Single-Step Extraction Coupled with Targeted HILIC-MS/MS Approach for Comprehensive Analysis of Human Plasma Lipidome and Polar Metabolome

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Sample Preparation Methods and Evaluation Workflow
2.2. Relative Signal Abundance
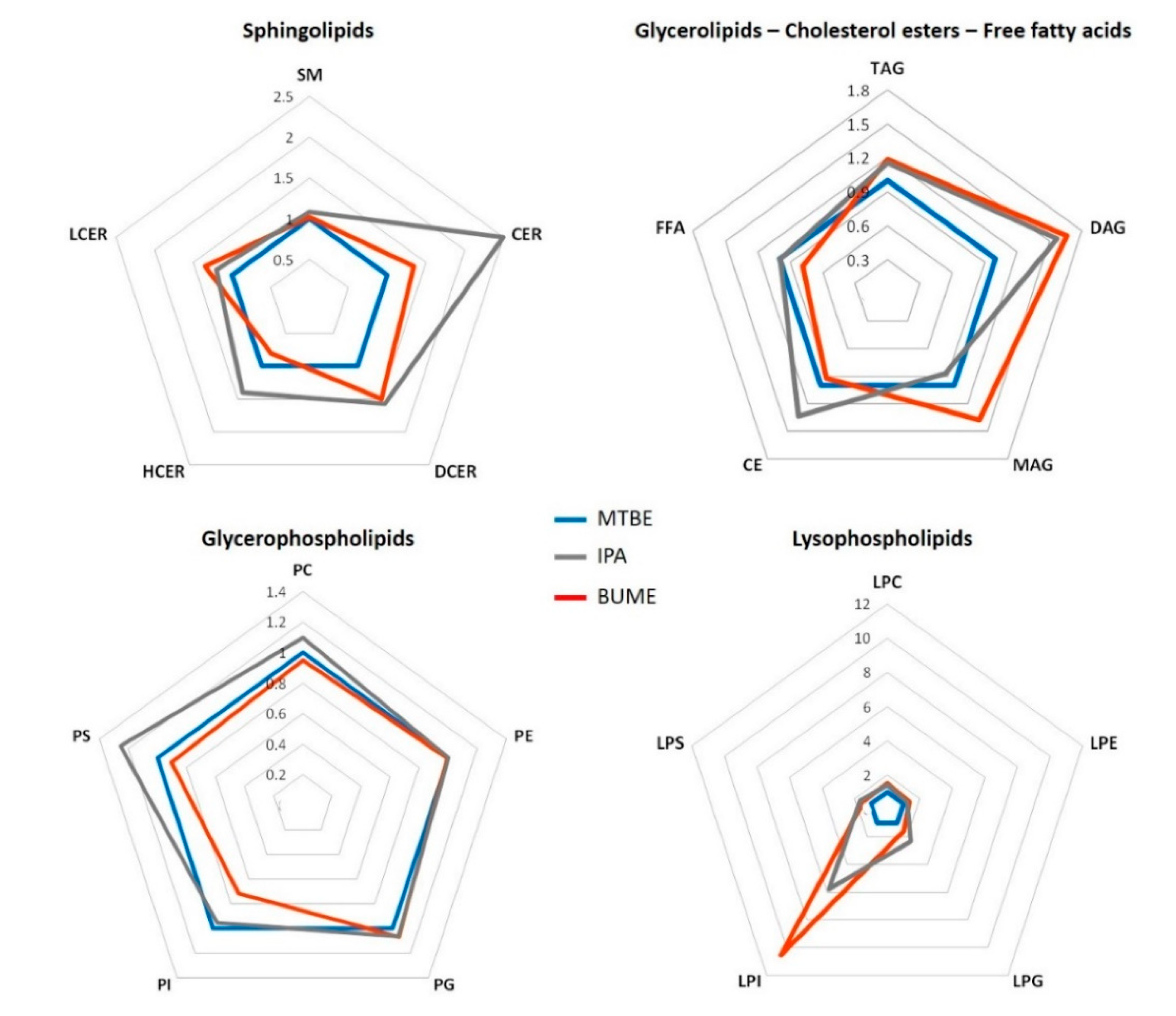
2.2.1. Complex Lipid Profile
2.2.2. Polar Metabolite Profile
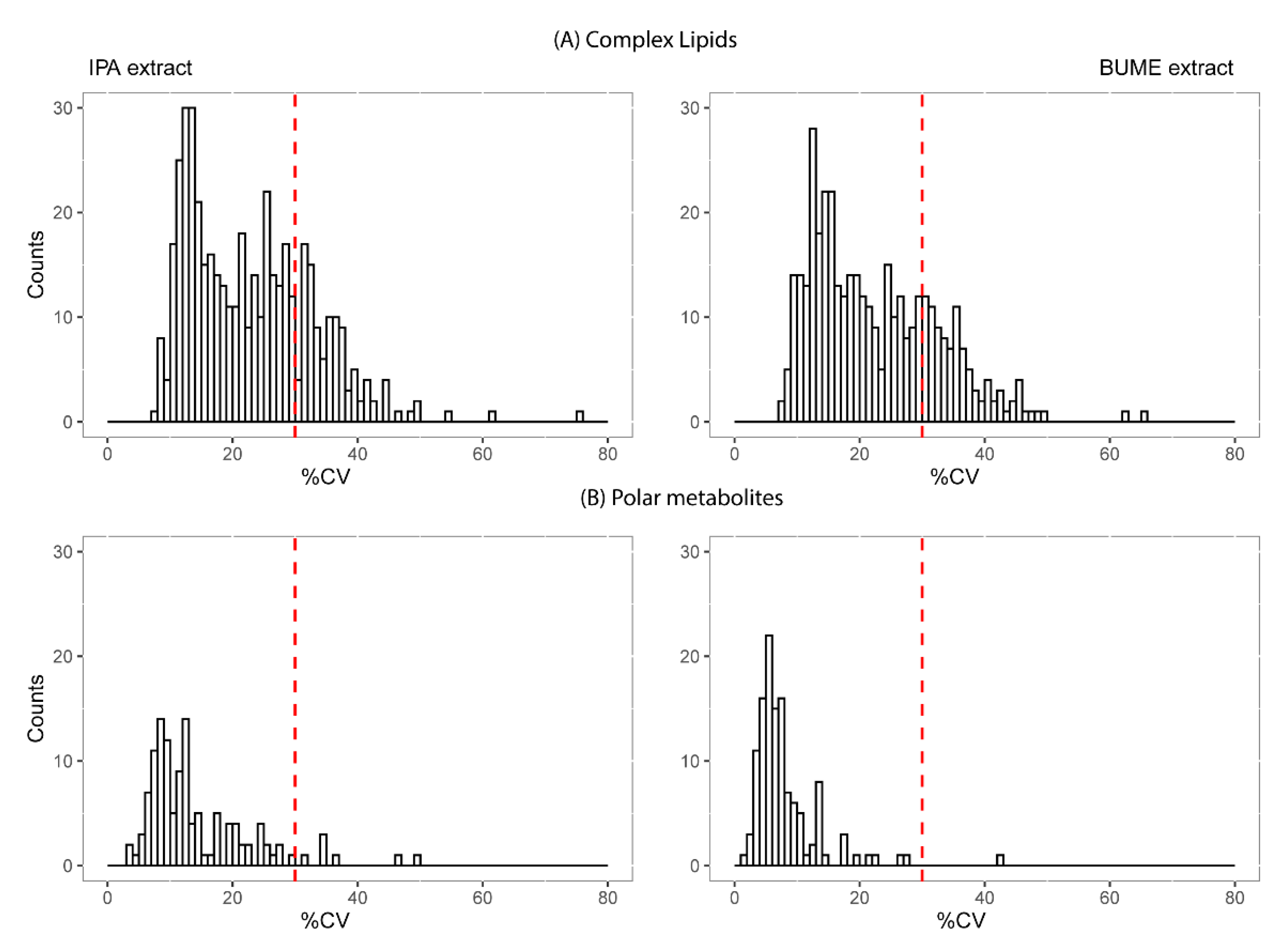
2.3. Reproducibility and Size of Measurable Lipidome and Metabolome
3. Discussion
3.1. Complex Lipid Extraction
3.2. Polar Metabolite Extraction
3.3. Extraction Method and Measurement Reproducibility
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Metabolite Extraction Protocols
4.3. Protein Quantification
4.4. LC-MS/MS Analysis
4.4.1. High-Coverage Targeted Lipidome Analysis
4.4.2. High-Coverage Targeted Metabolome Analysis
4.5. Data (Pre)Processing
4.6. Statistical Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Dennis, E.A. The Human Plasma Lipidome. N. Engl. J. Med. 2011, 365, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; Van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef]
- Holčapek, M.; Liebisch, G.; Ekroos, K. Lipidomic Analysis. Anal. Chem. 2018, 90, 4249–4257. [Google Scholar] [CrossRef]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Schooneman, M.G.; Vaz, F.M.; Houten, S.M.; Soeters, M.R. Acylcarnitines: Reflecting or inflicting insulin resistance? Diabetes 2013, 62, 1–8. [Google Scholar] [CrossRef]
- Bagheri, M.; Djazayery, A.; Farzadfar, F.; Qi, L.; Yekaninejad, M.S.; Aslibekyan, S.; Chamari, M.; Hassani, H.; Koletzko, B.; Uhl, O. Plasma metabolomic profiling of amino acids and polar lipids in Iranian obese adults. Lipids Health Dis. 2019, 18, 1–9. [Google Scholar] [CrossRef]
- Liu, J.; Liu, X.; Xiao, Z.; Locasale, J.W. Quantitative evaluation of a high resolution lipidomics platform. bioRxiv 2019, 627687. [Google Scholar] [CrossRef]
- Ivanisevic, J.; Want, E.J. From samples to insights into metabolism: Uncovering biologically relevant information in LC- HRMS metabolomics data. Metabolites 2019, 9, 308. [Google Scholar] [CrossRef]
- Gallart-Ayala, H.; Konz, I.; Mehl, F.; Teav, T.; Oikonomidi, A.; Peyratout, G.; van der Velpen, V.; Popp, J.; Ivanisevic, J. A global HILIC-MS approach to measure polar human cerebrospinal fluid metabolome: Exploring gender-associated variation in a cohort of elderly cognitively healthy subjects. Anal. Chim. Acta 2018, 1037, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite measurement: Pitfalls to avoid and practices to follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Ascoli, I.; Lees, M.; Meath, J.A.; LeBaron, F.N. Preparation of Lipide Extracts From Brain Tisuue. J. Biol. Chem. 1951, 191, 833–841. [Google Scholar] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Ulmer, C.Z.; Jones, C.M.; Yost, R.A.; Garrett, T.J.; Bowden, J.A. Optimization of Folch, Bligh-Dyer, and Matyash sample-to-extraction solvent ratios for human plasma-based lipidomics studies. Anal. Chim. Acta 2018, 1037, 351–357. [Google Scholar] [CrossRef]
- Vvedenskaya, O.; Wang, Y.; Ackerman, J.M.; Knittelfelder, O.; Shevchenko, A. Analytical challenges in human plasma lipidomics: A winding path towards the truth. TrAC Trends Anal. Chem. 2019, 120, 115277. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl- tert -butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef]
- Alshehry, Z.H.; Barlow, C.K.; Weir, J.M.; Zhou, Y.; McConville, M.J.; Meikle, P.J. An efficient single phase method for the extraction of plasma lipids. Metabolites 2015, 5, 389–403. [Google Scholar] [CrossRef]
- Sarafian, M.H.; Gaudin, M.; Lewis, M.R.; Martin, F.P.; Holmes, E.; Nicholson, J.K.; Dumas, M.E. Objective set of criteria for optimization of sample preparation procedures for ultra-high throughput untargeted blood plasma lipid profiling by ultra performance liquid chromatography-mass spectrometry. Anal. Chem. 2014, 86, 5766–5774. [Google Scholar] [CrossRef]
- Löfgren, L.; Ståhlman, M.; Forsberg, G.B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef]
- Cai, X.; Li, R. Concurrent profiling of polar metabolites and lipids in human plasma using HILIC-FTMS. Sci. Rep. 2016, 6, 36490. [Google Scholar] [CrossRef]
- Patterson, R.E.; Ducrocq, A.J.; McDougall, D.J.; Garrett, T.J.; Yost, R.A. Comparison of blood plasma sample preparation methods for combined LC-MS lipidomics and metabolomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 1002, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, 527–532. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef]
- Snyder, L.R. Classification off the solvent properties of common liquids. J. Chromatogr. Sci. 1978, 16, 223–234. [Google Scholar] [CrossRef]
- Lange, M.; Fedorova, M. Evaluation of lipid quantification accuracy using HILIC and RPLC MS on the example of NIST® SRM® 1950 metabolites in human plasma. Anal. Bioanal. Chem. 2020, 412, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950–Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef]
- Simón-Manso, Y.; Lowenthal, M.S.; Kilpatrick, L.E.; Sampson, M.L.; Telu, K.H.; Rudnick, P.A.; Mallard, W.G.; Bearden, D.W.; Schock, T.B.; Tchekhovskoi, D.V.; et al. Metabolite profiling of a NIST standard reference material for human plasma (SRM 1950): GC-MS, LC-MS, NMR, and clinical laboratory analyses, libraries, and web-based resources. Anal. Chem. 2013, 85, 11725–11731. [Google Scholar] [CrossRef]
- Lee, J.W.; Nagai, T.; Gotoh, N.; Fukusaki, E.; Bamba, T. Profiling of regioisomeric triacylglycerols in edible oils by supercritical fluid chromatography/tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 966, 193–199. [Google Scholar] [CrossRef]
- Lee, D.Y.; Kind, T.; Yoon, Y.R.; Fiehn, O.; Liu, K.H. Comparative evaluation of extraction methods for simultaneous mass-spectrometric analysis of complex lipids and primary metabolites from human blood plasma. Anal. Bioanal. Chem. 2014, 406, 7275–7286. [Google Scholar] [CrossRef]
- Ivanisevic, J.; Zhu, Z.-J.; Plate, L.; Tautenhahn, R.; Chen, S.; O’Brien, P.J.; Johnson, C.H.; Marletta, M.A.; Patti, G.J.; Siuzdak, G. Toward ‘{Omic} {Scale} {Metabolite} {Profiling}: {A} {Dual} {Separation}–{Mass} {Spectrometry} {Approach} for {Coverage} of {Lipid} and {Central} {Carbon} {Metabolism}. Anal. Chem. 2013, 85, 6876–6884. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Zhang, W.; Wolters, J.C.; Permentier, H.; Boer, T.; Horvatovich, P.; Heiner-Fokkema, M.R.; Reijngoud, D.J.; Bischoff, R. One- vs two-phase extraction: Re-evaluation of sample preparation procedures for untargeted lipidomics in plasma samples. Anal. Bioanal. Chem. 2018, 410, 5859–5870. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.W.K.; Braidy, N.; Pickford, R.; Sachdev, P.S.; Poljak, A. Comparison of single phase and biphasic extraction protocols for lipidomic studies using human plasma. Front. Neurol. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.; Wang, M.; Frisch-Daiello, J.; Han, X. Improved Butanol–Methanol (BUME) Method by Replacing Acetic Acid for Lipid Extraction of Biological Samples. Lipids 2016, 51, 887–896. [Google Scholar] [CrossRef]
- Sitnikov, D.G.; Monnin, C.S.; Vuckovic, D. Systematic Assessment of Seven Solvent and Solid-Phase Extraction Methods for Metabolomics Analysis of Human Plasma by LC-MS. Sci. Rep. 2016, 6, 38885. [Google Scholar] [CrossRef]
- Satomi, Y.; Hirayama, M.; Kobayashi, H. One-step lipid extraction for plasma lipidomics analysis by liquid chromatography mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1063, 93–100. [Google Scholar] [CrossRef]
- Calderón, C.; Sanwald, C.; Schlotterbeck, J.; Drotleff, B.; Lämmerhofer, M. Comparison of simple monophasic versus classical biphasic extraction protocols for comprehensive UHPLC-MS/MS lipidomic analysis of Hela cells. Anal. Chim. Acta 2019, 1048, 66–74. [Google Scholar] [CrossRef]
- Alshehry, Z.H.; Mundra, P.A.; Barlow, C.K.; Mellett, N.A.; Wong, G.; McConville, M.J.; Simes, J.; Tonkin, A.M.; Sullivan, D.R.; Barnes, E.H.; et al. Plasma Lipidomic Profiles Improve on Traditional Risk Factors for the Prediction of Cardiovascular Events in Type 2 Diabetes Mellitus. Circulation 2016, 134, 1637–1650. [Google Scholar] [CrossRef]
- Saw, W.Y.; Tantoso, E.; Begum, H.; Zhou, L.; Zou, R.; He, C.; Chan, S.L.; Tan, L.W.L.; Wong, L.P.; Xu, W.; et al. Establishing multiple omics baselines for three Southeast Asian populations in the Singapore Integrative Omics Study. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Huynh, K.; Barlow, C.K.; Jayawardana, K.S.; Weir, J.M.; Mellett, N.A.; Cinel, M.; Magliano, D.J.; Shaw, J.E.; Drew, B.G.; Meikle, P.J. High-Throughput Plasma Lipidomics: Detailed Mapping of the Associations with Cardiometabolic Risk Factors. Cell Chem. Biol. 2019, 26, 71–84.e4. [Google Scholar] [CrossRef]
- Lamour, S.D.; Gomez-romero, M.; Vorkas, P.A.; Alibu, V.P.; Saric, J.; Holmes, E.; Sternberg, J.M. Discovery of Infection Associated Metabolic Markers in Human African Trypanosomiasis. PLoS Negl. Trop. Dis. 2015, 9, e0004200. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, X.; Nijman, R.; Camuzeaux, S.; Sands, C.; Jackson, H.; Kaforou, M.; Emonts, M.; Herberg, J.A.; Maconochie, I.; Carrol, E.D.; et al. Plasma lipid profiles discriminate bacterial from viral infection in febrile children. Sci. Rep. 2019, 9, 17714. [Google Scholar] [CrossRef]
- Bhattacharya, S.K. Lipidomics: Methods and Protocols, Methods in Molecular Biology. Methods Mol. Biol. 2017, 1609, 91–106. [Google Scholar] [CrossRef]
- Cannon, W.B. Organization for Physiological Homeostasis. Physiol. Rev. 1929, 9, 399–431. [Google Scholar] [CrossRef]
- Cao, Z.; Schmitt, T.C.; Varma, V.; Sloper, D.; Beger, R.D.; Sun, J. Evaluation of the Performance of Lipidyzer Platform and Its Application in the Lipidomics Analysis in Mouse Heart and Liver. J. Proteome Res. 2020, 19, 2742–2749. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Ivanisevic, J.; Thomas, A.; Sidibé, J.; Teav, T.; Guijas, C.; Aisporna, A.E.; Rinehart, D.; Hoang, L.; et al. XCMS-MRM and METLIN-MRM: A cloud library and public resource for targeted analysis of small molecules. Nat. Methods 2018, 15, 681–684. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina, J.; van der Velpen, V.; Teav, T.; Guitton, Y.; Gallart-Ayala, H.; Ivanisevic, J. Single-Step Extraction Coupled with Targeted HILIC-MS/MS Approach for Comprehensive Analysis of Human Plasma Lipidome and Polar Metabolome. Metabolites 2020, 10, 495. https://doi.org/10.3390/metabo10120495
Medina J, van der Velpen V, Teav T, Guitton Y, Gallart-Ayala H, Ivanisevic J. Single-Step Extraction Coupled with Targeted HILIC-MS/MS Approach for Comprehensive Analysis of Human Plasma Lipidome and Polar Metabolome. Metabolites. 2020; 10(12):495. https://doi.org/10.3390/metabo10120495
Chicago/Turabian StyleMedina, Jessica, Vera van der Velpen, Tony Teav, Yann Guitton, Hector Gallart-Ayala, and Julijana Ivanisevic. 2020. "Single-Step Extraction Coupled with Targeted HILIC-MS/MS Approach for Comprehensive Analysis of Human Plasma Lipidome and Polar Metabolome" Metabolites 10, no. 12: 495. https://doi.org/10.3390/metabo10120495
APA StyleMedina, J., van der Velpen, V., Teav, T., Guitton, Y., Gallart-Ayala, H., & Ivanisevic, J. (2020). Single-Step Extraction Coupled with Targeted HILIC-MS/MS Approach for Comprehensive Analysis of Human Plasma Lipidome and Polar Metabolome. Metabolites, 10(12), 495. https://doi.org/10.3390/metabo10120495