
In Utero Exposure to Persistent Organic Pollutants and Childhood Lipid Levels



Abstract
1. Introduction
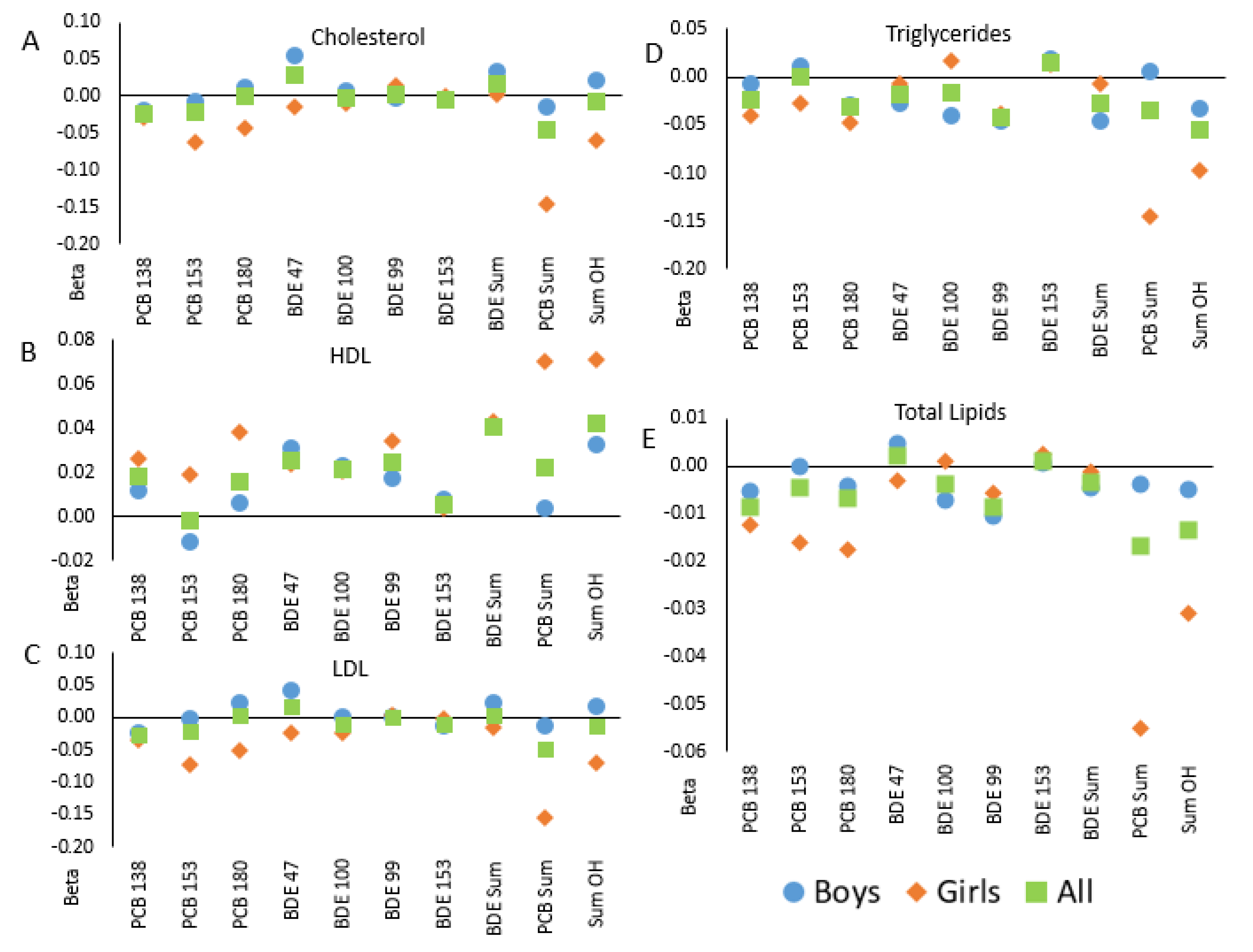
2. Results
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Data Collection
4.3. Exposure Assessment–PBDEs and PCBs in Utero and at 6–7
4.4. Outcome Assessment–Lipid Profiles in Children at Age 6–7
4.5. Additional Covariates in GESTE
4.6. Data Management and Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hites, R.A. Polybrominated diphenyl ethers in the environment and in people: A meta-analysis of concentrations. Environ. Sci. Technol. 2004, 38, 945–956. [Google Scholar] [CrossRef]
- Hurley, S.; Goldberg, D.; Nelson, D.O.; Guo, W.; Wang, Y.; Baek, H.G.; Park, J.S.; Petreas, M.; Bernstein, L.; Anton-Culver, H.; et al. Temporal Evaluation of Polybrominated Diphenyl Ether (PBDE) Serum Levels in Middle-Aged and Older California Women, 2011–2015. Environ. Sci. Technol. 2017, 51, 4697–4704. [Google Scholar] [CrossRef]
- Geyer, H.J.; Schramm, K.W.; Feicht, E.A.; Fried, K.W.; Henkelmann, B.; Lenoir, D.; Darnerud, P.O.; Aune, M.; Schmid, P.; McDonald, T.A. Terminal Elimination Half-lives of the Brominated Flame Retardants TBBPA, HBCD, and Lower Brominated PBDEs in Humans. Organohalogen Compd. 2004, 66, 3820–3825. [Google Scholar]
- Szabo, D.T.; Diliberto, J.J.; Hakk, H.; Huwe, J.K.; Birnbaum, L.S. Toxicokinetics of the flame retardant hexabromocyclododecane alpha: Effect of dose, timing, route, repeated exposure, and metabolism. Toxicol. Sci. 2011, 121, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Antignac, J.P.; Cariou, R.; Maume, D.; Marchand, P.; Monteau, F.; Zalko, D.; Berrebi, A.; Cravedi, J.-P.; Andre, F.; Le Bizec, B. Exposure assessment of fetus and newborn to brominated flame retardants in France: Preliminary data. Mol. Nutr. Food Res. 2008, 52, 258–265. [Google Scholar] [CrossRef]
- Herbstman, J.B.; Sjodin, A.; Apelberg, B.J.; Witter, F.R.; Patterson, D.G.; Halden, R.U.; Jones, R.S.; Park, A.; Zhang, Y.; Heidler, J.; et al. Determinants of prenatal exposure to polychlorinated biphenyls (PCBs) and polybrominated diphenyl ethers (PBDEs) in an urban population. Environ. Health Perspect. 2007, 115, 1794–1800. [Google Scholar] [CrossRef]
- Doucet, J.; Tague, B.; Arnold, D.L.; Cooke, G.M.; Hayward, S.; Goodyer, C.G. Persistent organic pollutant residues in human fetal liver and placenta from Greater Montreal, Quebec: A longitudinal study from 1998 through 2006. Environ. Health Perspect. 2009, 117, 605–610. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Kublbeck, J.; Niskanen, J.; Honkakoski, P. Metabolism-Disrupting Chemicals and the Constitutive Androstane Receptor CAR. Cells 2020, 9, 2306. [Google Scholar] [CrossRef]
- Helsley, R.N.; Sui, Y.; Ai, N.; Park, S.H.; Welsh, W.J.; Zhou, C. Pregnane X receptor mediates dyslipidemia induced by the HIV protease inhibitor amprenavir in mice. Mol. Pharmacol. 2013, 83, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Krausz, K.W.; Tanaka, N.; Gonzalez, F.J. Chronic exposure to rifaximin causes hepatic steatosis in pregnane X receptor-humanized mice. Toxicol. Sci. 2012, 129, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Spruiell, K.; Richardson, R.M.; Cullen, J.M.; Awumey, E.M.; Gonzalez, F.J.; Gyamfi, M.A. Role of pregnane X receptor in obesity and glucose homeostasis in male mice. J. Biol. Chem. 2014, 289, 3244–3261. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Sunaga, H.; Miyata, K.; Shirasaki, H.; Uchiyama, Y.; Shimba, S. Aryl Hydrocarbon Receptor Plays Protective Roles against High Fat Diet (HFD)-induced Hepatic Steatosis and the Subsequent Lipotoxicity via Direct Transcriptional Regulation of Socs3 Gene Expression. J. Biol. Chem. 2016, 291, 7004–7016. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.L.; Zhang, C.W.; Wang, L.; Tang, K.L.; Tanaka, N.; Gonzalez, F.J.; Xu, Y.; Fang, Z.-Z. Lipidomics reveal aryl hydrocarbon receptor (Ahr)-regulated lipid metabolic pathway in alpha-naphthyl isothiocyanate (ANIT)-induced intrahepatic cholestasis. Xenobiotica 2019, 49, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Clodfelter, K.H.; Holloway, M.G.; Hodor, P.; Park, S.H.; Ray, W.J.; Waxman, D.J. Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5b-dependent activation of male genes and repression of female genes revealed by microarray analysis. Mol. Endocrinol. 2006, 20, 1333–1351. [Google Scholar] [CrossRef]
- Rinn, J.L.; Rozowsky, J.S.; Laurenzi, I.J.; Petersen, P.H.; Zou, K.; Zhong, W.; Gerstein, W.; Snyder, M. Major molecular differences between mammalian sexes are involved in drug metabolism and renal function. Dev. Cell 2004, 6, 791–800. [Google Scholar] [CrossRef]
- Kwekel, J.C.; Desai, V.G.; Moland, C.L.; Branham, W.S.; Fuscoe, J.C. Age and sex dependent changes in liver gene expression during the life cycle of the rat. BMC Genom. 2010, 11, 675. [Google Scholar] [CrossRef]
- Zhang, Y.; Klein, K.; Sugathan, A.; Nassery, N.; Dombkowski, A.; Zanger, U.M.; Waxman, D.J. Transcriptional profiling of human liver identifies sex-biased genes associated with polygenic dyslipidemia and coronary artery disease. PLoS ONE 2011, 6, e23506. [Google Scholar] [CrossRef] [PubMed]
- Lau-Corona, D.; Suvorov, A.; Waxman, D.J. Feminization of male mouse liver by persistent growth hormone stimulation: Activation of sex-biased transcriptional networks and dynamic changes in chromatin states. Mol. Cell. Biol. 2017, 37, e00301-17. [Google Scholar] [CrossRef]
- Waxman, D.J.; Holloway, M.G. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol. Pharmacol. 2009, 76, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Dingemans, M.M.; Kock, M.; van den Berg, M. Mechanisms of Action Point Towards Combined PBDE/NDL-PCB Risk Assessment. Toxicol. Sci. 2016, 153, 215–224. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Khalil, A.; Parker, M.; Mpanga, R.; Cevik, E.C.; Thorburn, C.; Suvorov, A. Developmental Exposure to 2,2′,4,4′–Tetrabromodiphenyl Ether Induces Long-Lasting Changes in Liver Metabolism in Male Mice. J. Endocr. Soc. 2017, 1, 323–344. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yan, J.; Teng, M.; Yan, S.; Zhou, Z.; Zhu, W. In utero and lactational exposure to BDE-47 promotes obesity development in mouse offspring fed a high-fat diet: Impaired lipid metabolism and intestinal dysbiosis. Arch. Toxicol. 2018, 92, 1847–1860. [Google Scholar] [CrossRef]
- Suvorov, A.; Naumov, V.; Shtratnikova, V.; Logacheva, M.; Shershebnev, A.; Wu, H.; Gerasimov, E.; Zheludkevich, A.; Pilsner, J.R.; Sergeyev, O. Rat liver epigenome programing by perinatal exposure to 2,2′,4′4′-tetrabromodiphenyl ether. Epigenomics 2020, 12, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Cevik, S.E.; Hung, S.; Kolla, S.; Roy, M.A.; Suvorov, A. Developmental Exposure to 2,2′,4,4′-Tetrabromodiphenyl Ether Permanently Alters Blood-Liver Balance of Lipids in Male Mice. Front. Endocrinol. (Lausanne) 2018, 9, 548. [Google Scholar] [CrossRef]
- Suvorov, A.; Takser, L. Global Gene Expression Analysis in the Livers of Rat Offspring Perinatally Exposed to Low Doses of 2,2′,4,4′-tetrabromodiphenyl ether. Environ. Health Perspect. 2010, 118, 97–102. [Google Scholar] [CrossRef]
- Gomez, M.V.; Dutta, M.; Suvorov, A.; Shi, X.; Gu, H.; Mani, S.; Cui, J.Y. Early life exposure to environmental contaminants (BDE-47, TBBPA, and BPS) produced persistent alterations in fecal microbiome in adult male mice. Toxicol. Sci. 2020, 179, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Chowen, J.A.; Frago, L.M.; Argente, J. The regulation of GH secretion by sex steroids. Eur. J. Endocrinol. 2004, 151 (Suppl. 3), U95–U100. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Gemma, C.; Rakyan, V.K.; Holland, M.L. Sexually dimorphic gene expression emerges with embryonic genome activation and is dynamic throughout development. BMC Genom. 2015, 16, 295. [Google Scholar] [CrossRef] [PubMed]
- Serme-Gbedo, Y.K.; Abdelouahab, N.; Pasquier, J.C.; Cohen, A.A.; Takser, L. Maternal levels of endocrine disruptors, polybrominated diphenyl ethers, in early pregnancy are not associated with lower birth weight in the Canadian birth cohort GESTE. Environ. Health 2016, 15, 49. [Google Scholar] [CrossRef]
- Abdelouahab, N.; Langlois, M.F.; Lavoie, L.; Corbin, F.; Pasquier, J.C.; Takser, L. Maternal and cord-blood thyroid hormone levels and exposure to polybrominated diphenyl ethers and polychlorinated biphenyls during early pregnancy. Am. J. Epidemiol. 2013, 178, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Needham, L.L.; Barr, D.B.; Caudill, S.P.; Pirkle, J.L.; Turner, W.E.; Osterloh, J.; Jones, R.L.; Sampson, E.J. Concentrations of environmental chemicals associated with neurodevelopmental effects in U.S. population. Neurotoxicology 2005, 26, 531–545. [Google Scholar] [CrossRef]
- Shan, Q.; Li, H.; Chen, N.; Qu, F.; Guo, J. Understanding the Multiple Effects of PCBs on Lipid Metabolism. Diabetes Metab. Syndr. Obes. 2020, 13, 3691–3702. [Google Scholar] [CrossRef]
- De Boever, P.; Wens, B.; Boix, J.; Felipo, V.; Schoeters, G. Perinatal exposure to purity-controlled polychlorinated biphenyl 52, 138, or 180 alters toxicogenomic profiles in peripheral blood of rats after 4 months. Chem. Res. Toxicol. 2013, 26, 1159–1167. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Wahlang, B.; Falkner, K.C.; Clair, H.B.; Al-Eryani, L.; Prough, R.A.; States, J.C.; Coslo, D.M.; Omiecinski, C.J.; Cave, M.C. Human receptor activation by aroclor 1260, a polychlorinated biphenyl mixture. Toxicol. Sci. 2014, 140, 283–297. [Google Scholar] [CrossRef]
- Lin, Y.S.; Yasuda, K.; Assem, M.; Cline, C.; Barber, J.; Li, C.W.; Kholodovych, V.; Ai, N.; Chen, J.D.; Welsh, W.J.; et al. The major human pregnane X receptor (PXR) splice variant, PXR.2, exhibits significantly diminished ligand-activated transcriptional regulation. Drug Metab. Dispos. 2009, 37, 1295–1304. [Google Scholar] [CrossRef]
- Hardesty, J.E.; Wahlang, B.; Falkner, K.C.; Clair, H.B.; Clark, B.J.; Ceresa, B.P.; Prough, R.A.; Cave, M.C. Polychlorinated biphenyls disrupt hepatic epidermal growth factor receptor signaling. Xenobiotica 2017, 47, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Gigli, S.; Seguella, L.; Nobile, N.; D’Alessandro, A.; Pesce, M.; Capoccia, E.; Steardo, L.; Cirillo, C.; Cirillo, C.; et al. Rifaximin, a non-absorbable antibiotic, inhibits the release of pro-angiogenic mediators in colon cancer cells through a pregnane X receptor-dependent pathway. Int. J. Oncol. 2016, 49, 639–645. [Google Scholar] [CrossRef]
- Patton, H.M.; Sirlin, C.; Behling, C.; Middleton, M.; Schwimmer, J.B.; Lavine, J.E. Pediatric nonalcoholic fatty liver disease: A critical appraisal of current data and implications for future research. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 413–427. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef]
- Kohli, R.; Boyd, T.; Lake, K.; Dietrich, K.; Nicholas, L.; Balistreri, W.F.; Ebach, D.; Shashidhar, H.; Xanthakos, S.A. Rapid progression of NASH in childhood. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Xanthakos, S.A.; Lavine, J.E.; Yates, K.P.; Schwimmer, J.B.; Molleston, J.P.; Rosenthal, P.; Murray, K.F.; Vos, M.B.; Jain, A.K.; Scheimann, A.O.; et al. Progression of Fatty Liver Disease in Children Receiving Standard of Care Lifestyle Advice. Gastroenterology 2020, 159, 1731–1751.e10. [Google Scholar] [CrossRef]
- Holterman, A.X.; Guzman, G.; Fantuzzi, G.; Wang, H.; Aigner, K.; Browne, A.; Holterman, M. Nonalcoholic fatty liver disease in severely obese adolescent and adult patients. Obesity 2013, 21, 591–597. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Charatcharoenwitthaya, P.; Treeprasertsuk, S.; Benson, J.T.; Enders, F.B.; Angulo, P. The natural history of non-alcoholic fatty liver disease in children: A follow-up study for up to 20 years. Gut 2009, 58, 1538–1544. [Google Scholar] [CrossRef]
- Schisterman, E.F.; Whitcomb, B.W.; Louis, G.M.; Louis, T.A. Lipid adjustment in the analysis of environmental contaminants and human health risks. Environ. Health Perspect. 2005, 113, 853–857. [Google Scholar] [CrossRef]
- 52. Goncharov, A.; Haase, R.F.; Santiago-Rivera, A.; Morse, G.; Akwesasne Task Force on the Environment; McCaffrey, R.J.; Rej, R.; Carpenter, D.A. High serum PCBs are associated with elevation of serum lipids and cardiovascular disease in a Native American population. Environ. Res. 2008, 106, 226–239. [Google Scholar] [CrossRef]
- Lee, D.H.; Steffes, M.W.; Sjodin, A.; Jones, R.S.; Needham, L.L.; Jacobs, D.R., Jr. Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS ONE 2011, 6, e15977. [Google Scholar] [CrossRef]
- Hennig, B.; Reiterer, G.; Toborek, M.; Matveev, S.V.; Daugherty, A.; Smart, E.; Robertson, L.W. Dietary fat interacts with PCBs to induce changes in lipid metabolism in mice deficient in low-density lipoprotein receptor. Environ. Health Perspect. 2005, 113, 83–87. [Google Scholar] [CrossRef]
- Qin, W.P.; Li, C.H.; Guo, L.H.; Ren, X.M.; Zhang, J.Q. Binding and activity of polybrominated diphenyl ether sulfates to thyroid hormone transport proteins and nuclear receptors. Environ. Sci. Process. Impacts 2019, 21, 950–956. [Google Scholar] [CrossRef]
- Meerts, I.A.; van Zanden, J.J.; Luijks, E.A.; van Leeuwen-Bol, I.; Marsh, G.; Jakobsson, E.; Bergman, Å.; Brouwer, A. Potent competitive interactions of some brominated flame retardants and related compounds with human transthyretin in vitro. Toxicol. Sci. 2000, 56, 95–104. [Google Scholar] [CrossRef]
- Hamers, T.; Kamstra, J.H.; Sonneveld, E.; Murk, A.J.; Kester, M.H.; Andersson, P.L.; Legler, J.; Brouwer, A. In vitro profiling of the endocrine-disrupting potency of brominated flame retardants. Toxicol. Sci. 2006, 92, 157–173. [Google Scholar] [CrossRef]
- Staskal, D.F.; Hakk, H.; Bauer, D.; Diliberto, J.J.; Birnbaum, L.S. Toxicokinetics of polybrominated diphenyl ether congeners 47, 99, 100, and 153 in mice. Toxicol. Sci. 2006, 94, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Langsted, A.; Freiberg, J.J.; Nordestgaard, B.G. Fasting and nonfasting lipid levels: Influence of normal food intake on lipids, lipoproteins, apolipoproteins, and cardiovascular risk prediction. Circulation 2008, 118, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Mora, S.; Rifai, N.; Buring, J.E.; Ridker, P.M. Fasting compared with nonfasting lipids and apolipoproteins for predicting incident cardiovascular events. Circulation 2008, 118, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.F.; Cohn, J.S. Whither the lipid profile: Feast, famine, or no free lunch? Clin. Chem. 2011, 57, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M. Should we fast before we measure our lipids? Arch. Intern. Med. 2012, 172, 1705–1706. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Mora, S. Fasting for lipid testing: Is it worth the trouble? Arch. Intern. Med. 2012, 172, 1710–1712. [Google Scholar] [CrossRef]
- Sidhu, D.; Naugler, C. Fasting time and lipid levels in a community-based population: A cross-sectional study. Arch. Intern. Med. 2012, 172, 1707–1710. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Hilsted, L.; Stender, S. Plasma lipids in non-fasting patients and signal values of laboratory results. Ugeskr. Laeger. 2009, 171, 1093. [Google Scholar]
- Langsted, A.; Nordestgaard, B.G. Nonfasting lipids, lipoproteins, and apolipoproteins in individuals with and without diabetes: 58,434 individuals from the Copenhagen General Population Study. Clin. Chem. 2011, 57, 482–489. [Google Scholar] [CrossRef]
- National Clinical Guideline Centre (UK). Lipid Modification: Cardiovascular Risk Assessment and the Modification of Blood Lipids for the Primary and Secondary Prevention of Cardiovascular Disease. National Institute for Health and Care Excellence (UK); 2014. Available online: http://www.ncbi.nlm.nih.gov/books/NBK248067/ (accessed on 2 September 2021).
- Nordestgaard, B.G.; Langsted, A.; Mora, S.; Kolovou, G.; Baum, H.; Bruckert, E.; Watts, G.F.; Sypniewska, G.; Wiklund, O.; Borén, J.; et al. Fasting is not routinely required for determination of a lipid profile: Clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur. Heart J. 2016, 37, 1944–1958. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Staedelin, L.; Takser, L.; Abdelouahab, N.; Zhu, J. Assessment of selected chlorinated and brominated flame retardants in human plasma samples among co-residing family members. Environ. Pollut. 2019, 252, 1035–1041. [Google Scholar] [CrossRef]
- Laue, H.E.; Cassoulet, R.; Abdelouahab, N.; Serme-Gbedo, Y.K.; Desautels, A.S.; Brennan, K.J.M.; Bellenger, J.-P.; Burris, H.H.; Coull, B.A.; Weisskopf, M.G.; et al. Association between Meconium Acetaminophen and Childhood Neurocognitive Development in GESTE, a Canadian Cohort Study. Toxicol. Sci. 2019, 167, 138–144. [Google Scholar] [CrossRef]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009, 42, 377–381. [Google Scholar] [CrossRef]
- Covaci, A.; Voorspoels, S. Optimization of the determination of polybrominated diphenyl ethers in human serum using solid-phase extraction and gas chromatography-electron capture negative ionization mass spectrometry. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2005, 827, 216–223. [Google Scholar] [CrossRef]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef]
- Phillips, D.L.; Pirkle, J.L.; Burse, V.W.; Bernert, J.T., Jr.; Henderson, L.O.; Needham, L.L. Chlorinated hydrocarbon levels in human serum: Effects of fasting and feeding. Arch. Environ. Contam. Toxicol. 1989, 18, 495–500. [Google Scholar] [CrossRef]
- Bergonzi, R.; De Palma, G.; Tomasi, C.; Ricossa, M.C.; Apostoli, P. Evaluation of different methods to determine total serum lipids for normalization of circulating organochlorine compounds. Int. Arch. Occup. Environ. Health 2009, 82, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Planchart, A.; Green, A.; Hoyo, C.; Mattingly, C.J. Heavy Metal Exposure and Metabolic Syndrome: Evidence from Human and Model System Studies. Curr. Environ. Health Rep. 2018, 5, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Iglewicz, B.; Hoaglin, D. Volume 16, How to Detect and Handle Outliers. In The ASQC Basic References in Quality Control. Statistical Techniques; Mykytka, E.F., Ed.; ASQC Quality Press: Milwaukee, WI, USA, 1993; pp. 1–85. [Google Scholar]


{kind=link}
| Tertile 1, n = 51 (Lower-Upper) + | Tertile 2, n = 45 (Lower-Upper) + | Tertile 3, n = 51 (Lower-Upper) + | |||||
|---|---|---|---|---|---|---|---|
| 4.05 | 0.35 | 4.72 | 0.15 | 5.55 | 0.46 | ||
| Mean * | (SD) | Mean * | (SD) | Mean * | (SD) | p # | |
| Maternal age (years) at pregnancy | 30.3 | (3.9) | 30.0 | (4.4) | 27.8 | (4.9) | 0.009 |
| Maternal BMI after delivery | 26.7 | (4.5) | 26.6 | (5.3) | 27.4 | (6.2) | 0.76 |
| Child BMI at age 6–7 years | 15.8 | (1.4) | 15.9 | (1.5) | 15.9 | (1.9) | 0.95 |
| PCB Sum (ng/mL) | 0.090 | (3.34) | 0.086 | (2.71) | 0.066 | (4.63) | 0.43 |
| BDE Sum (ng/mL) | 0.135 | (3.56) | 0.123 | (3.66) | 0.155 | (3.93) | 0.68 |
| Triglycerides (mmol/L) | 0.697 | (1.39) | 0.794 | (1.36) | 1.393 | (1.43) | <0.0001 |
| Maternal serum lead levels at delivery (umol/L) | 0.037 | (1.46) | 0.037 | (1.62) | 0.041 | (1.71) | 0.35 |
| Maternal serum manganese levels at delivery (nmol/L) | 247.0 | (1.27) | 253.3 | (1.35) | 282.0 | (1.29) | 0.03 |
| Maternal serum cadmium levels at delivery (nmol/L) | 2.008 | (2.09) | 1.716 | (1.65) | 2.117 | (2.02) | 0.29 |
| Maternal serum mercury levels at delivery (μg/L) | 2.585 | (2.00) | 2.740 | (1.67) | 2.351 | (2.04) | 0.51 |
| Child serum lead levels at 6–7 years (umol/L) | 0.033 | (1.58) | 0.037 | (1.52) | 0.035 | (1.53) | 0.54 |
| Child serum manganese levels at 6–7 years (nmol/L) | 172.2 | (1.32) | 185.6 | (1.30) | 178.8 | (1.27) | 0.37 |
| Child serum cadmium levels at 6–7 years (nmol/L) | 0.800 | (1.41) | 0.795 | (1.31) | 0.803 | (1.27) | 0.99 |
| Child serum mercury levels at 6–7 years (μg/L) | 1.569 | (1.83) | 1.474 | (1.88) | 1.512 | (1.84) | 0.88 |
| % | % | % | |||||
| % Female child | 35.3% | 48.9% | 49.0% | 0.28 | |||
| % Reported smoking during pregnancy a | 6.0% | 0.0% | 7.8% | 0.18 | |||
| % Reported alcohol use during pregnancy a | 30.0% | 25.0% | 25.5% | 0.83 | |||
| % Vaginal delivery a | 82.4% | 75.6% | 84.3% | 0.53 | |||
| Maternal educational level | 0.42 | ||||||
| Primary | 0.0% | 0.0% | 2.0% | ||||
| Secondary | 9.8% | 8.9% | 19.6% | ||||
| College | 19.6% | 24.4% | 29.4% | ||||
| University | 35.3% | 42.2% | 27.5% | ||||
| Other | 31.3% | 22.2% | 21.6% | ||||
| Missing | 3.9% | 2.2% | 0.0% | ||||
| % below LOC | Minimum | 25% Q1 | 50% Median | 75% Q3 | Maximum | Q3 ÷ Q1 | |
|---|---|---|---|---|---|---|---|
| BDE 47 | 0% | 0.00035 | 0.020 | 0.05 | 0.11 | 1.05 | 5.6 |
| BDE 99 | 13% | 0.00005 | 0.004 | 0.02 | 0.06 | 0.64 | 14.1 |
| BDE 100 | 12% | 0.00005 | 0.010 | 0.02 | 0.06 | 0.43 | 5.8 |
| BDE 153 | 16% | 0.00005 | 0.008 | 0.03 | 0.06 | 0.87 | 8.3 |
| Sum BDEs | -- | 0.00241 | 0.064 | 0.14 | 0.34 | 2.49 | 5.3 |
| PCB 138 | 20% | 0.00001 | 0.002 | 0.02 | 0.04 | 0.27 | 18.1 |
| PCB 153 | 5% | 0.00001 | 0.024 | 0.05 | 0.07 | 0.41 | 3.0 |
| PCB 180 | 10% | 0.00001 | 0.009 | 0.03 | 0.05 | 0.26 | 4.8 |
| Sum PCBs | -- | 0.00003 | 0.039 | 0.09 | 0.16 | 0.91 | 4.1 |
| Sum of All Organohalogens | -- | 0.00244 | 0.14 | 0.27 | 0.55 | 2.64 | 3.9 |
| Bivariate Model | Multivariate Model + | ||||||
|---|---|---|---|---|---|---|---|
| Chemical | Beta | SE | p | Beta | SE | p | |
| Cholesterol a | PCB 138 | −0.028 | (0.014) | 0.04 | −0.024 | (0.014) | 0.09 |
| PCB 153 | −0.029 | (0.022) | ** | −0.022 | (0.022) | ** | |
| PCB 180 | −0.003 | (0.019) | ** | 0.001 | (0.018) | ** | |
| Sum PCB | −0.062 | (0.037) | 0.09 | −0.046 | (0.037) | ** | |
| BDE 47 | 0.020 | (0.032) | ** | 0.029 | (0.031) | ** | |
| BDE 100 | −0.005 | (0.020) | ** | −0.004 | (0.019) | ** | |
| BDE 99 | 0.002 | (0.018) | ** | 0.003 | (0.018) | ** | |
| BDE 153 | −0.009 | (0.017) | ** | −0.006 | (0.017) | ** | |
| Sum BDE | 0.011 | (0.036) | ** | 0.015 | (0.035) | ** | |
| Sum OH | −0.021 | (0.045) | ** | −0.007 | (0.044) | ** | |
| HDL b | PCB 138 | 0.018 | (0.010) | 0.06 | 0.018 | (0.010) | 0.07 |
| PCB 153 | −0.001 | (0.016) | ** | −0.002 | (0.016) | ** | |
| PCB 180 | 0.015 | (0.013) | ** | 0.015 | (0.013) | ** | |
| Sum PCB | 0.023 | (0.026) | ** | 0.022 | (0.027) | ** | |
| BDE 47 | 0.026 | (0.022) | ** | 0.025 | (0.022) | ** | |
| BDE 100 | 0.021 | (0.013) | ** | 0.021 | (0.014) | ** | |
| BDE 99 | 0.024 | (0.013) | 0.06 | 0.024 | (0.013) | 0.06 | |
| BDE 153 | 0.006 | (0.012) | ** | 0.005 | (0.012) | ** | |
| Sum BDE | 0.041 | (0.025) | ** | 0.041 | (0.025) | ** | |
| Sum OH | 0.043 | (0.031) | ** | 0.042 | (0.031) | ** | |
| LDL a | PCB 138 | −0.026 | (0.014) | 0.06 | −0.022 | (0.013) | ** |
| PCB 153 | −0.026 | (0.022) | ** | −0.022 | (0.022) | ** | |
| PCB 180 | 0.011 | (0.018) | ** | 0.014 | (0.018) | ** | |
| Sum PCB | −0.049 | (0.036) | ** | −0.037 | (0.036) | ** | |
| BDE 47 | 0.012 | (0.031) | ** | 0.022 | (0.030) | ** | |
| BDE 100 | −0.010 | (0.019) | ** | −0.007 | (0.019) | ** | |
| BDE 99 | 0.012 | (0.018) | ** | 0.014 | (0.018) | ** | |
| BDE 153 | −0.017 | (0.017) | ** | −0.013 | (0.016) | ** | |
| Sum BDE | 0.005 | (0.035) | ** | 0.011 | (0.034) | ** | |
| Sum OH | −0.008 | (0.043) | ** | 0.004 | (0.043) | ** | |
| Triglycerides b | PCB 138 | −0.026 | (0.011) | 0.02 | −0.023 | (0.011) | 0.04 |
| PCB 153 | −0.008 | (0.018) | ** | 0.000 | (0.018) | ** | |
| PCB 180 | −0.033 | (0.015) | 0.02 | −0.032 | (0.015) | 0.03 | |
| Sum PCB | −0.046 | (0.029) | ** | −0.035 | (0.030) | ** | |
| BDE 47 | −0.015 | (0.025) | ** | −0.018 | (0.025) | ** | |
| BDE 100 | −0.014 | (0.016) | ** | −0.016 | (0.015) | ** | |
| BDE 99 | −0.041 | (0.014) | 0.01 | −0.043 | (0.014) | 0.003 | |
| BDE 153 | 0.016 | (0.014) | ** | 0.015 | (0.014) | ** | |
| Sum BDE | −0.024 | (0.029) | ** | −0.028 | (0.028) | ** | |
| Sum OH | −0.060 | (0.035) | 0.09 | −0.055 | (0.035) | ** | |
| Total lipids b | PCB 138 | −0.010 | (0.004) | 0.01 | −0.009 | (0.004) | 0.02 |
| PCB 153 | −0.007 | (0.006) | ** | −0.004 | (0.006) | ** | |
| PCB 180 | −0.008 | (0.005) | ** | −0.007 | (0.005) | ** | |
| Sum PCB | −0.022 | (0.010) | 0.02 | −0.017 | (0.010) | 0.09 | |
| BDE 47 | 0.001 | (0.008) | ** | 0.002 | (0.008) | ** | |
| BDE 100 | −0.004 | (0.005) | ** | −0.004 | (0.005) | ** | |
| BDE 99 | −0.008 | (0.005) | 0.09 | −0.009 | (0.005) | 0.07 | |
| BDE 153 | 0.001 | (0.005) | ** | 0.001 | (0.005) | ** | |
| Sum BDE | −0.004 | (0.010) | ** | −0.004 | (0.009) | ** | |
| Sum OH | −0.017 | (0.012) | ** | −0.013 | (0.012) | ** | |
| Bivariate Model | Multivariate Model + | ||
|---|---|---|---|
| Chemical | p * | p * | |
| Cholesterol a | PCB 138 | 0.66 | 0.75 |
| PCB 153 | 0.36 | 0.34 | |
| PCB 180 | 0.34 | 0.22 | |
| Sum PCB | 0.16 | 0.14 | |
| BDE 47 | 0.16 | 0.18 | |
| BDE 100 | 0.47 | 0.55 | |
| BDE 99 | 0.98 | 0.75 | |
| BDE 153 | 0.94 | 0.99 | |
| Sum BDE | 0.48 | 0.52 | |
| Sum OH | 0.27 | 0.33 | |
| HDL b | PCB 138 | 0.56 | 0.56 |
| PCB 153 | 0.45 | 0.46 | |
| PCB 180 | 0.29 | 0.29 | |
| Sum PCB | 0.35 | 0.35 | |
| BDE 47 | 0.85 | 0.85 | |
| BDE 100 | 0.93 | 0.93 | |
| BDE 99 | 0.48 | 0.50 | |
| BDE 153 | 0.85 | 0.85 | |
| Sum BDE | 0.97 | 0.96 | |
| Sum OH | 0.62 | 0.63 | |
| LDL a | PCB 138 | 0.69 | 0.77 |
| PCB 153 | 0.24 | 0.21 | |
| PCB 180 | 0.16 | 0.08 | |
| Sum PCB | 0.15 | 0.13 | |
| BDE 47 | 0.17 | 0.19 | |
| BDE 100 | 0.34 | 0.41 | |
| BDE 99 | 0.70 | 0.92 | |
| BDE 153 | 0.91 | 0.85 | |
| Sum BDE | 0.42 | 0.46 | |
| Sum OH | 0.28 | 0.34 | |
| Triglycerides b | PCB 138 | 0.12 | 0.16 |
| PCB 153 | 0.25 | 0.32 | |
| PCB 180 | 0.46 | 0.56 | |
| Sum PCB | 0.02 | 0.02 | |
| BDE 47 | 0.76 | 0.76 | |
| BDE 100 | 0.07 | 0.08 | |
| BDE 99 | 0.996 | 0.88 | |
| BDE 153 | 0.86 | 0.80 | |
| Sum BDE | 0.50 | 0.55 | |
| Sum OH | 0.38 | 0.36 | |
| Total lipids b | PCB 138 | 0.28 | 0.37 |
| PCB 153 | 0.26 | 0.28 | |
| PCB 180 | 0.30 | 0.25 | |
| Sum PCB | 0.03 | 0.03 | |
| BDE 47 | 0.47 | 0.51 | |
| BDE 100 | 0.59 | 0.53 | |
| BDE 99 | 0.95 | 0.70 | |
| BDE 153 | 0.97 | 0.96 | |
| Sum BDE | 0.99 | 0.99 | |
| Sum OH | 0.23 | 0.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boutot, M.E.; Whitcomb, B.W.; Abdelouahab, N.; Baccarelli, A.A.; Boivin, A.; Caku, A.; Gillet, V.; Martinez, G.; Pasquier, J.-C.; Zhu, J.; et al. In Utero Exposure to Persistent Organic Pollutants and Childhood Lipid Levels. Metabolites 2021, 11, 657. https://doi.org/10.3390/metabo11100657
Boutot ME, Whitcomb BW, Abdelouahab N, Baccarelli AA, Boivin A, Caku A, Gillet V, Martinez G, Pasquier J-C, Zhu J, et al. In Utero Exposure to Persistent Organic Pollutants and Childhood Lipid Levels. Metabolites. 2021; 11(10):657. https://doi.org/10.3390/metabo11100657
Chicago/Turabian StyleBoutot, Maegan E., Brian W. Whitcomb, Nadia Abdelouahab, Andrea A. Baccarelli, Amélie Boivin, Artuela Caku, Virginie Gillet, Guillaume Martinez, Jean-Charles Pasquier, Jiping Zhu, and et al. 2021. "In Utero Exposure to Persistent Organic Pollutants and Childhood Lipid Levels" Metabolites 11, no. 10: 657. https://doi.org/10.3390/metabo11100657
APA StyleBoutot, M. E., Whitcomb, B. W., Abdelouahab, N., Baccarelli, A. A., Boivin, A., Caku, A., Gillet, V., Martinez, G., Pasquier, J.-C., Zhu, J., Takser, L., St-Cyr, L., & Suvorov, A. (2021). In Utero Exposure to Persistent Organic Pollutants and Childhood Lipid Levels. Metabolites, 11(10), 657. https://doi.org/10.3390/metabo11100657