
Metabolic Tumor Microenvironment Characterization of Contrast Enhancing Brain Tumors Using Physiologic MRI

 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
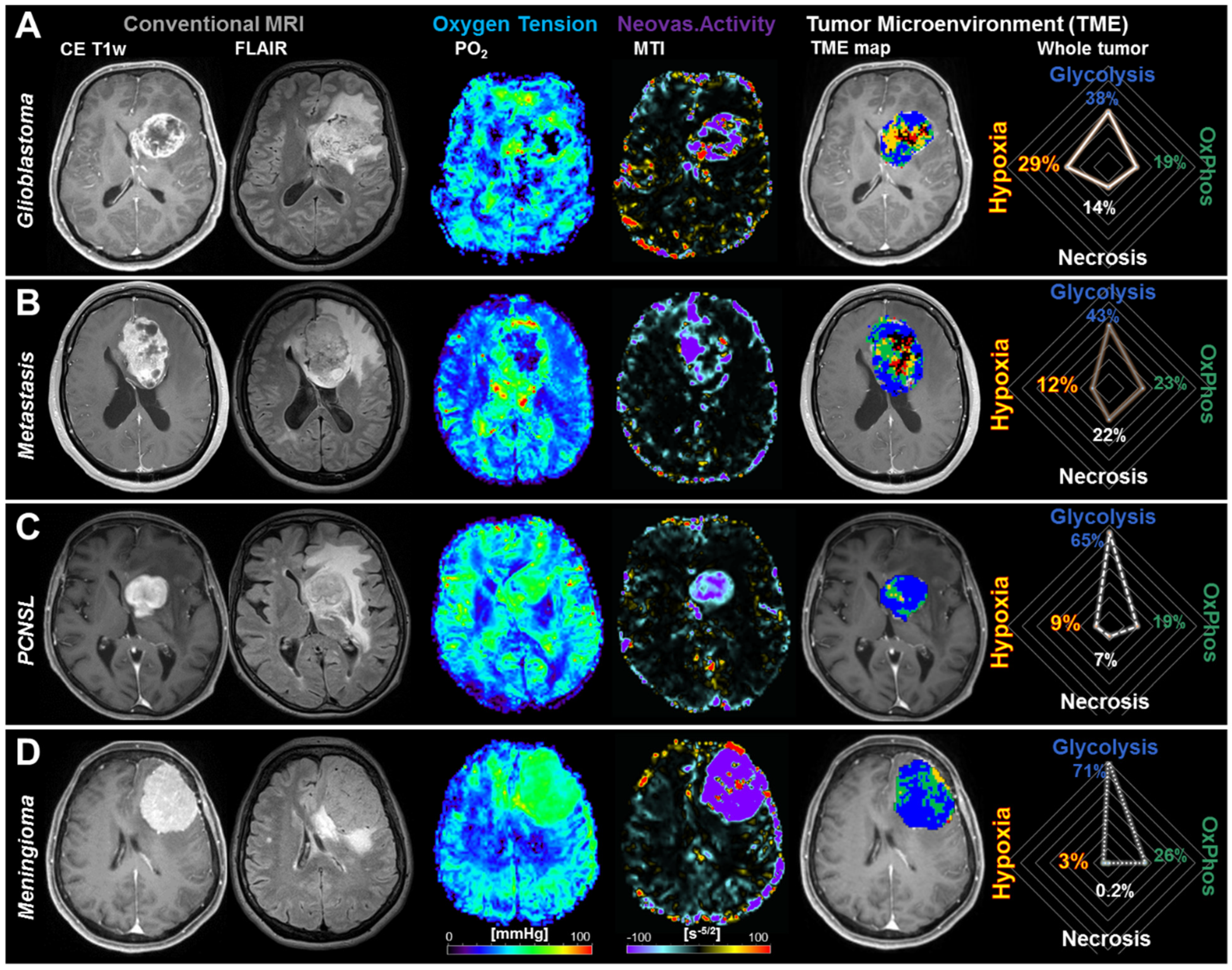
2.2. Physiological MRI and TME Mapping of Contrast-Enhancing Brain Tumors
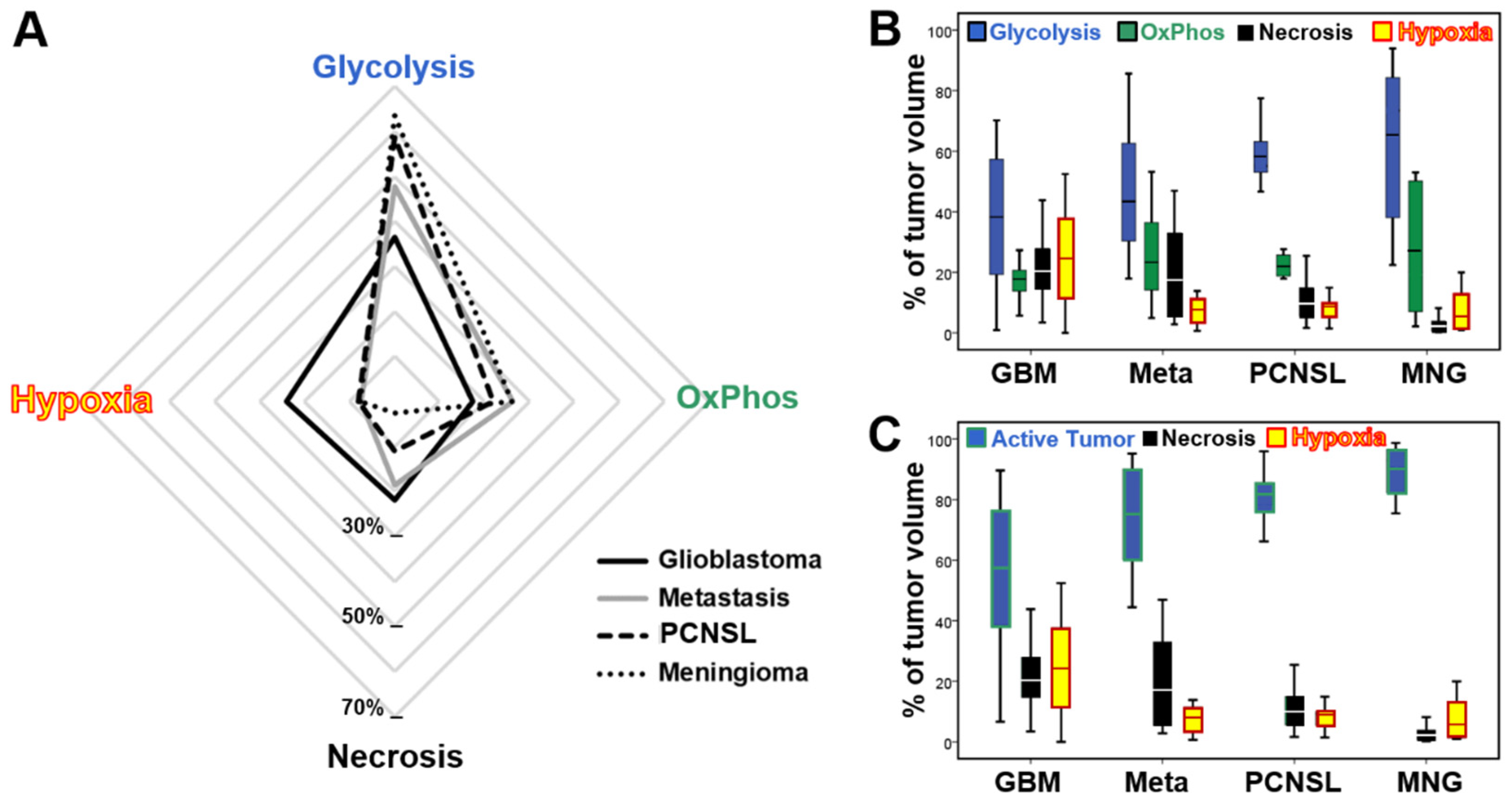
2.3. Differences in the Tumor Microenvironment between Contrast-Enhancing Brain Tumors
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. MRI Data Acquisition
4.3. MRI Data Processing
4.4. Calculation of MRI Biomarkers
4.5. Tumor Microenvironment Mapping
- Hypoxia without neovascularization or with dysfunctional tumor vasculature for voxels with high OEF, normal CMRO2 (associated with a low PO2 accordingly to Equation (4)), and low MTI: Red diamonds in the OEF-CMRO2-scatterplot and red voxels in the TME map (right-hand side in Figure 3).
- Hypoxia combined with neovascularization activity for voxels with normal to low OEF, high CMRO2 (associated with a low PO2), and high MTI: yellow diamonds in the OEF-CMRO2-scatterplot and yellow voxels in the TME map (right-hand side in Figure 3).
- Necrosis for voxels with very low CMRO2 and high OEF combined with highly defective tumor vasculature: Black crosses in the OEF-CMRO2-scatterplot and black voxels in the TME map (right-hand side in Figure 3).
- OxPhos for voxels with normal to low OEF, high CMRO2 (associated with normal PO2), and functional tumor neovasculature, under the assumption of predominantly mitochondrial oxidative phosphorylation for energy production: Green squares in the OEF-CMRO2-scatterplot and green voxels in the TME map (right-hand side in Figure 3).
- Glycolysis for voxels with low OEF, low CMRO2, (associated with high PO2), and functional tumor neovasculature, under the assumption of predominantly cytosolic aerobe glycolysis by the Warburg effect for energy production: Blue circles in the OEF-CMRO2-scatterplot and blue voxels in the TME map (right-hand side in Figure 3).
4.6. Quantitative and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- George, D.N.; Jehlol, H.B.; Oleiwi, S.A. Brain Tumor Detection Using Shape features and Machine Learning Algorithms Diagnosing ASD using machine learning View project Brain Tumor Detection Using Shape features and Machine Learning Algorithms. Int. J. Sci. Eng. Res. 2015, 6, 454–459. [Google Scholar]
- Villano, J.L.; Koshy, M.; Shaikh, H.; Dolecek, T.A.; McCarthy, B.J. Age, gender, and racial differences in incidence and survival in primary CNS lymphoma. Br. J. Cancer 2011, 105, 1414–1418. [Google Scholar] [CrossRef]
- Bernstein, S.H.; Unger, J.M.; LeBlanc, M.; Friedberg, J.; Miller, T.P.; Fisher, R.I. Natural history of cns relapse in patients with aggressive non-hodgkin’s lymphoma: A 20-year follow-up analysis of swog 8516-the southwest oncology group. J. Clin. Oncol. 2009, 27, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, T.; Abrey, L.E. Current Management of Metastatic Brain Disease. Neurotherapeutics 2009, 6, 598–603. [Google Scholar] [CrossRef] [Green Version]
- Sherwood, P.R.; Stommel, M.; Murman, D.L.; Given, C.W.; Given, B.A. Primary malignant brain tumor incidence and Medicaid enrollment. Neurology 2004, 62, 1788–1793. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; McCulloh, C.; Chen, Y.; Devine, K.; Wolinsky, Y.; Davitkov, P.; Robbins, S.; Cherukuri, R.; Patel, A.; Gupta, R.; et al. Family History of Cancer in Benign Brain Tumor Subtypes Versus Gliomas. Front. Oncol. 2012, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holleczek, B.; Zampella, D.; Urbschat, S.; Sahm, F.; von Deimling, A.; Oertel, J.; Ketter, R. Incidence, mortality and outcome of meningiomas: A population-based study from Germany. Cancer Epidemiol. 2019, 62, 101562. [Google Scholar] [CrossRef]
- Abe, T.; Mizobuchi, Y.; Nakajima, K.; Otomi, Y.; Irahara, S.; Obama, Y.; Majigsuren, M.; Khashbat, D.; Kageji, T.; Nagahiro, S.; et al. Diagnosis of brain tumors using dynamic contrast-enhanced perfusion imaging with a short acquisition time. Springerplus 2015, 4, 88. [Google Scholar] [CrossRef] [Green Version]
- Mukundan, S.; Holder, C.; Olson, J.J. Neuroradiological assessment of newly diagnosed glioblastoma. J. Neurooncol. 2008, 89, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, K.; Gideon, P.; Wagn, P.; Hansen, U.; Thomsen, C.; Madsen, F. MR diffusion imaging of human intracranial tumours. Neuroradiology 1997, 39, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Bataille, B.; Delwail, V.; Menet, E.; Vandermarcq, P.; Ingrand, P.; Wager, M.; Guy, G.; Lapierre, F. Primary intracerebral malignant lymphoma: Report of 248 cases. J. Neurosurg. 2000, 92, 261–266. [Google Scholar] [CrossRef]
- Marko, N.F.; Weil, R.J.; Schroeder, J.L.; Lang, F.F.; Suki, D.; Sawaya, R.E. Extent of resection of glioblastoma revisited: Personalized survival modeling facilitates more accurate survival prediction and supports a maximum-safe-resection approach to surgery. J. Clin. Oncol. 2014, 32, 774–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; van den Bent, M.; Hopkins, K.; Tonn, J.C.; Stupp, R.; Falini, A.; Cohen-Jonathan-Moyal, E.; Frappaz, D.; Henriksson, R.; Balana, C.; et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014, 15, e395–e403. [Google Scholar] [CrossRef] [Green Version]
- Cha, S. Update on brain tumor imaging: From anatomy to physiology. AJNR Am. J. Neuroradiol. 2006, 27, 475–487. [Google Scholar]
- Pasricha, S.; Gupta, A.; Gawande, J.; Trivedi, P.; Patel, D. Primary central nervous system lymphoma: A study of clinicopathological features and trend in western India. Indian J. Cancer 2011, 48, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Sperduto, P.W.; Chao, S.T.; Sneed, P.K.; Luo, X.; Suh, J.; Roberge, D.; Bhatt, A.; Jensen, A.W.; Brown, P.D.; Shih, H.; et al. Diagnosis-Specific Prognostic Factors, Indexes, and Treatment Outcomes for Patients With Newly Diagnosed Brain Metastases: A Multi-Institutional Analysis of 4259 Patients. Int. J. Radiat. Oncol. 2010, 77, 655–661. [Google Scholar] [CrossRef]
- Omuro, A. Glioblastoma and Other Malignant Gliomas. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Gaudy-Marqueste, C.; Carron, R.; Delsanti, C.; Loundou, A.; Monestier, S.; Archier, E.; Richard, M.A.; Regis, J.; Grob, J.J. On demand Gamma-Knife strategy can be safely combined with BRAF inhibitors for the treatment of melanoma brain metastases. Ann. Oncol. 2014, 25, 2086–2091. [Google Scholar] [CrossRef] [PubMed]
- Olivero, W.C.; Lister, J.R.; Elwood, P.W. The natural history and growth rate of asymptomatic meningiomas: A review of 60 patients. J. Neurosurg. 1995, 83, 222–224. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Vartanian, A.; Singh, S.K.; Agnihotri, S.; Jalali, S.; Burrell, K.; Aldape, K.D.; Zadeh, G. GBM’s multifaceted landscape: Highlighting regional and microenvironmental heterogeneity. Neuro. Oncol. 2014, 16, 1167–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.; Cantley, L.; Thompson, C. Understanding the Warburg effect: The metabolic Requiremetns of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Walker-Samuel, S.; Ramasawmy, R.; Torrealdea, F.; Rega, M.; Rajkumar, V.; Johnson, S.P.; Richardson, S.; Gonçalves, M.; Parkes, H.G.; Årstad, E.; et al. In vivo imaging of glucose uptake and metabolism in tumors. Nat. Med. 2013, 19, 1067–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hangel, G.; Jain, S.; Springer, E.; Hečková, E.; Strasser, B.; Považan, M.; Gruber, S.; Widhalm, G.; Kiesel, B.; Furtner, J.; et al. High-resolution metabolic mapping of gliomas via patch-based super-resolution magnetic resonance spectroscopic imaging at 7T. Neuroimage 2019, 191, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, A.; Zimmermann, M.; Doerfler, A.; Oberndorfer, S.; Buchfelder, M.; Coras, R.; Kitzwögerer, M.; Roessler, K. Intratumoral heterogeneity of oxygen metabolism and neovascularization uncovers 2 survival-relevant subgroups of IDH1 wild-type glioblastoma. Neuro. Oncol. 2018, 20, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Schulze, A.; Warburg, O.; Flier, J.; Mueckler, M.; Usher, P.; Lodish, H.; Osthus, R.; Shim, H.; Kim, S.; et al. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Svolos, P.; Tsolaki, E.; Kapsalaki, E.; Theodorou, K.; Fountas, K.; Fezoulidis, I.; Tsougos, I. Investigating brain tumor differentiation with diffusion and perfusion metrics at 3T MRI using pattern recognition techniques. Magn. Reson. Imaging 2013, 31, 1567–1577. [Google Scholar] [CrossRef]
- Tsougos, I.; Svolos, P.; Kousi, E.; Fountas, K.; Theodorou, K.; Fezoulidis, I.; Kapsalaki, E. Differentiation of glioblastoma multiforme from metastatic brain tumor using proton magnetic resonance spectroscopy, diffusion and perfusion metrics at 3 T. Cancer Imaging 2012, 12, 423–436. [Google Scholar] [CrossRef]
- Chiang, I.C.; Kuo, Y.T.; Lu, C.Y.; Yeung, K.W.; Lin, W.C.; Sheu, F.O.; Liu, G.C. Distinction between high-grade gliomas and solitary metastases using peritumoral 3-T magnetic resonance spectroscopy, diffusion, and perfusion imagings. Neuroradiology 2004, 46, 619–627. [Google Scholar] [CrossRef]
- Yamasaki, F.; Kurisu, K.; Satoh, K.; Arita, K.; Sugiyama, K.; Ohtaki, M.; Takaba, J.; Tominaga, A.; Hanaya, R.; Yoshioka, H.; et al. Apparent diffusion coefficient of human brain tumors at MR imaging. Radiology 2005, 235, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Heiland, S.; Harting, I.; Tronnier, V.M.; Sommer, C.; Ludwig, R.; Sartor, K. Distinguishing of primary cerebral lymphoma from high-grade glioma with perfusion-weighted magnetic resonance imaging. Neurosci. Lett. 2003, 338, 119–122. [Google Scholar] [CrossRef]
- Xing, Z.; You, R.X.; Li, J.; Liu, Y.; Cao, D.R. Differentiation of Primary Central Nervous System Lymphomas from High-Grade Gliomas by rCBV and Percentage of Signal Intensity Recovery Derived from Dynamic Susceptibility-Weighted Contrast-Enhanced Perfusion MR Imaging. Clin. Neuroradiol. 2014, 24, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.D.; Baird, G.L.; Bell, L.C.; Quarles, C.C.; Boxerman, J.L. Utility of percentage signal recovery and baseline signal in DSC-MRI optimized for relative cbv measurement for differentiating glioblastoma, lymphoma, metastasis, and meningioma. Am. J. Neuroradiol. 2019, 40, 1445–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangla, R.; Kolar, B.; Zhu, T.; Zhong, J.; Almast, J.; Ekholm, S. Percentage signal recovery derived from MR dynamic susceptibility contrast imaging is useful to differentiate common enhancing malignant lesions of the brain. Am. J. Neuroradiol. 2011, 32, 1004–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, L.; Crasto, S.G.; Moruno, P.G.; Cassoni, P.; Rudà, R.; Boccaletti, R.; Brosio, M.; De Lucchi, R.; Fava, C. Impiego delle tecniche di diffusione e di perfusione con RM nella caratterizzazione dei tumori cerebrali. Radiol. Medica 2009, 114, 645–659. [Google Scholar] [CrossRef]
- Cindil, E.; Sendur, H.N.; Cerit, M.N.; Dag, N.; Erdogan, N.; Celebi, F.E.; Oner, Y.; Tali, T. Validation of combined use of DWI and percentage signal recovery-optimized protocol of DSC-MRI in differentiation of high-grade glioma, metastasis, and lymphoma. Neuroradiology 2021, 63, 331–342. [Google Scholar] [CrossRef]
- Iv, M.; Yoon, B.C.; Heit, J.J.; Fischbein, N.; Wintermark, M. Current Clinical State of Advanced Magnetic Resonance Imaging for Brain Tumor Diagnosis and Follow Up. Semin. Roentgenol. 2018, 53, 45–61. [Google Scholar] [CrossRef]
- Bisdas, S.; Kirkpatrick, M.; Giglio, P.; Welsh, C.; Spampinato, M.V.; Rumboldt, Z. Cerebral Blood Volume Measurements by Perfusion-Weighted MR Imaging in Gliomas: Ready for Prime Time in Predicting Short-Term Outcome and Recurrent Disease? Am. J. Neuroradiol. 2009, 30, 681–688. [Google Scholar] [CrossRef] [Green Version]
- Mikkilineni, L.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Sprandio, J.; Avena, P.; Cotzia, P.; Dulau-Florea, A.; Gong, J.; Uppal, G.; Zhan, T.; et al. Hodgkin lymphoma: A complex metabolic ecosystem with glycolytic reprogramming of the tumor microenvironment. Semin. Oncol. 2017, 44, 218–225. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Weis, S.; Mayr, J.A.; Zimmermann, F.A.; Bogner, B.; Sperl, W.; Kofler, B. Alterations of oxidative phosphorylation in meningiomas and peripheral nerve sheath tumors. Neuro. Oncol. 2016, 18, 184–194. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R.; Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef] [Green Version]
- Stadlbauer, A.; Oberndorfer, S.; Zimmermann, M.; Renner, B.; Buchfelder, M.; Heinz, G.; Doerfler, A.; Kleindienst, A.; Roessler, K. Physiologic MR imaging of the tumor microenvironment revealed switching of metabolic phenotype upon recurrence of glioblastoma in humans. J. Cereb. Blood Flow Metab. 2020, 40, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, A.; Roessler, K.; Zimmermann, M.; Buchfelder, M.; Kleindienst, A.; Doerfler, A.; Heinz, G.; Oberndorfer, S. Predicting Glioblastoma Response to Bevacizumab Through MRI Biomarkers of the Tumor Microenvironment. Mol. Imaging Biol. 2019, 21, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Yablonskiy, D.A.; Haacke, E.M. Theory of NMR signal behavior in magnetically inhomogeneous tissues: The static dephasing regime. Magn. Reson. Med. 1994, 32, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarz, S.; Göttler, J.; Zimmer, C.; Hyder, F.; Preibisch, C. Characterizing white matter fiber orientation effects on multi-parametric quantitative BOLD assessment of oxygen extraction fraction. J. Cereb. Blood Flow Metab. 2020, 40, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, N.M.; Toth, V.; Förschler, A.; Kooijman, H.; Zimmer, C.; Preibisch, C. Technical considerations on the validity of blood oxygenation level-dependent-based MR assessment of vascular deoxygenation. NMR Biomed. 2014, 27, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Tóth, V.; Förschler, A.; Hirsch, N.M.; Den Hollander, J.; Kooijman, H.; Gempt, J.; Ringel, F.; Schlegel, J.; Zimmer, C.; Preibisch, C. MR-based hypoxia measures in human glioma. J. Neurooncol. 2013, 115, 197–207. [Google Scholar] [CrossRef]
- Vajkoczy, P.; Schilling, L.; Ullrich, A.; Schmiedek, P.; Menger, M.D. Characterization of angiogenesis and microcirculation of high-grade glioma: An intravital multifluorescence microscopic approach in the athymic nude mouse. J. Cereb. Blood Flow Metab. 1998, 18, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Valable, S.; Corroyer-Dulmont, A.; Chakhoyan, A.; Durand, L.; Toutain, J.; Divoux, D.; Barré, L.; MacKenzie, E.T.; Petit, E.; Bernaudin, M.; et al. Imaging of brain oxygenation with magnetic resonance imaging: A validation with positron emission tomography in the healthy and tumoural brain. J. Cereb. Blood Flow Metab. 2017, 37, 2584–2597. [Google Scholar] [CrossRef] [Green Version]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Christen, T.; Schmiedeskamp, H.; Straka, M.; Bammer, R.; Zaharchuk, G. Measuring brain oxygenation in humans using a multiparametric quantitative blood oxygenation level dependent MRI approach. Magn. Reson. Med. 2012, 68, 905–911. [Google Scholar] [CrossRef]
- Stadlbauer, A.; Zimmermann, M.; Heinz, G.; Oberndorfer, S.; Doerfler, A.; Buchfelder, M.; Rössler, K. Magnetic resonance imaging biomarkers for clinical routine assessment of microvascular architecture in glioma. J. Cereb. Blood Flow Metab. 2017, 37, 632–643. [Google Scholar] [CrossRef] [Green Version]
- Stadlbauer, A.; Zimmermann, M.; Kitzwögerer, M.; Oberndorfer, S.; Rössler, K.; Dörfler, A.; Buchfelder, M.; Heinz, G. MR Imaging–derived Oxygen Metabolism and Neovascularization Characterization for Grading and IDH Gene Mutation Detection of Gliomas. Radiology 2017, 283, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Stadlbauer, A.; Mouridsen, K.; Doerfler, A.; Bo Hansen, M.; Oberndorfer, S.; Zimmermann, M.; Buchfelder, M.; Heinz, G.; Roessler, K. Recurrence of glioblastoma is associated with elevated microvascular transit time heterogeneity and increased hypoxia. J. Cereb. Blood Flow Metab. 2018, 38, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essig, M.; Wenz, F.; Scholdei, R.; Brüning, R.; Berchtenbreiter, C.; Meurer, M.; Knopp, M.V. Dynamic susceptibility contrast-enhanced echo-planar imaging of cerebral gliomas. Effect of contrast medium extravasation. Acta Radiol. 2002, 43, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Bjornerud, A.; Emblem, K.E. A fully automated method for quantitative cerebral hemodynamic analysis using DSC-MRI. J. Cereb Blood Flow Metab 2010, 30, 1066–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; Grandin, C.B.; Duprez, T.; Mataigne, F.; Cosnard, G. Whole Brain Quantitative CBF, CBV, and MTT Measurements Using MRI Bolus Tracking: Implementation and Application to Data Acquired From Hyperacute Stroke Patients. J. Magn. Reson. Imaging 2000, 12, 400–410. [Google Scholar] [CrossRef]
- Preibisch, C.; Volz, S.; Anti, S.; Deichmann, R. Exponential excitation pulses for improved water content mapping in the presence of background gradients. Magn. Reson. Med. 2008, 60, 908–916. [Google Scholar] [CrossRef]
- Prasloski, T.; Mädler, B.; Xiang, Q.S.; MacKay, A.; Jones, C. Applications of stimulated echo correction to multicomponent T2 analysis. Magn. Reson. Med. 2012, 67, 1803–1814. [Google Scholar] [CrossRef]
- Boxerman, J.L.; Prah, D.E.; Paulson, E.S.; Machan, J.T.; Bedekar, D.; Schmainda, K.M. The role of preload and leakage correction in gadolinium-based cerebral blood volume estimation determined by comparison with MION as a criterion standard. Am. J. Neuroradiol. 2012, 33, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Boxerman, J.L.; Schmainda, K.M.; Weisskoff, R.M. Relative cerebral blood volume maps corrected for contrast agent extravasation significantly correlate with glioma tumor grade, whereas uncorrected maps do not. Am. J. Neuroradiol. 2006, 27, 859–867. [Google Scholar]
- Ducreux, D.; Buvat, I.; Meder, J.F.; Mikulis, D.; Crawley, A.; Fredy, D.; TerBrugge, K.; Lasjaunias, P.; Bittoun, J. Perfusion-weighted MR imaging studies in brain hypervascular diseases: Comparison of arterial input function extractions for perfusion measurement. AJNR Am. J. Neuroradiol 2006, 27, 1059–1069. [Google Scholar]
- Xu, C.; Kiselev, V.G.; Möller, H.E.; Fiebach, J.B. Dynamic hysteresis between gradient echo and spin echo attenuations in dynamic susceptibility contrast imaging. Magn. Reson. Med. 2013, 69, 981–991. [Google Scholar] [CrossRef]
- Stadlbauer, A.; Zimmermann, M.; Oberndorfer, S.; Dörfler, A.; Buchfelder, M.; Heinz, G.; Rössler, K. Vascular Hysteresis Loops and Vascular Architecture Mapping in Patients with Glioblastoma treated with Antiangiogenic Therapy. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gjedde, A. Cerebral blood flow change in arterial hypoxemia is consistent with negligible oxygen tension in brain mitochondria. Neuroimage 2002, 17, 1876–1881. [Google Scholar] [CrossRef]
- Vafaee, M.S.; Vang, K.; Bergersen, L.H.; Gjedde, A. Oxygen consumption and blood flow coupling in human motor cortex during intense finger tapping: Implication for a role of lactate. J. Cereb. Blood Flow Metab. 2012, 32, 1859–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennan, R.P.; Zhong, J.; Gore, J.C. Intravascular susceptibility contrast mechanisms in tissues. Magn. Reson. Med. 1994, 31, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Vafaee, M.S.; Gjedde, A. Model of blood-brain transfer of oxygen explains nonlinear flow-metabolism coupling during stimulation of visual cortex. J. Cereb. Blood Flow Metab. 2000, 20, 747–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, J.H.; Lu, H.; Inglese, M. Microvessel density estimation in the human brain by means of dynamic contrast-enhanced echo-planar imaging. Magn. Reson. Med. 2006, 56, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Emblem, K.E.; Mouridsen, K.; Bjornerud, A.; Farrar, C.T.; Jennings, D.; Borra, R.J.; Wen, P.Y.; Ivy, P.; Batchelor, T.T.; Rosen, B.R.; et al. Vessel architectural imaging identifies cancer patient responders to anti-angiogenic therapy. Nat. Med. 2013, 19, 1178–1183. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Availability, not respiratory capacity governs oxygen consumption of solid tumors. Int. J. Biochem. Cell Biol. 2012, 44, 1477–1481. [Google Scholar] [CrossRef]
- Combs, S.E.; Schmid, T.E.; Vaupel, P.; Multhoff, G. Stress Response Leading to Resistance in Glioblastoma-The Need for Innovative Radiotherapy (iRT) Concepts. Cancers 2016, 8, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreau, A.; Hafny-Rahbi, B.E.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Glioblastoma | Metastasis | PCNSL | Meningioma | |
|---|---|---|---|---|
| Aerobic Glycolysis | 37% ± 22% 1–70% | 48% ± 25% 18–86% | 59% ± 10% 47–77% | 63% ± 27% 22–94% |
| Oxidative Phosphorylation | 17% ± 6% 1–27% | 26% ± 17% 5–53% | 22% ± 4% 18–28% | 26% ± 22% 2–53% |
| Vital Tumor | 54% ± 24% 7–90% | 74% ± 18% 44–95% | 81% ± 10% 66–96% | 90% ± 9% 75–99% |
| Necrosis | 22% ± 11% 3–44% | 19% ± 17% 3–47% | 11% ± 8% 2–25% | 3% ± 3% 0–8% |
| Hypoxia with Neovascularization | 15% ± 10% 0–36% | 5% ± 4% 0–14% | 5% ± 4% 0–11% | 7% ± 7% 1–18% |
| Hypoxia without Neovascularization | 9% ± 7% 0–27% | 2% ± 2% 0–6% | 3% ± 2% 0–6% | 1% ± 1% 0–2% |
| Total Hypoxia | 24% ± 16% 0–52% | 7% ± 5% 1–14% | 8% ± 5% 1–15% | 8% ± 7% 1–20% |
| Conventional MRI Sequences | Physiological MRI Sequences | ||||||
|---|---|---|---|---|---|---|---|
| FLAIR | MPRAGE | DWI | GE-DSC | SE-DSC | R2 * Mapping | R2 Mapping | |
| In-plane resolution | 0.45 × 0.45 | 1.0 × 1.0 | 1.2 × 1.2 | 1.8 × 1.8 | 1.8 × 1.8 | 1.8 × 1.8 | 1.8 × 1.8 |
| Slice thickness [mm] | 3.0 | 1.0 | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 |
| Number of slices | 48 | 176 | 29 | 29 | 29 | 29 | 29 |
| TR [ms] | 5000 | 2100 | 5300 | 1740 | 1740 | 1210 | 3260 |
| TE [ms] | 460 | 2.3 | 98 | 22 | 33 | 5–40 ms | 13–104 ms |
| Flip angle * [°] | 120 | 12 | 90 | 90 | 90 | 90 | 90 |
| GRAPPA | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| other | TI = 1800 ms | b = 0 and 1000 s/mm2 | 60 dynamic volumes | 60 dynamic volumes | 8 echoes | 8 echoes | |
| Description | Color Code | CMRO2 Range | OEF Range | MTI Limit | MVD Limit | PO2 Limit |
|---|---|---|---|---|---|---|
| in TME Map | [µmol/100 g·min] | [%] | [s-5/2] | [mm-2] | [mmHg] | |
| Hypoxia without NV | red | >80 and <150 | >50 | >−5.0 and <5.0 | <250 | <10 |
| Hypoxia with NV | yellow | >150 | <50 | <−5.0 and >5.0 | >250 | <10 |
| Necrosis | black | <130 | >75 | >−5.0 and <5.0 | <250 | n.a. |
| OxPhos with NV | green | >70 | <50 | <−5.0 and >5.0 | >250 | 10−60 |
| Glycolysis with NV | blue | <150 | <20 | <−5.0 and >5.0 | >250 | >60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stadlbauer, A.; Marhold, F.; Oberndorfer, S.; Heinz, G.; Zimmermann, M.; Buchfelder, M.; Heynold, E.; Kinfe, T.M. Metabolic Tumor Microenvironment Characterization of Contrast Enhancing Brain Tumors Using Physiologic MRI. Metabolites 2021, 11, 668. https://doi.org/10.3390/metabo11100668
Stadlbauer A, Marhold F, Oberndorfer S, Heinz G, Zimmermann M, Buchfelder M, Heynold E, Kinfe TM. Metabolic Tumor Microenvironment Characterization of Contrast Enhancing Brain Tumors Using Physiologic MRI. Metabolites. 2021; 11(10):668. https://doi.org/10.3390/metabo11100668
Chicago/Turabian StyleStadlbauer, Andreas, Franz Marhold, Stefan Oberndorfer, Gertraud Heinz, Max Zimmermann, Michael Buchfelder, Elisabeth Heynold, and Thomas M. Kinfe. 2021. "Metabolic Tumor Microenvironment Characterization of Contrast Enhancing Brain Tumors Using Physiologic MRI" Metabolites 11, no. 10: 668. https://doi.org/10.3390/metabo11100668
APA StyleStadlbauer, A., Marhold, F., Oberndorfer, S., Heinz, G., Zimmermann, M., Buchfelder, M., Heynold, E., & Kinfe, T. M. (2021). Metabolic Tumor Microenvironment Characterization of Contrast Enhancing Brain Tumors Using Physiologic MRI. Metabolites, 11(10), 668. https://doi.org/10.3390/metabo11100668