Lipidomics Issues on Human Positive ssRNA Virus Infection: An Update




Abstract
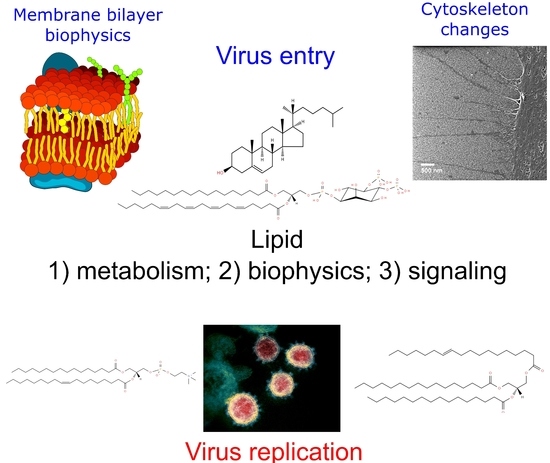
{kind=link}
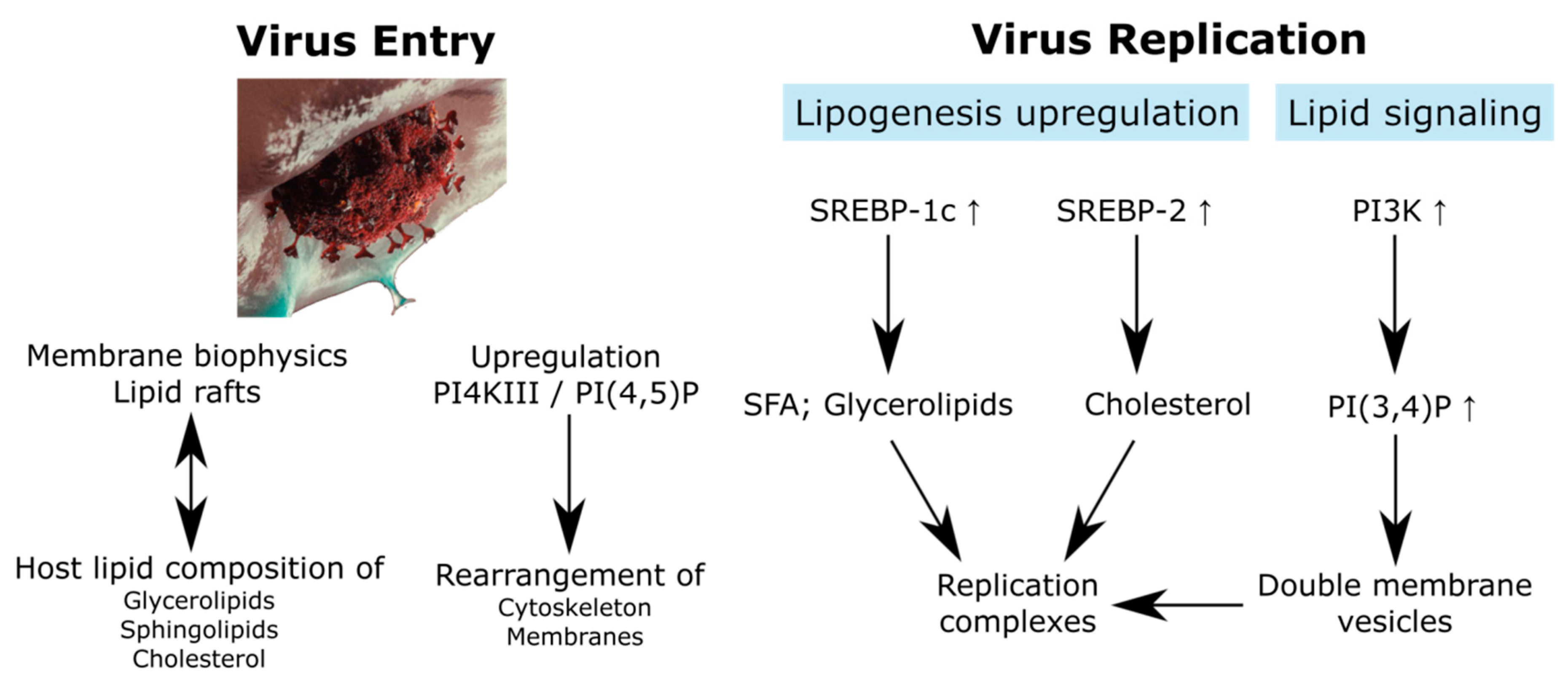{kind=link}
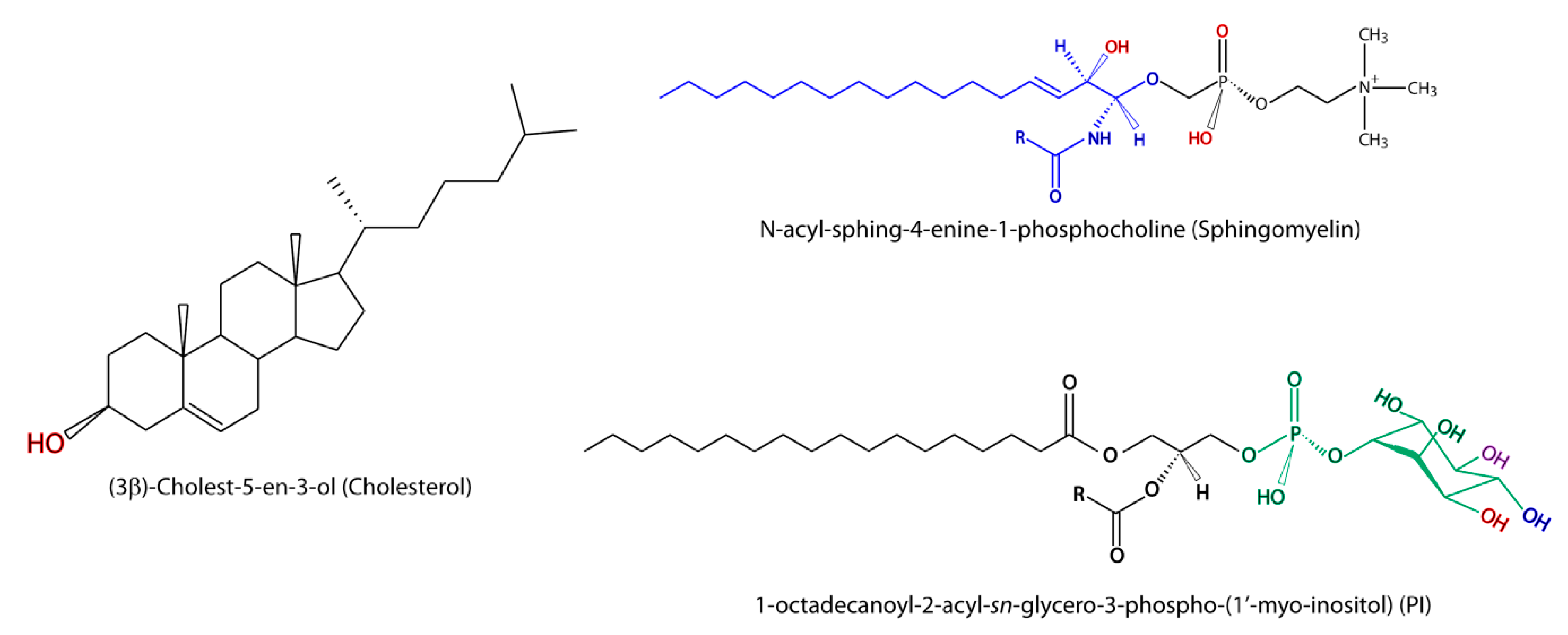{kind=link}
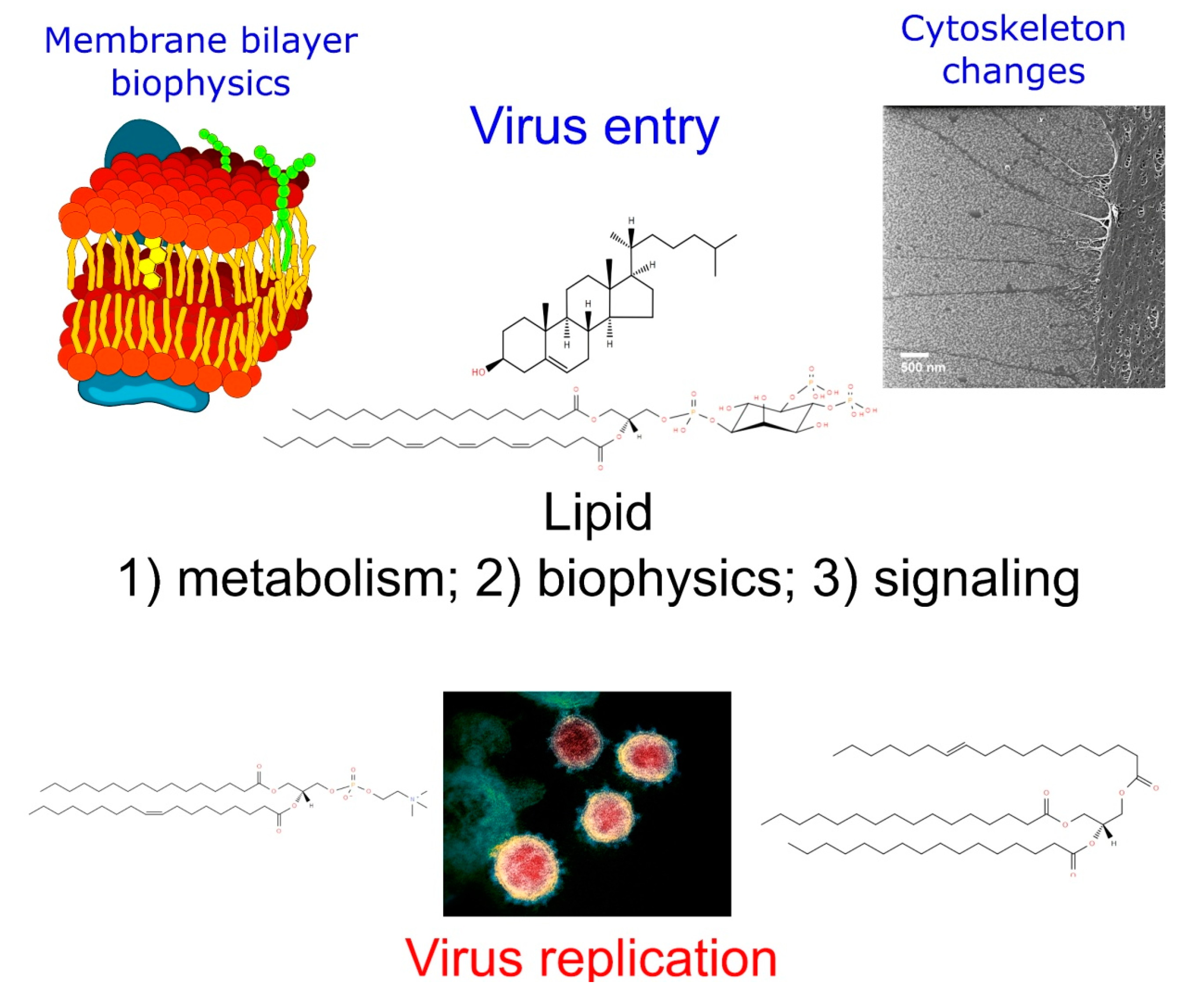{kind=link}
1. Introduction
2. Virus Entry: Lipid Rafts and Membrane Domains
2.1. Membrane Mechanical Properties Required for Virus Infection
2.2. Raft Lipids Related to Virus Entry
3. Lipid Regulation in Virus Replication: Viral Factories
3.1. Viral Replication Complexes
3.2. Lipid-Related Host Factors Associated to the RCs’ Buildup
4. Additional Pathways of Lipid Metabolism Affected in Virus Infection
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- World Health Organization. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 9 June 2020).
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Methods in Molecular Biology; Maier, H.J., Bickerton, E., Britton, P., Eds.; Springer: New York, NY, USA, 2015; Volume 1282, pp. 1–23. ISBN 978-1-4939-2437-0. [Google Scholar]
- Reid, C.R.; Airo, A.M.; Hobman, T.C. The Virus-Host Interplay: Biogenesis of +RNA Replication Complexes. Viruses 2015, 7, 4385–4413. [Google Scholar] [CrossRef] [PubMed]
- Novoa, R.R.; Calderita, G.; Arranz, R.; Fontana, J.; Granzow, H.; Risco, C. Virus factories: Associations of cell organelles for viral replication and morphogenesis. Biol. Cell 2005, 97, 147–172. [Google Scholar] [CrossRef]
- Marsh, M.; Helenius, A. Virus Entry: Open Sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Zheng, W.; Shan, Y.; Mu, Z.; Dominguez, S.R.; Holmes, K.V.; Qian, Z. Identification of the Fusion Peptide-Containing Region in Betacoronavirus Spike Glycoproteins. J. Virol. 2016, 90, 5586–5600. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, Q.; Yan, M.; Chen, X.-B.; Yang, H.; Zhou, W.; Rao, Z. Structural characterization of the HCoV-229E fusion core. Biochem. Biophys. Res. Commun. 2018, 497, 705–712. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Felmlee, D.J.; Baumert, T.F. Hepatitis C Virus Entry. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Bartenschlager, R., Ed.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2013; Volume 369, pp. 87–112. ISBN 978-3-642-27339-1. [Google Scholar]
- Zhu, Y.-Z.; Qian, X.-J.; Zhao, P.; Qi, Z.-T. How hepatitis C virus invades hepatocytes: The mystery of viral entry. World J. Gastroenterol. 2014, 20, 3457. [Google Scholar] [CrossRef]
- Lakadamyali, M.; Rust, M.J.; Zhuang, X. Endocytosis of influenza viruses. Microbes Infect. 2004, 6, 929–936. [Google Scholar] [CrossRef]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus Entry by Endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef]
- Robinson, M.; Schor, S.; Barouch-Bentov, R.; Einav, S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018, 75, 3693–3714. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yuan, X.; Sun, Y.; Mao, X.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Ding, C.; Liao, Y. Infectious bronchitis virus entry mainly depends on clathrin mediated endocytosis and requires classical endosomal/lysosomal system. Virology 2019, 528, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Che, B.; Guo, Z.; Li, C.; Liu, Y.; Wu, W.; Wang, S.; Li, D.; Cui, Z.; Liang, M. Single virus tracking of Ebola virus entry through lipid rafts in living host cells. Biosaf. Health 2020, 2, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.T.; Navis, G.; Van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Chigbu, D.G.I.; Loonawat, R.; Sehgal, M.; Patel, D.; Jain, P. Hepatitis C Virus Infection: Host–Virus Interaction and Mechanisms of Viral Persistence. Cells 2019, 8, 376. [Google Scholar] [CrossRef]
- Plemper, R.K. Cell entry of enveloped viruses. Curr. Opin. Virol. 2011, 1, 92–100. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef]
- Benhaim, M.A.; Lee, K.K. New Biophysical Approaches Reveal the Dynamics and Mechanics of Type I Viral Fusion Machinery and Their Interplay with Membranes. Viruses 2020, 12, 413. [Google Scholar] [CrossRef]
- Ivanovic, T.; Choi, J.L.; Whelan, S.P.; Van Oijen, A.M.; Harrison, S.C. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife 2013, 2, e00333. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.H.; Klein, D.E.; Schmidt, A.G.; Peña, J.M.; Harrison, S.C. Sequential conformational rearrangements in flavivirus membrane fusion. eLife 2014, 3, e04389. [Google Scholar] [CrossRef]
- Aeffner, S.; Reusch, T.; Weinhausen, B.; Salditt, T. Energetics of stalk intermediates in membrane fusion are controlled by lipid composition. Proc. Natl. Acad. Sci. USA 2012, 109, E1609–E1618. [Google Scholar] [CrossRef]
- Gui, L.; Ebner, J.L.; Mileant, A.; Williams, J.A.; Lee, K.K. Visualization and Sequencing of Membrane Remodeling Leading to Influenza Virus Fusion. J. Virol. 2016, 90, 6948–6962. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Freed, J.H. Two Conserved Residues Are Important for Inducing Highly Ordered Membrane Domains by the Transmembrane Domain of Influenza Hemagglutinin. Biophys. J. 2011, 100, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Basso, L.G.M.; Vicente, E.F.; Crusca, E.; Cilli, E.M.; Costa-Filho, A. SARS-CoV fusion peptides induce membrane surface ordering and curvature. Sci. Rep. 2016, 6, 37131. [Google Scholar] [CrossRef]
- Kuhl, T.; Guo, Y.; Alderfer, J.L.; Berman, A.D.; Leckband, D.; Israelachvili, J.; Hui, S.W. Direct Measurement of Polyethylene Glycol Induced Depletion Attraction between Lipid Bilayers. Langmuir 1996, 12, 3003–3014. [Google Scholar] [CrossRef]
- Mahajan, M.; Bhattacharjya, S. NMR structures and localization of the potential fusion peptides and the pre-transmembrane region of SARS-CoV: Implications in membrane fusion. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 721–730. [Google Scholar] [CrossRef]
- Carravilla, P.; Nieva, J.L.; Eggeling, C. Fluorescence Microscopy of the HIV-1 Envelope. Viruses 2020, 12, 348. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef]
- Earnest, J.T.; Hantak, M.P.; Park, J.-E.; Gallagher, T. Coronavirus and Influenza Virus Proteolytic Priming Takes Place in Tetraspanin-Enriched Membrane Microdomains. J. Virol. 2015, 89, 6093–6104. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Huang, M.; Yuan, Q.; Wei, Y.; Gao, Y.; Mao, L.; Gu, L.; Tan, Y.W.; Zhong, Y.; Liu, D.; et al. The Important Role of Lipid Raft-Mediated Attachment in the Infection of Cultured Cells by Coronavirus Infectious Bronchitis Virus Beaudette Strain. PLoS ONE 2017, 12, e0170123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pekosz, A.; Lamb, R.A. Influenza Virus Assembly and Lipid Raft Microdomains: A Role for the Cytoplasmic Tails of the Spike Glycoproteins. J. Virol. 2000, 74, 4634–4644. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Leser, G.P.; Russell, C.J.; Lamb, R.A. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. USA 2003, 100, 14610–14617. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Levental, I.; Levental, K.R.; Heberle, F.A. Lipid Rafts: Controversies Resolved, Mysteries Remain. Trends Cell Biol. 2020, 30, 341–353. [Google Scholar] [CrossRef]
- Biswas, S.; Yin, S.-R.; Blank, P.S.; Zimmerberg, J. Cholesterol Promotes Hemifusion and Pore Widening in Membrane Fusion Induced by Influenza Hemagglutinin. J. Gen. Physiol. 2008, 131, 503–513. [Google Scholar] [CrossRef]
- Chlanda, P.; Mekhedov, E.; Waters, H.; Schwartz, C.L.; Fischer, E.R.; Ryham, R.J.; Cohen, F.S.; Blank, P.S.; Zimmerberg, J. The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat. Microbiol. 2016, 1, 16050. [Google Scholar] [CrossRef]
- Zaitseva, E.; Yang, S.-T.; Melikov, K.; Pourmal, S.; Chernomordik, L.V. Dengue Virus Ensures Its Fusion in Late Endosomes Using Compartment-Specific Lipids. PLoS Pathog. 2010, 6, e1001131. [Google Scholar] [CrossRef]
- Meher, G.; Bhattacharjya, S.; Chakraborty, H. Membrane Cholesterol Modulates Oligomeric Status and Peptide-Membrane Interaction of Severe Acute Respiratory Syndrome Coronavirus Fusion Peptide. J. Phys. Chem. B 2019, 123, 10654–10662. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.N.; Boxer, S.G. Target Membrane Cholesterol Modulates Single Influenza Virus Membrane Fusion Efficiency but Not Rate. Biophys. J. 2020, 118, 2426–2433. [Google Scholar] [CrossRef] [PubMed]
- Masters, P.S. The Molecular Biology of Coronaviruses. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 2006; Volume 66, pp. 193–292. ISBN 978-0-12-039869-0. [Google Scholar]
- Munro, S.P. Lipid Rafts. Cell 2003, 115, 377–388. [Google Scholar] [CrossRef]
- Garcia-Arribas, A.B.; Alonso, A.; Goñi, F.M. Cholesterol interactions with ceramide and sphingomyelin. Chem. Phys. Lipids 2016, 199, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Corver, J.; Moesby, L.; Erukulla, R.K.; Reddy, K.C.; Bittman, R.; Wilschut, J. Sphingolipid-dependent fusion of Semliki Forest virus with cholesterol-containing liposomes requires both the 3-hydroxyl group and the double bond of the sphingolipid backbone. J. Virol. 1995, 69, 3220–3223. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, N.; Sakata, M.; Saito, K.; Okamoto, K.; Mori, Y.; Hanada, K.; Takeda, M. Both Sphingomyelin and Cholesterol in the Host Cell Membrane Are Essential for Rubella Virus Entry. J. Virol. 2017, 92, e01130-17. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.E.; Adhikary, S.; Kolokoltsov, A.A.; Davey, R.A. Ebolavirus Requires Acid Sphingomyelinase Activity and Plasma Membrane Sphingomyelin for Infection. J. Virol. 2012, 86, 7473–7483. [Google Scholar] [CrossRef]
- Pastenkos, G.; Miller, J.L.; Pritchard, S.M.; Nicola, A.V. Role of Sphingomyelin in Alphaherpesvirus Entry. J. Virol. 2018, 93, e01547-18. [Google Scholar] [CrossRef]
- Soudani, N.; Hage-Sleiman, R.; Karam, W.; Dbaibo, G.; Zaraket, H. Ceramide Suppresses Influenza A Virus Replication In Vitro. J. Virol. 2019, 93, e00053-19. [Google Scholar] [CrossRef]
- Grassmé, H.; Riehle, A.; Wilker, B.; Gulbins, E. Rhinoviruses Infect Human Epithelial Cells via Ceramide-enriched Membrane Platforms. J. Biol. Chem. 2005, 280, 26256–26262. [Google Scholar] [CrossRef]
- Aktepe, T.E.; Pham, H.; MacKenzie, J.M. Differential utilisation of ceramide during replication of the flaviviruses West Nile and dengue virus. Virology 2015, 484, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Drews, K.; Calgi, M.P.; Harrison, W.C.; Drews, C.M.; Costa-Pinheiro, P.; Shaw, J.J.P.; Jobe, K.A.; Nelson, E.A.; Han, J.D.; Fox, T.; et al. Glucosylceramidase Maintains Influenza Virus Infection by Regulating Endocytosis. J. Virol. 2019, 93, e00017-19. [Google Scholar] [CrossRef]
- Sieben, C.; Sezgin, E.; Eggeling, C.; Manley, S. Influenza A viruses use multivalent sialic acid clusters for cell binding and receptor activation. PLoS Pathog. 2020, 16, e1008656. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.-J.; Bosch, B.-J.; Rey, F.A.; De Groot, R.J.; et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef]
- Mazzon, M.; Mercer, J. Lipid interactions during virus entry and infection. Cell Microbiol. 2014, 16, 1493–1502. [Google Scholar] [CrossRef]
- Ewers, H.; Römer, W.; Smith, A.E.; Bacia, K.; Dmitrieff, S.; Chai, W.; Mancini, R.; Kartenbeck, J.; Chambon, V.; Berland, L.; et al. GM1 structure determines SV40-induced membrane invagination and infection. Nat. Cell Biol. 2010, 12, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Moller-Tank, S.; Kondratowicz, A.S.; Davey, R.A.; Rennert, P.D.; Maury, W.J. Role of the Phosphatidylserine Receptor TIM-1 in Enveloped-Virus Entry. J. Virol. 2013, 87, 8327–8341. [Google Scholar] [CrossRef] [PubMed]
- Jemielity, S.; Wang, J.J.; Chan, Y.K.; Ahmed, A.A.; Li, W.; Monahan, S.; Bu, X.; Farzan, M.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-family Proteins Promote Infection of Multiple Enveloped Viruses through Virion-associated Phosphatidylserine. PLoS Pathog. 2013, 9, e1003232. [Google Scholar] [CrossRef]
- Nanbo, A.; Maruyama, J.; Imai, M.; Ujie, M.; Fujioka, Y.; Nishide, S.; Takada, A.; Ohba, Y.; Kawaoka, Y. Ebola virus requires a host scramblase for externalization of phosphatidylserine on the surface of viral particles. PLoS Pathog. 2018, 14, e1006848. [Google Scholar] [CrossRef]
- Cheshenko, N.; Pierce, C.; Herold, B.C. Herpes simplex viruses activate phospholipid scramblase to redistribute phosphatidylserines and Akt to the outer leaflet of the plasma membrane and promote viral entry. PLoS Pathog. 2018, 14, e1006766. [Google Scholar] [CrossRef]
- Das, A.; Maury, W.; Lemon, S.M. TIM1 (HAVCR1): An Essential “Receptor” or an “Accessory Attachment Factor” for Hepatitis A Virus? J. Virol. 2019, 93, e01793-18. [Google Scholar] [CrossRef] [PubMed]
- Van den Bout, I.; Divecha, N. PIP5K-driven PtdIns(4,5)P2 synthesis: Regulation and cellular functions. J. Cell Sci. 2009, 122, 3837–3850. [Google Scholar] [CrossRef]
- Vicinanza, M.; D’Angelo, G.; Di Campli, A.; De Matteis, M.A. Function and dysfunction of the PI system in membrane trafficking. EMBO J. 2008, 27, 2457–2470. [Google Scholar] [CrossRef]
- Perugini, V.R.; Gordón-Alonso, M.; Sánchez-Madrid, F. PIP2: Choreographer of actin-adaptor proteins in the HIV-1 dance. Trends Microbiol. 2014, 22, 379–388. [Google Scholar] [CrossRef][Green Version]
- Muecksch, F.; Laketa, V.; Muller, B.; Schultz, C.; Kräusslich, H.-G. Synchronized HIV assembly by tunable PIP2 changes reveals PIP2 requirement for stable Gag anchoring. eLife 2017, 6, e25287. [Google Scholar] [CrossRef] [PubMed]
- Mücksch, F.; Citir, M.; Lüchtenborg, C.; Glass, B.; Traynor-Kaplan, A.; Schultz, C.; Brügger, B.; Kräusslich, H.-G. Quantification of phosphoinositides reveals strong enrichment of PIP2 in HIV-1 compared to producer cell membranes. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wijesinghe, K.J.; Stahelin, R.V. Investigation of the Lipid Binding Properties of the Marburg Virus Matrix Protein VP40. J. Virol. 2016, 90, 3074–3085. [Google Scholar] [CrossRef]
- Gonzales, B.; De Rocquigny, H.; Beziau, A.; Durand, S.; Burlaud-Gaillard, J.; Lefebvre, A.; Krull, S.; Emond, P.; Brand, D.; Piver, E. Type I phosphatidylinositol-4-phosphate 5-kinase α and γ play a key role in targeting HIV-1 Pr55Gag to the plasma membrane. J. Virol. 2020, 94, e00189-20. [Google Scholar] [CrossRef]
- Diehl, N.; Schaal, H. Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef]
- Wu, S.-X.; Chen, W.-N.; Jing, Z.-T.; Liu, W.; Lin, X.-J.; Lin, X. Hepatitis B Spliced Protein (HBSP) Suppresses Fas-Mediated Hepatocyte Apoptosis via Activation of PI3K/Akt Signaling. J. Virol. 2018, 92, e01273-18. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Harak, C.; Lohmann, V. Ultrastructure of the replication sites of positive-strand RNA viruses. Virology 2015, 479–480, 418–433. [Google Scholar] [CrossRef] [PubMed]
- Neuman, B.W.; Angelini, M.M.; Buchmeier, M.J. Does form meet function in the coronavirus replicative organelle? Trends Microbiol. 2014, 22, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and Biochemical Characterization of the Membranous Hepatitis C Virus Replication Compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Van Den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; Van Der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-Coronavirus Replication Is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef]
- Netherton, C.L.; Wileman, T. Virus factories, double membrane vesicles and viroplasm generated in animal cells. Curr. Opin. Virol. 2011, 1, 381–387. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.-Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-Dimensional Architecture and Biogenesis of Membrane Structures Associated with Hepatitis C Virus Replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Wölk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of Hepatitis C Virus Proteins Induces Distinct Membrane Alterations Including a Candidate Viral Replication Complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef]
- Wang, H.; Tai, A.W. Mechanisms of Cellular Membrane Reorganization to Support Hepatitis C Virus Replication. Viruses 2016, 8, 142. [Google Scholar] [CrossRef]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Proteins 3, 4, and 6 Induce Double-Membrane Vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef]
- Brügger, B.; Glass, B.; Haberkant, P.; Leibrecht, I.; Wieland, F.T.; Kräusslich, H.-G. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. USA 2006, 103, 2641–2646. [Google Scholar] [CrossRef] [PubMed]
- Martín-Acebes, M.A.; Merino-Ramos, T.; Blazquez, A.; Casas, J.; Escribano-Romero, E.; Sobrino, F.; Sáiz, J.-C. The Composition of West Nile Virus Lipid Envelope Unveils a Role of Sphingolipid Metabolism in Flavivirus Biogenesis. J. Virol. 2014, 88, 12041–12054. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Perry, J.W.; Lauring, A.S.; Neddermann, P.; De Francesco, R.; Tai, A.W. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 2014, 146, 1373–1385. [Google Scholar] [CrossRef]
- Khan, I.; Katikaneni, D.S.; Han, Q.; Sanchez-Felipe, L.; Hanada, K.; Ambrose, R.L.; MacKenzie, J.M.; Konan, K. Modulation of Hepatitis C Virus Genome Replication by Glycosphingolipids and Four-Phosphate Adaptor Protein 2. J. Virol. 2014, 88, 12276–12295. [Google Scholar] [CrossRef]
- Delang, L.; Harak, C.; Benkheil, M.; Khan, H.; Leyssen, P.; Andrews, M.; Lohmann, V.; Neyts, J. PI4KIII inhibitor enviroxime impedes the replication of the hepatitis C virus by inhibiting PI3 kinases. J. Antimicrob. Chemother. 2018, 73, 3375–3384. [Google Scholar] [CrossRef]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Phosphatidylinositol 4-Kinase III Beta Is a Target of Enviroxime-Like Compounds for Antipoliovirus Activity. J. Virol. 2011, 85, 2364–2372. [Google Scholar] [CrossRef]
- Martín-Acebes, M.A.; Blazquez, A.; De Oya, N.J.; Escribano-Romero, E.; Sáiz, J.-C. West Nile Virus Replication Requires Fatty Acid Synthesis but Is Independent on Phosphatidylinositol-4-Phosphate Lipids. PLoS ONE 2011, 6, e24970. [Google Scholar] [CrossRef]
- Yang, N.; Ma, P.; Lang, J.; Zhang, Y.; Deng, J.; Ju, X.; Zhang, G.; Jiang, C. Phosphatidylinositol 4-Kinase IIIβ Is Required for Severe Acute Respiratory Syndrome Coronavirus Spike-mediated Cell Entry. J. Biol. Chem. 2012, 287, 8457–8467. [Google Scholar] [CrossRef]
- De Wilde, A.H.; Wannee, K.F.; Scholte, F.E.M.; Goeman, J.J.; Dijke, P.T.; Snijder, E.J.; Kikkert, M.; Van Hemert, M.J. A Kinome-Wide Small Interfering RNA Screen Identifies Proviral and Antiviral Host Factors in Severe Acute Respiratory Syndrome Coronavirus Replication, Including Double-Stranded RNA-Activated Protein Kinase and Early Secretory Pathway Proteins. J. Virol. 2015, 89, 8318–8333. [Google Scholar] [CrossRef]
- De Wilde, A.H.; Snijder, E.J.; Kikkert, M.; Van Hemert, M.J. Host Factors in Coronavirus Replication. In Roles of Host Gene and Non-Coding RNA Expression in Virus Infection; Tripp, R.A., Tompkins, S.M., Eds.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Switzerland, 2017; Volume 419, pp. 1–42. ISBN 978-3-030-05368-0. [Google Scholar]
- Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent Roles of Autophagy in Virus Infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 114, E3462–E3471. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Dengue Virus-Induced Autophagy Regulates Lipid Metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Dengue Virus and Autophagy. Viruses 2011, 3, 1332–1341. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Bosc, C.; Broin, N.; Fanjul, M.; Saland, E.; Farge, T.; Courdy, C.; Batut, A.; Masoud, R.; Larrue, C.; Skuli, S.; et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses Exploit the Lipid Droplet Protein AUP1 to Trigger Lipophagy and Drive Virus Production. Cell Host Microbe 2018, 23, 819–831. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Balgoma, D.; Pettersson, C.; Hedeland, M. Common Fatty Markers in Diseases with Dysregulated Lipogenesis. Trends Endocrinol. Metab. 2019, 30, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chu, H.; Chan, J.F.-W.; Ye, Z.-W.; Wen, L.; Yan, B.; Lai, P.-M.; Tee, K.-M.; Huang, J.; Chen, D.; et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 2019, 10, 120. [Google Scholar] [CrossRef]
- Petersen, J.; Drake, M.J.; Bruce, E.A.; Riblett, A.M.; Didigu, C.A.; Wilen, C.B.; Malani, N.; Male, F.; Lee, F.-H.; Bushman, F.D.; et al. The Major Cellular Sterol Regulatory Pathway Is Required for Andes Virus Infection. PLoS Pathog. 2014, 10, e1003911. [Google Scholar] [CrossRef]
- Kleinfelter, L.M.; Jangra, R.K.; Jae, L.T.; Herbert, A.S.; Mittler, E.; Stiles, K.M.; Wirchnianski, A.S.; Kielian, M.; Brummelkamp, T.R.; Dye, J.M.; et al. Haploid Genetic Screen Reveals a Profound and Direct Dependence on Cholesterol for Hantavirus Membrane Fusion. mBio 2015, 6, e00801-15. [Google Scholar] [CrossRef]
- Blanc, M.; Hsieh, W.Y.; Robertson, K.A.; Watterson, S.; Shui, G.; Lacaze, P.; Khondoker, M.; Dickinson, P.; Sing, G.; Rodríguez-Martín, S.; et al. Host Defense against Viral Infection Involves Interferon Mediated Down-Regulation of Sterol Biosynthesis. PLoS Biol. 2011, 9, e1000598. [Google Scholar] [CrossRef]
- Robertson, K.A.; Hsieh, W.Y.; Forster, T.; Blanc, M.; Lu, H.; Crick, P.J.; Yutuc, E.; Watterson, S.; Martin, K.; Griffiths, S.J.; et al. An Interferon Regulated MicroRNA Provides Broad Cell-Intrinsic Antiviral Immunity through Multihit Host-Directed Targeting of the Sterol Pathway. PLoS Biol. 2016, 14, e1002364. [Google Scholar] [CrossRef]
- Oudshoorn, D.; Van Der Hoeven, B.; Limpens, R.W.A.L.; Beugeling, C.; Snijder, E.J.; Bárcena, M.; Kikkert, M. Antiviral Innate Immune Response Interferes with the Formation of Replication-Associated Membrane Structures Induced by a Positive-Strand RNA Virus. mBio 2016, 7, e01991-16. [Google Scholar] [CrossRef]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.-C.; Farzan, M.; Jung, J.U. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef]
- Cui, H.L.; Grant, A.; Mukhamedova, N.; Pushkarsky, T.; Jennelle, L.; Dubrovsky, L.; Gaus, K.; Fitzgerald, M.L.; Sviridov, D.; Bukrinsky, M. HIV-1 Nef mobilizes lipid rafts in macrophages through a pathway that competes with ABCA1-dependent cholesterol efflux. J. Lipid Res. 2012, 53, 696–708. [Google Scholar] [CrossRef]
- Tang, H.-B.; Lu, Z.-L.; Wei, X.-K.; Zhong, T.-Z.; Zhong, Y.-Z.; Ouyang, L.-X.; Luo, Y.; Xing, X.-W.; Liao, F.; Peng, K.-K.; et al. Viperin inhibits rabies virus replication via reduced cholesterol and sphingomyelin and is regulated upstream by TLR4. Sci. Rep. 2016, 6, 30529. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.-A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal Proteome and Lipidome Profiles Reveal Hepatitis C Virus-Associated Reprogramming of Hepatocellular Metabolism and Bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, L.; Sun, X.; Yan, Z.; Hu, C.; Wu, J.; Xu, L.; Li, X.; Liu, H.; Yin, P.; et al. Altered Lipid Metabolism in Recovered SARS Patients Twelve Years after Infection. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Vijay, R.; Hua, X.; Meyerholz, D.K.; Miki, Y.; Yamamoto, K.; Gelb, M.; Murakami, M.; Perlman, S. Critical role of phospholipase A2 group IID in age-related susceptibility to severe acute respiratory syndrome–CoV infection. J. Exp. Med. 2015, 212, 1851–1868. [Google Scholar] [CrossRef]
- Vijay, R.; Fehr, A.R.; Janowski, A.M.; Athmer, J.; Wheeler, D.L.; Grunewald, M.; Sompallae, R.; Kurup, S.P.; Meyerholz, D.K.; Sutterwala, F.S.; et al. Virus-induced inflammasome activation is suppressed by prostaglandin D2/DP1 signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E5444–E5453. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; Macary, P.A.; Ng, L.F. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Yan, B.; Chu, H.; Yang, D.; Sze, K.H.; Lai, P.-M.; Yuan, S.; Shuai, H.; Wang, Y.; Kao, R.Y.; Chan, J.F.-W.; et al. Characterization of the Lipidomic Profile of Human Coronavirus-Infected Cells: Implications for Lipid Metabolism Remodeling upon Coronavirus Replication. Viruses 2019, 11, 73. [Google Scholar] [CrossRef]
- Grant, W.B.; Lahore, H.; McDonnell, S.; Baggerly, C.A.; French, C.B.; Aliano, J.L.; Bhattoa, H.P. Evidence that Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths. Nutrients 2020, 12, 988. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am. J. Clin. Nutr. 2004, 80, 1678S–1688S. [Google Scholar] [CrossRef]
- Martínez-Moreno, J.; Hernández, J.C.; Urcuqui-Inchima, S. Effect of high doses of vitamin D supplementation on dengue virus replication, Toll-like receptor expression, and cytokine profiles on dendritic cells. Mol. Cell. Biochem. 2020, 464, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Sze, C.W.; Tan, Y.-J. Viral Membrane Channels: Role and Function in the Virus Life Cycle. Viruses 2015, 7, 3261–3284. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Hyser, J.M.; Utama, B.; Estes, M.K. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase- signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. USA 2012, 109, E3405–E3413. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balgoma, D.; Gil-de-Gómez, L.; Montero, O. Lipidomics Issues on Human Positive ssRNA Virus Infection: An Update. Metabolites 2020, 10, 356. https://doi.org/10.3390/metabo10090356
Balgoma D, Gil-de-Gómez L, Montero O. Lipidomics Issues on Human Positive ssRNA Virus Infection: An Update. Metabolites. 2020; 10(9):356. https://doi.org/10.3390/metabo10090356
Chicago/Turabian StyleBalgoma, David, Luis Gil-de-Gómez, and Olimpio Montero. 2020. "Lipidomics Issues on Human Positive ssRNA Virus Infection: An Update" Metabolites 10, no. 9: 356. https://doi.org/10.3390/metabo10090356
APA StyleBalgoma, D., Gil-de-Gómez, L., & Montero, O. (2020). Lipidomics Issues on Human Positive ssRNA Virus Infection: An Update. Metabolites, 10(9), 356. https://doi.org/10.3390/metabo10090356