaert, J.C.; Grupping, A.Y.; Pipeleers, D.G. Low Density Lipoprotein Can Cause Death of Islet β-Cells by Its Cellular Uptake and Oxidative Modification. Endocrinology 2002, 143, 3449–3453. [Google Scholar] [CrossRef]
- El-Assaad, W.; Buteau, J.; Peyot, M.-L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated Fatty Acids Synergize with Elevated Glucose to Cause Pancreatic β-Cell Death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef]
- Kharroubi, I.; Ladrière, L.; Cardozo, A.K.; Dogusan, Z.; Cnop, M.; Eizirik, D.L. Free Fatty Acids and Cytokines Induce Pancreatic β-Cell Apoptosis by Different Mechanisms: Role of Nuclear Factor-κB and Endoplasmic Reticulum Stress. Endocrinology 2004, 145, 5087–5096. [Google Scholar] [CrossRef]
- Choi, S.-E.; Kim, H.-E.; Shin, H.-C.; Jang, H.-J.; Lee, K.-W.; Kim, Y.; Kang, S.S.; Chun, J.; Kang, Y. Involvement of Ca2+-mediated apoptotic signals in palmitate-induced MIN6N8a beta cell death. Mol. Cell. Endocrinol. 2007, 272, 50–62. [Google Scholar] [CrossRef]
- Ameisen, J.C. On the origin, evolution, and nature of programmed cell death: A timeline of four billion years. Cell Death Differ. 2002, 9, 367–393. [Google Scholar] [CrossRef]
- Buja, L.M.; Eigenbrodt, M.L.; Eigenbrodt, E.H. Apoptosis and necrosis. Basic types and mechanisms of cell death. Arch. Pathol. Lab. Med. 1993, 117, 1208–1214. [Google Scholar]
- Gerschenson, L.E.; Rotello, R.J. Apoptosis: A different type of cell death. FASEB J. 1992, 6, 2450–2455. [Google Scholar] [CrossRef]
- Hui, H.; Dotta, F.; Di Mario, U.; Perfetti, R. Role of caspases in the regulation of apoptotic pancreatic islet beta-cells death. J. Cell. Physiol. 2004, 200, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Del Guerra, S.; Marselli, L.; Lupi, R.; Masini, M.; Pollera, M.; Bugliani, M.; Boggi, U.; Vistoli, F.; Mosca, F.; et al. Pancreatic Islets from Type 2 Diabetic Patients Have Functional Defects and Increased Apoptosis That Are Ameliorated by Metformin. J. Clin. Endocrinol. Metab. 2004, 89, 5535–5541. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and autoimmune diseases. Ann. N. Y. Acad. Sci. 2010, 1209, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Tonnus, W.; Belavgeni, A.; Beuschlein, F.; Eisenhofer, G.; Fassnacht, M.; Kroiss, M.; Krone, N.P.; Reincke, M.; Bornstein, S.R.; Linkermann, A. The role of regulated necrosis in endocrine diseases. Nat. Rev. Endocrinol. 2021, 17, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; De Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.; Kuan, C.-Y.; et al. Apoptotic Caspases Prevent the Induction of Type I Interferons by Mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef]
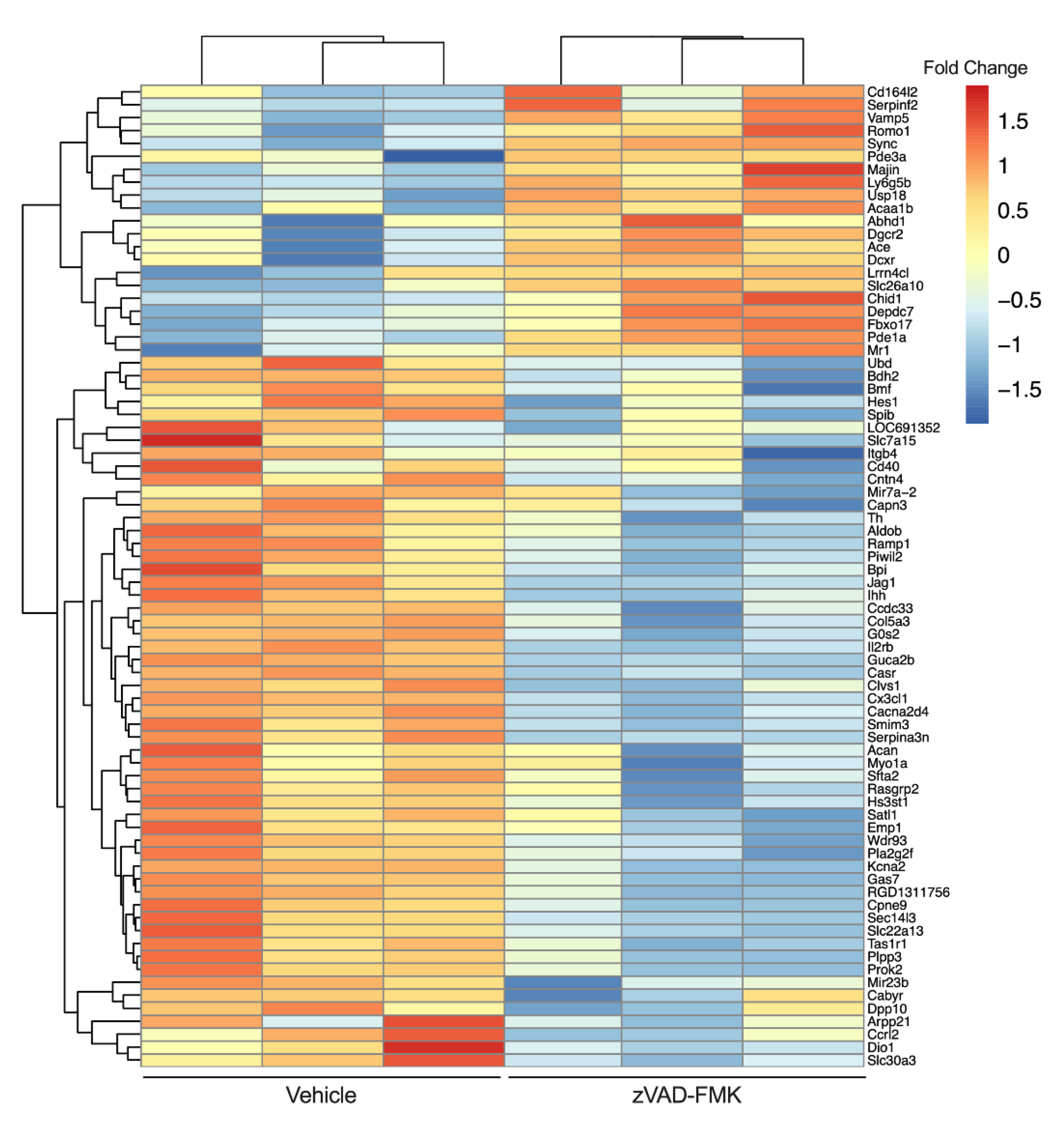
- Contreras, C.J.; Lin, L.; Hogan, M.F.; Oberst, A.; Kahn, S.E.; Templin, A.T. 294-OR: RIPK3-Mediated Necroptosis Is an Alternative Form of TNFa-Induced ß-Cell Death. Diabetes 2021, 70 (Suppl. S1), 294. [Google Scholar] [CrossRef]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res. 2003, 283, 1–16. [Google Scholar] [CrossRef]
- Rock, K.L.; Kono, H. The Inflammatory Response to Cell Death. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 99–126. [Google Scholar] [CrossRef]
- Fehsel, K.; Kolb-Bachofen, V.; Kröncke, K.-D. Necrosis is the predominant type of islet cell death during development of insulin-dependent diabetes mellitus in BB rats. Lab. Investig. 2003, 83, 549–559. [Google Scholar] [CrossRef][Green Version]
- Steer, S.A.; Scarim, A.L.; Chambers, K.T.; Corbett, J.A. Interleukin-1 Stimulates β-Cell Necrosis and Release of the Immunological Adjuvant HMGB. PLoS Med. 2005, 3, e17. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Van De Casteele, M.; Eizirik, D.L.; Pipeleers, D.G. Interleukin-1beta-induced alteration in a beta-cell phenotype can reduce cellular sensitivity to conditions that cause necrosis but not to cytokine-induced apoptosis. Diabetes 2000, 49, 340–345. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoorens, A.; Stangé, G.; Pavlovic, D.; Pipeleers, D. Distinction Between Interleukin-1-Induced Necrosis and Apoptosis of Islet Cells. Diabetes 2001, 50, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Bruni, A.; Pepper, A.R.; Pawlick, R.L.; Gala-Lopez, B.; Gamble, A.F.; Kin, T.; Seeberger, K.; Korbutt, G.S.; Bornstein, S.R.; Linkermann, A.; et al. Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis. 2018, 9, 595. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Templin, A.T.; Hogan, M.F.; Esser, N.; Zraika, S.; Hull, R.L.; Kahn, S.E. Evidence for Necroptosis as a Mechanism of Islet Amyloid–Induced Beta-Cell Death. Diabetes 2018, 67 (Suppl. S1), 82. [Google Scholar] [CrossRef]
- Sha, W.; Hu, F.; Xi, Y.; Chu, Y.; Bu, S. Mechanism of Ferroptosis and Its Role in Type 2 Diabetes Mellitus. J. Diabetes Res. 2021, 2021, e9999612. [Google Scholar] [CrossRef]
- Jehn, M.; Clark, J.M.; Guallar, E. Serum Ferritin and Risk of the Metabolic Syndrome in U.S. Adults. Diabetes Care 2004, 27, 2422–2428. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Berghe, T.V.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef]
- Yang, B.; Maddison, L.A.; Zaborska, K.E.; Dai, C.; Yin, L.; Tang, Z.; Zang, L.; Jacobson, D.A.; Powers, A.C.; Chen, W. RIPK3-mediated inflammation is a conserved β cell response to ER stress. Sci. Adv. 2020, 6, eabd7272. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Steinke, J.; Taylor, K.W. Viruses and the Etiology of Diabetes. Diabetes 1974, 23, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Filippi, C.M.; von Herrath, M.G. Viral Trigger for Type 1 Diabetes: Pros and Cons. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Cain, H.; Gerstenkorn, B. Karyological, karyometric and histochemical studies on cell death and necrosis in liver implant in rats. Beitr. Pathol. Anat. 1962, 126, 426–453. [Google Scholar] [PubMed]
- Patrlck, R.I.; Kroe, D.J.; Klavins, J.V. Renal papillary necrosis induced by heterologous serum. Arch. Pathol. 1964, 78, 108–113. [Google Scholar] [PubMed]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Programmed Cell Death (Apoptosis). Mol. Biol. Cell 4th Ed. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26873/ (accessed on 7 October 2021).
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Old, L.J. Tumor Necrosis Factor (TNF). Science 1985, 230, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, M.B. The role of signal transduction in the delayed necrosis of the hippocampal CA1 pyramidal cells following transient ischemia. Acta Neurol. Scand. Suppl. 1993, 143, 1–20. [Google Scholar] [PubMed]
- Kitanaka, C.; Kuchino, Y. Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ. 1999, 6, 508–515. [Google Scholar] [CrossRef]
- Kraupp, B.G.; Ruttkay-Nedecky, B.; Koudelka, H.; Bukowska, K.; Bursch, W.; Schulte-Hermann, R. In situ detection of fragmented dna (tunel assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: A cautionary note. Hepatology 1995, 21, 1465–1468. [Google Scholar] [CrossRef]
- Kelly, K.J.; Sandoval, R.M.; Dunn, K.W.; Molitoris, B.A.; Dagher, P.C. A novel method to determine specificity and sensitivity of the TUNEL reaction in the quantitation of apoptosis. Am. J. Physiol. Physiol. 2003, 284, C1309–C1318. [Google Scholar] [CrossRef] [PubMed]
- Atale, N.; Gupta, S.; Yadav, U.; Rani, V. Cell-death assessment by fluorescent and nonfluorescent cytosolic and nuclear staining techniques—ATALE—2014. J. Microsc. 2014, 255, 7–19. Available online: https://onlinelibrary.wiley.com/doi/10.1111/jmi.12133 (accessed on 7 October 2021). [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Galluzzi, L.; Aaronson, S.A.; Abrams, J.; Alnemri, E.S.; Andrews, D.W.; Baehrecke, E.H.; Bazan, N.G.; Blagosklonny, M.V.; Blomgren, K.; Borner, C.; et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009, 16, 1093–1107. [Google Scholar] [CrossRef]
- Orozco, S.L.; Yatim, N.; Werner, M.R.; Tran, H.; Gunja, S.Y.; Tait, S.W.G.; Albert, M.L.; Green, D.R.; Oberst, A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014, 21, 1511–1521. [Google Scholar] [CrossRef]
- Shi, H.; Kwok, R.T.K.; Liu, J.; Xing, B.; Tang, B.Z.; Liu, B. Real-Time Monitoring of Cell Apoptosis and Drug Screening Using Fluorescent Light-Up Probe with Aggregation-Induced Emission Characteristics. J. Am. Chem. Soc. 2012, 134, 17972–17981. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Cui, W.; Wang, W.; Zhang, H.; Liu, L.; Zhang, Z.; Li, Z.; Ying, G.; Zhang, N.; et al. Visualization of caspase-3-like activity in cells using a genetically encoded fluorescent biosensor activated by protein cleavage. Nat. Commun. 2013, 4, 2157. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Hamilton, C.A.; Bhojani, M.S.; Lee, K.C.; Dlugosz, A.; Ross, B.D.; Rehemtulla, A. A Transgenic Mouse for Imaging Caspase-Dependent Apoptosis within the Skin. J. Investig. Dermatol. 2010, 130, 1797–1806. [Google Scholar] [CrossRef]
- Tang, H.L.; Fung, M.C.; Hardwick, J.M. In vivo CaspaseTracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci. Rep. 2015, 5, 9015. [Google Scholar] [CrossRef] [PubMed]
- Murai, S.; Yamaguchi, Y.; Shirasaki, Y.; Yamagishi, M.; Shindo, R.; Hildebrand, J.M.; Miura, R.; Nakabayashi, O.; Totsuka, M.; Tomida, T.; et al. A FRET biosensor for necroptosis uncovers two different modes of the release of DAMPs. Nat. Commun. 2018, 9, 4457. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, N.; Lin, L.; Contreras, C.J.; Templin, A.T. β-Cell Death in Diabetes: Past Discoveries, Present Understanding, and Potential Future Advances. Metabolites 2021, 11, 796. https://doi.org/10.3390/metabo11110796
Mukherjee N, Lin L, Contreras CJ, Templin AT. β-Cell Death in Diabetes: Past Discoveries, Present Understanding, and Potential Future Advances. Metabolites. 2021; 11(11):796. https://doi.org/10.3390/metabo11110796
Chicago/Turabian StyleMukherjee, Noyonika, Li Lin, Christopher J. Contreras, and Andrew T. Templin. 2021. "β-Cell Death in Diabetes: Past Discoveries, Present Understanding, and Potential Future Advances" Metabolites 11, no. 11: 796. https://doi.org/10.3390/metabo11110796
APA StyleMukherjee, N., Lin, L., Contreras, C. J., & Templin, A. T. (2021). β-Cell Death in Diabetes: Past Discoveries, Present Understanding, and Potential Future Advances. Metabolites, 11(11), 796. https://doi.org/10.3390/metabo11110796