Electromembrane Extraction of Highly Polar Compounds: Analysis of Cardiovascular Biomarkers in Plasma

 , , ,
, , ,
Abstract
1. Introduction
2. Experimental
2.1. Chemical and Reagents
2.2. Preparation of Standard Solutions
2.3. Plasma Samples
2.4. Sample Preparation: Protein Precipitation
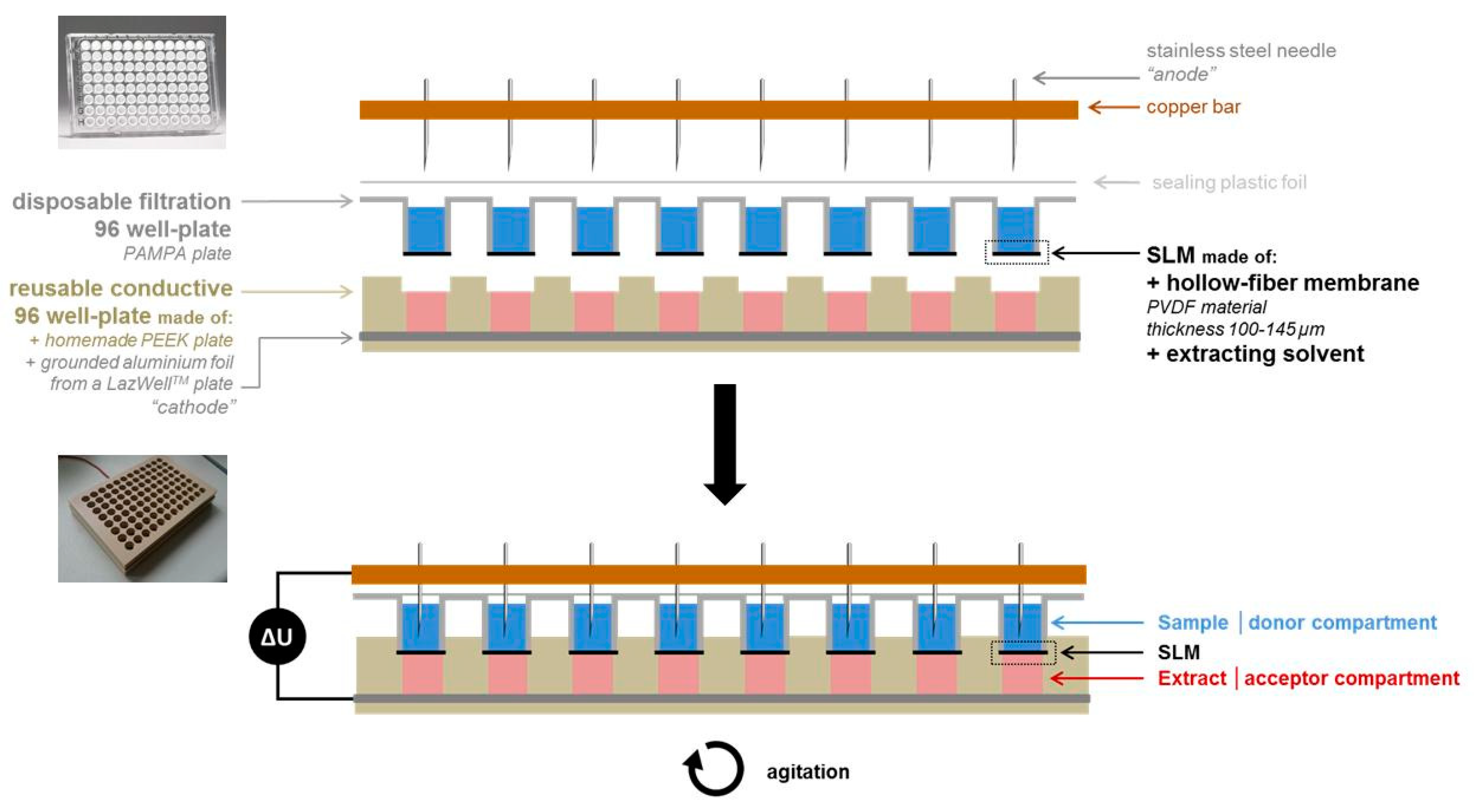
2.5. Pa-EME Set-Up and Procedure
2.6. Capillary Electrophoresis–Mass Spectrometry
2.7. Fast-Liquid Chromatography–Mass Spectrometry
2.8. Calculation of Extraction Yield, Process Efficiency, and Matrix Effect
3. Results and Discussion
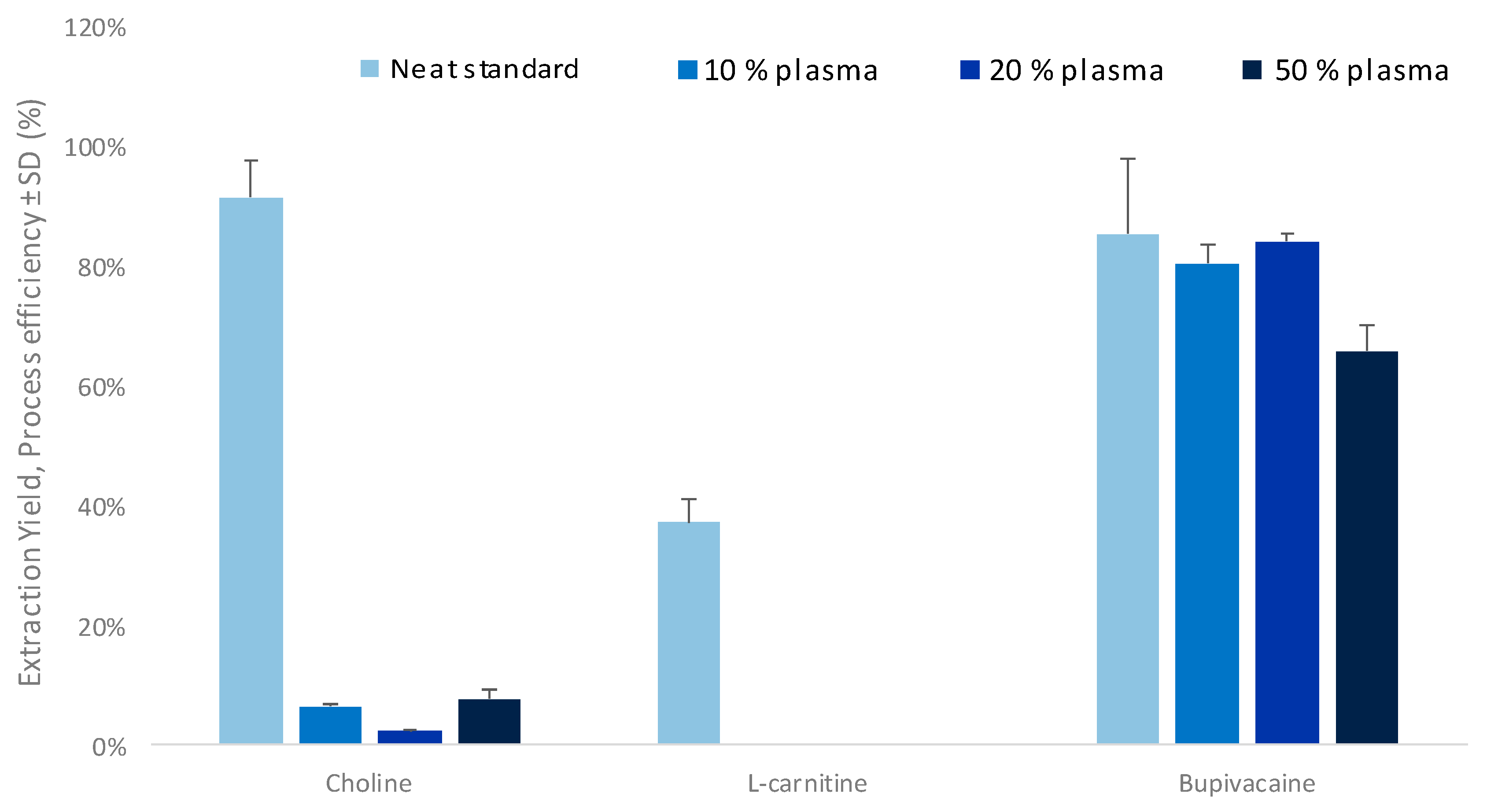
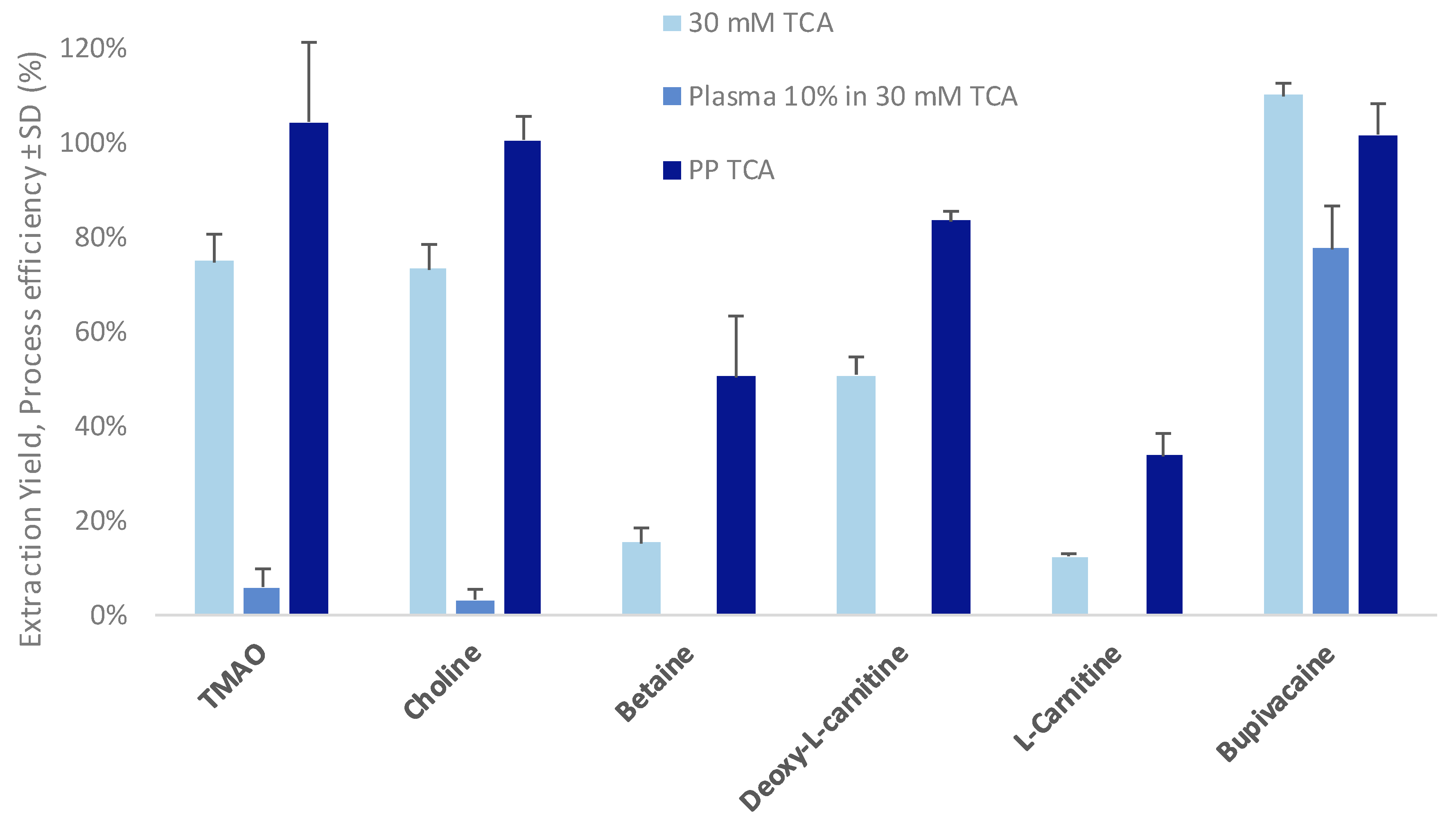
3.1. Optimization of the Parallel Electromembrane Extraction Set-Up
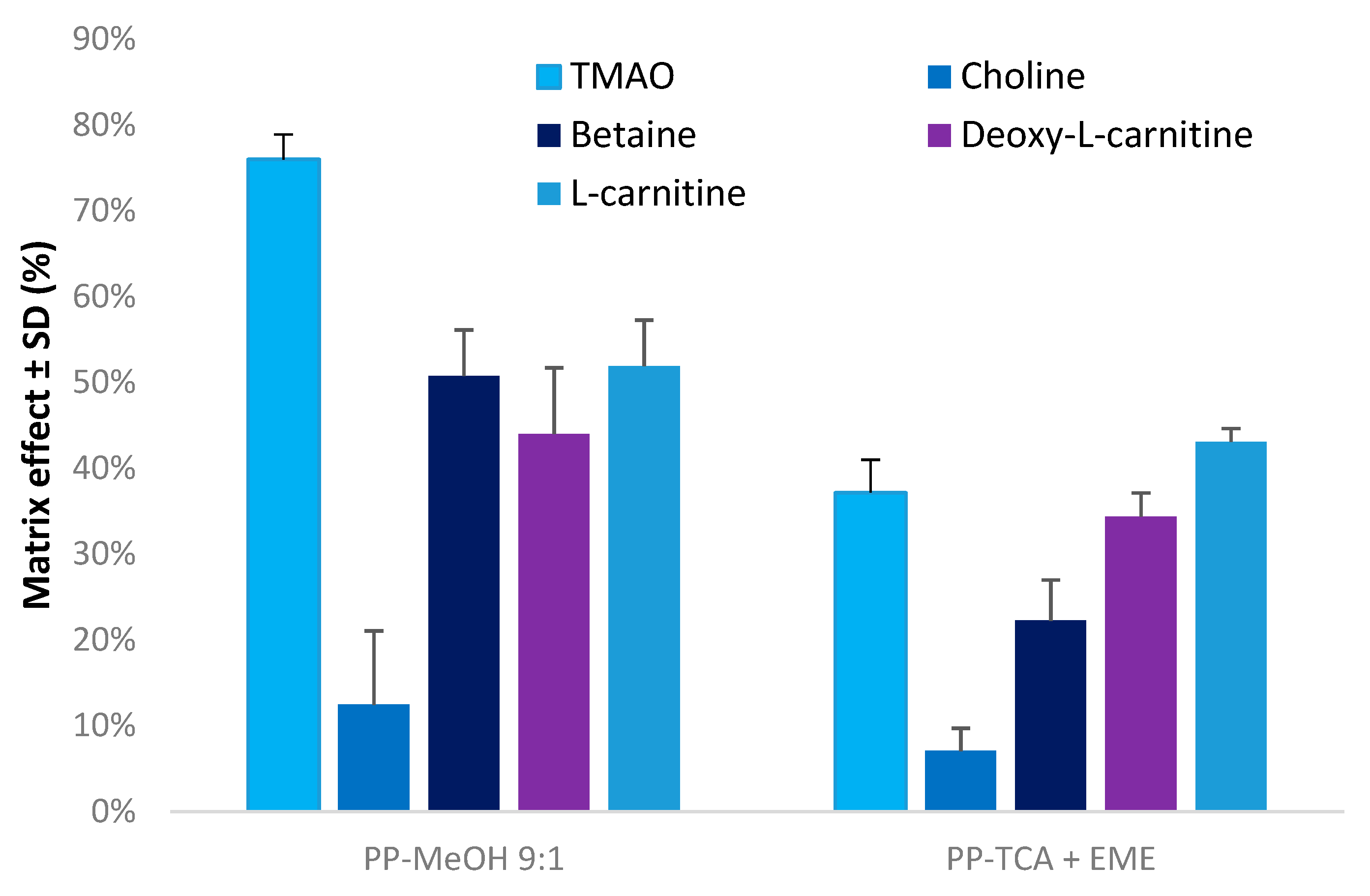
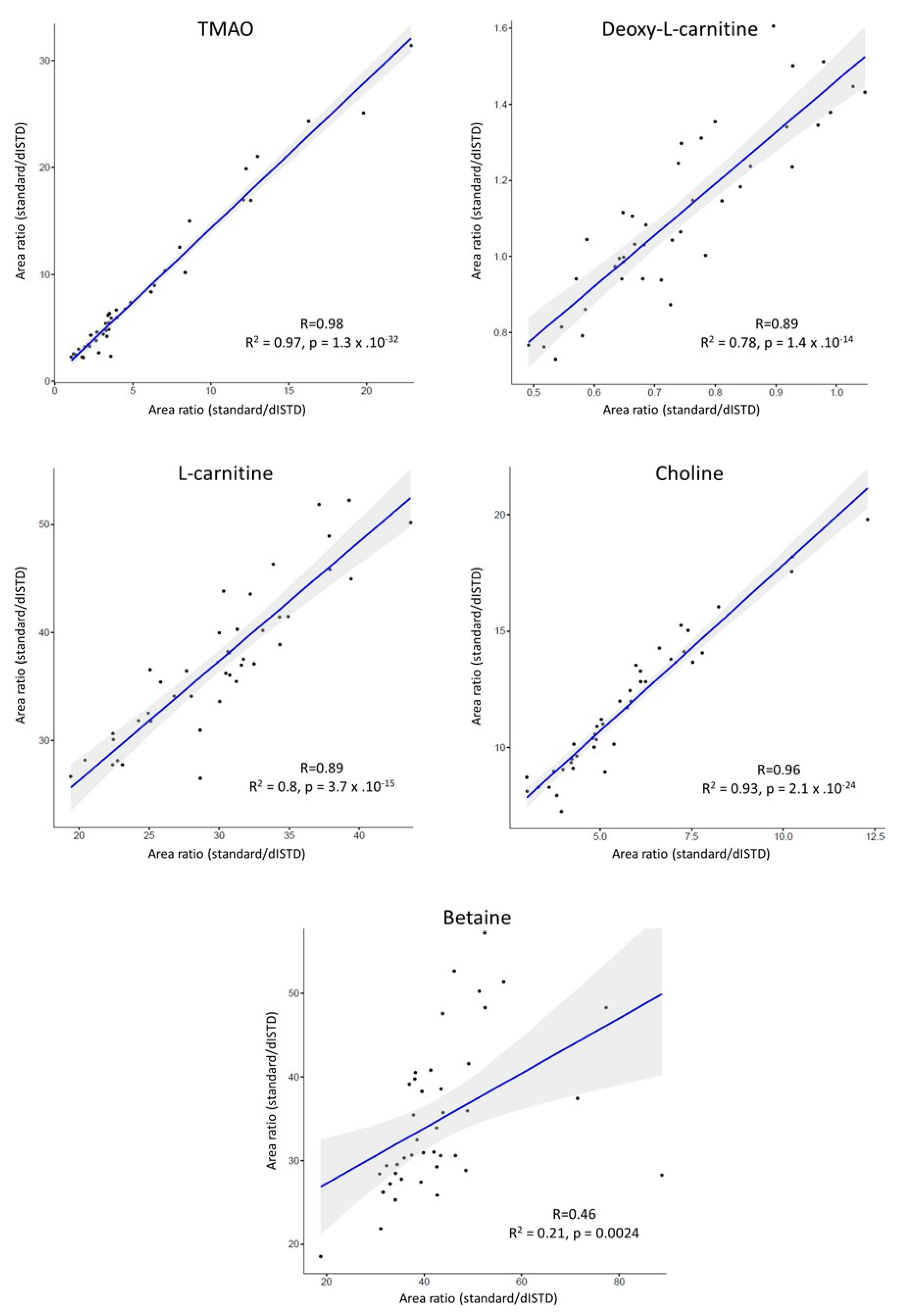
3.2. Evaluation of Matrix Effects
3.3. Application to Metabolomics Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CVD | cardiovascular disease |
| CE-MS | capillary electrophoresis–mass spectrometry |
| dISTD | deuterated internal standard |
| EME | electromembrane extraction |
| EY | extraction yield |
| FA | formic acid |
| Fast-LC-MS/MS | fast liquid chromatography–tandem mass spectrometry |
| HILIC | hydrophilic interaction chromatography |
| ME | matrix effect |
| MSI-CE-MS | multisegment injection–capillary electrophoresis–mass spectrometry |
| NPPE | 2-nitrophenylpentyl ether |
| MeCN | acetonitrile |
| MeOH | methanol |
| Pa-EME | parallel electromembrane extraction |
| PE | process efficiency |
| PEEK | polyether ether ketone |
| PP | protein precipitation |
| PP-MeOH | protein precipitation using methanol, ratio methanol/sample 9:1 (v/v) |
| PP-TCA | protein precipitation using trichloroacetic acid, ratio trichloroacetic acid/sample 0.05:1 (v/v) |
| PVDF | polyvinyldifluoride |
| SLM | supported liquid membrane |
| SRM | selected reaction monitoring |
| TCA | trichloroacetic acid |
| TMAO | trimethylamine N-oxide |
| UHPLC | ultra-high pressure liquid chromatography |
References
- Mortality, G.B.D.; Causes of Death, C. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Shrestha, K.; Borowski, A.G.; Wu, Y.; Troughton, R.W.; Klein, A.L.; Hazen, S.L. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J. Card. Fail. 2015, 21, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Dambrova, M.; Skapare-Makarova, E.; Konrade, I.; Pugovics, O.; Grinberga, S.; Tirzite, D.; Petrovska, R.; Kalvins, I.; Liepins, E. Meldonium decreases the diet-increased plasma levels of trimethylamine N-oxide, a metabolite associated with atherosclerosis. J. Clin. Pharmacol. 2013, 53, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Steuer, C.; Schutz, P.; Bernasconi, L.; Huber, A.R. Simultaneous determination of phosphatidylcholine-derived quaternary ammonium compounds by a LC-MS/MS method in human blood plasma, serum and urine samples. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1008, 206–211. [Google Scholar] [CrossRef]
- Gagnebin, Y.; Pezzatti, J.; Lescuyer, P.; Boccard, J.; Ponte, B.; Rudaz, S. Toward a better understanding of chronic kidney disease with complementary chromatographic methods hyphenated with mass spectrometry for improved polar metabolome coverage. J. Chromatogr. B 2019. [Google Scholar] [CrossRef] [PubMed]
- Lomonaco, T.; Ghimenti, S.; Piga, I.; Onor, M.; Melai, B.; Fuoco, R.; Di Francesco, F. Determination of total and unbound warfarin and warfarin alcohols in human plasma by high performance liquid chromatography with fluorescence detection. J. Chromatogr. A 2013, 1314, 54–62. [Google Scholar] [CrossRef]
- Drouin, N.; Rudaz, S.; Schappler, J. Sample preparation for polar metabolites in bioanalysis. Analyst 2017, 143, 16–20. [Google Scholar] [CrossRef]
- van der Laan, T.; Kloots, T.; Beekman, M.; Kindt, A.; Dubbelman, A.C.; Harms, A.; van Duijn, C.M.; Slagboom, P.E.; Hankemeier, T. Fast LC-ESI-MS/MS analysis and influence of sampling conditions for gut metabolites in plasma and serum. Sci. Rep. 2019, 9, 12370. [Google Scholar] [CrossRef] [PubMed]
- Pedersen-Bjergaard, S.; Rasmussen, K.E. Electrokinetic migration across artificial liquid membranes. New concept for rapid sample preparation of biological fluids. J. Chromatogr. A 2006, 1109, 183–190. [Google Scholar] [CrossRef]
- Drouin, N.; Kubáň, P.; Rudaz, S.; Pedersen-Bjergaard, S.; Schappler, J. Electromembrane extraction: Overview of the last decade. TrAC Trends Anal. Chem. 2018. [Google Scholar] [CrossRef]
- Oedit, A.; Ramautar, R.; Hankemeier, T.; Lindenburg, P.W. Electroextraction and electromembrane extraction: Advances in hyphenation to analytical techniques. Electrophoresis 2016, 37, 1170–1186. [Google Scholar] [CrossRef]
- Drouin, N.; Rudaz, S.; Schappler, J. Dynamic-Electromembrane Extraction: A Technical Development for the Extraction of Neuropeptides. Anal. Chem. 2016, 88, 5308–5315. [Google Scholar] [CrossRef]
- Balchen, M.; Gjelstad, A.; Rasmussen, K.E.; Pedersen-Bjergaard, S. Electrokinetic migration of acidic drugs across a supported liquid membrane. J. Chromatogr. A 2007, 1152, 220–225. [Google Scholar] [CrossRef]
- Gjelstad, A.; Rasmussen, K.E.; Pedersen-Bjergaard, S. Electromembrane extraction of basic drugs from untreated human plasma and whole blood under physiological pH conditions. Anal. Bioanal. Chem. 2009, 393, 921–928. [Google Scholar] [CrossRef]
- Vardal, L.; Gjelstad, A.; Huang, C.; Oiestad, E.L.; Pedersen-Bjergaard, S. Efficient discrimination and removal of phospholipids during electromembrane extraction from human plasma samples. Bioanalysis 2017, 9, 631–641. [Google Scholar] [CrossRef]
- Seip, K.F.; Jensen, H.; Kieu, T.E.; Gjelstad, A.; Pedersen-Bjergaard, S. Salt effects in electromembrane extraction. J. Chromatogr. A 2014, 1347, 1–7. [Google Scholar] [CrossRef]
- Huang, C.; Seip, K.F.; Gjelstad, A.; Pedersen-Bjergaard, S. Electromembrane extraction of polar basic drugs from plasma with pure bis(2-ethylhexyl) phosphite as supported liquid membrane. Anal. Chim. Acta 2016, 934, 80–87. [Google Scholar] [CrossRef]
- Drouin, N.; Rudaz, S.; Schappler, J. New supported liquid membrane for electromembrane extraction of polar basic endogenous metabolites. J. Pharm. Biomed. Anal. 2018, 159, 53–59. [Google Scholar] [CrossRef]
- Fernandez, E.; Vardal, L.; Vidal, L.; Canals, A.; Gjelstad, A.; Pedersen-Bjergaard, S. Complexation-mediated electromembrane extraction of highly polar basic drugs-a fundamental study with catecholamines in urine as model system. Anal. Bioanal. Chem. 2017, 409, 4215–4223. [Google Scholar] [CrossRef]
- Huang, C.X.; Shen, X.T.; Gjelstad, A.; Pedersen-Bjergaard, S. Investigation of alternative supported liquid membranes in electromembrane extraction of basic drugs from human plasma. J. Membr. Sci. 2018, 548, 176–183. [Google Scholar] [CrossRef]
- Lindenburg, P.W.; Tjaden, U.R.; van der Greef, J.; Hankemeier, T. Feasibility of electroextraction as versatile sample preconcentration for fast and sensitive analysis of urine metabolites, demonstrated on acylcarnitines. Electrophoresis 2012, 33, 2987–2995. [Google Scholar] [CrossRef]
- Raterink, R.J.; Lindenburg, P.W.; Vreeken, R.J.; Hankemeier, T. Three-phase electroextraction: A new (online) sample purification and enrichment method for bioanalysis. Anal. Chem. 2013, 85, 7762–7768. [Google Scholar] [CrossRef]
- Schoonen, J.W.; van Duinen, V.; Oedit, A.; Vulto, P.; Hankemeier, T.; Lindenburg, P.W. Continuous-flow microelectroextraction for enrichment of low abundant compounds. Anal. Chem. 2014, 86, 8048–8056. [Google Scholar] [CrossRef]
- Schoeman, J.C.; Hou, J.; Harms, A.C.; Vreeken, R.J.; Berger, R.; Hankemeier, T.; Boonstra, A. Metabolic characterization of the natural progression of chronic hepatitis B. Genome Med. 2016, 8, 64. [Google Scholar] [CrossRef]
- Noga, M.J.; Dane, A.; Shi, S.; Attali, A.; van Aken, H.; Suidgeest, E.; Tuinstra, T.; Muilwijk, B.; Coulier, L.; Luider, T.; et al. Metabolomics of cerebrospinal fluid reveals changes in the central nervous system metabolism in a rat model of multiple sclerosis. Metab. Off. J. Metab. Soc. 2012, 8, 253–263. [Google Scholar] [CrossRef] [PubMed]
- van de Rest, O.; Schutte, B.A.; Deelen, J.; Stassen, S.A.; van den Akker, E.B.; van Heemst, D.; Dibbets-Schneider, P.; van Dipten-van der Veen, R.A.; Kelderman, M.; Hankemeier, T.; et al. Metabolic effects of a 13-weeks lifestyle intervention in older adults: The Growing Old Together Study. Aging 2016, 8, 111–126. [Google Scholar] [CrossRef]
- Drouin, N.; Mandscheff, J.F.; Rudaz, S.; Schappler, J. Development of a New Extraction Device Based on Parallel-Electromembrane Extraction. Anal. Chem. 2017, 89, 6346–6350. [Google Scholar] [CrossRef]
- Marchi, I.; Viette, V.; Badoud, F.; Fathi, M.; Saugy, M.; Rudaz, S.; Veuthey, J.L. Characterization and classification of matrix effects in biological samples analyses. J. Chromatogr. A 2010, 1217, 4071–4078. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Beale, R.; Airs, R. Quantification of glycine betaine, choline and trimethylamine N-oxide in seawater particulates: Minimisation of seawater associated ion suppression. Anal. Chim. Acta 2016, 938, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Drouin, N.; Rudaz, S.; Schappler, J. New Trends in Sample Preparation for Bioanalysis. Am. Pharm. Rev. 2016, 16, 62–66. [Google Scholar]
- Geiser, L.; Rudaz, S.; Veuthey, J.L. Decreasing analysis time in capillary electrophoresis: Validation and comparison of quantitative performances in several approaches. Electrophoresis 2005, 26, 2293–2302. [Google Scholar] [CrossRef]
- Kuehnbaum, N.L.; Kormendi, A.; Britz-McKibbin, P. Multisegment injection-capillary electrophoresis-mass spectrometry: A high-throughput platform for metabolomics with high data fidelity. Anal. Chem. 2013, 85, 10664–10669. [Google Scholar] [CrossRef]
- DiBattista, A.; Rampersaud, D.; Lee, H.; Kim, M.; Britz-McKibbin, P. High Throughput Screening Method for Systematic Surveillance of Drugs of Abuse by Multisegment Injection-Capillary Electrophoresis-Mass Spectrometry. Anal. Chem. 2017, 89, 11853–11861. [Google Scholar] [CrossRef]
- Gonzalez-Ruiz, V.; Gagnebin, Y.; Drouin, N.; Codesido, S.; Rudaz, S.; Schappler, J. ROMANCE: A new software tool to improve data robustness and feature identification in CE-MS metabolomics. Electrophoresis 2018. [Google Scholar] [CrossRef]
- Drouin, N.; Pezzatti, J.; Gagnebin, Y.; Gonzalez-Ruiz, V.; Schappler, J.; Rudaz, S. Effective mobility as a robust criterion for compound annotation and identification in metabolomics: Toward a mobility-based library. Anal. Chim. Acta 2018, 1032, 178–187. [Google Scholar] [CrossRef]
- Seip, K.F.; Jensen, H.; Sonsteby, M.H.; Gjelstad, A.; Pedersen-Bjergaard, S. Electromembrane extraction: Distribution or electrophoresis? Electrophoresis 2013, 34, 792–799. [Google Scholar] [CrossRef]
- Seip, K.F.; Gjelstad, A.; Pedersen-Bjergaard, S. Electromembrane extraction from aqueous samples containing polar organic solvents. J. Chromatogr. A 2013, 1308, 37–44. [Google Scholar] [CrossRef]
- Blanchard, J. Evaluation of the Relative Efficacy of Various Techniques for Deproteinizing Plasma Samples Prior to High-Performance Liquid-Chromatographic Analysis. J. Chromatogr. 1981, 226, 455–460. [Google Scholar] [CrossRef]
- Polson, C.; Sarkar, P.; Incledon, B.; Raguvaran, V.; Grant, R. Optimization of protein precipitation based upon effectiveness of protein removal and ionization effect in liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2003, 785, 263–275. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Weight (Da) | LogP * | pKa * | Structure | ChEBI | |
|---|---|---|---|---|---|
| TMAO | 75.11 | −0.9 | 4.7 |  | 15724 |
| Choline | 104.17 | −4.7 | 14.1 |  | 15354 |
| Betaine | 117.15 | −4.5 | 2.3 |  | 17750 |
| Deoxy-l-carnitine(4-trimethylammoniobutanoic acid) | 145.20 | −4.0 | 4.5 |  | 16244 |
| l-carnitine | 161.20 | −4.9 | 4.2 |  | 17126 |
| Bupivacaine | 288.43 | 4.5 | 13.6 |  | 3215 |
| PE (RSD) in % | Dynamic Range (µM) | R2 | |||
|---|---|---|---|---|---|
| 10% Plasma 30 mM TCA | 20% Plasma 60 mM TCA | 50% Plasma 150 mM TCA | |||
| TMAO | 104 (16) | 96 (13) | 94 (13) | 0.27–43.19 | 0.997 |
| Choline | 100 (5) | 84 (4) | 89 (4) | 1.8–286.5 | 0.996 |
| Betaine | 55 (12) | 66 (14) | 71 (9) | 4.6–716.1 | 0.994 |
| Deoxy-l-carnitine | 84 (2) | 60 (14) | 60 (10) | 0.1–17.6 | 0.997 |
| l-carnitine | 34 (14) | 18 (54) | 12 (6) | 3.5–556.5 | 0.995 |
| Bupivacaine | 102 (7) | 101 (3) | 105 (3) | n.d. | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drouin, N.; Kloots, T.; Schappler, J.; Rudaz, S.; Kohler, I.; Harms, A.; Wilhelmus Lindenburg, P.; Hankemeier, T. Electromembrane Extraction of Highly Polar Compounds: Analysis of Cardiovascular Biomarkers in Plasma. Metabolites 2020, 10, 4. https://doi.org/10.3390/metabo10010004
Drouin N, Kloots T, Schappler J, Rudaz S, Kohler I, Harms A, Wilhelmus Lindenburg P, Hankemeier T. Electromembrane Extraction of Highly Polar Compounds: Analysis of Cardiovascular Biomarkers in Plasma. Metabolites. 2020; 10(1):4. https://doi.org/10.3390/metabo10010004
Chicago/Turabian StyleDrouin, Nicolas, Tim Kloots, Julie Schappler, Serge Rudaz, Isabelle Kohler, Amy Harms, Petrus Wilhelmus Lindenburg, and Thomas Hankemeier. 2020. "Electromembrane Extraction of Highly Polar Compounds: Analysis of Cardiovascular Biomarkers in Plasma" Metabolites 10, no. 1: 4. https://doi.org/10.3390/metabo10010004
APA StyleDrouin, N., Kloots, T., Schappler, J., Rudaz, S., Kohler, I., Harms, A., Wilhelmus Lindenburg, P., & Hankemeier, T. (2020). Electromembrane Extraction of Highly Polar Compounds: Analysis of Cardiovascular Biomarkers in Plasma. Metabolites, 10(1), 4. https://doi.org/10.3390/metabo10010004