A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics

 ,
, 

 , ,
, , 
Abstract
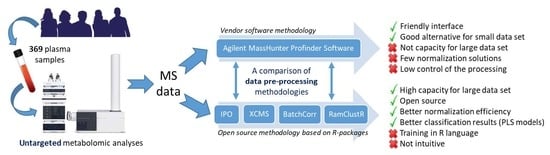
1. Introduction
2. Results and Discussion
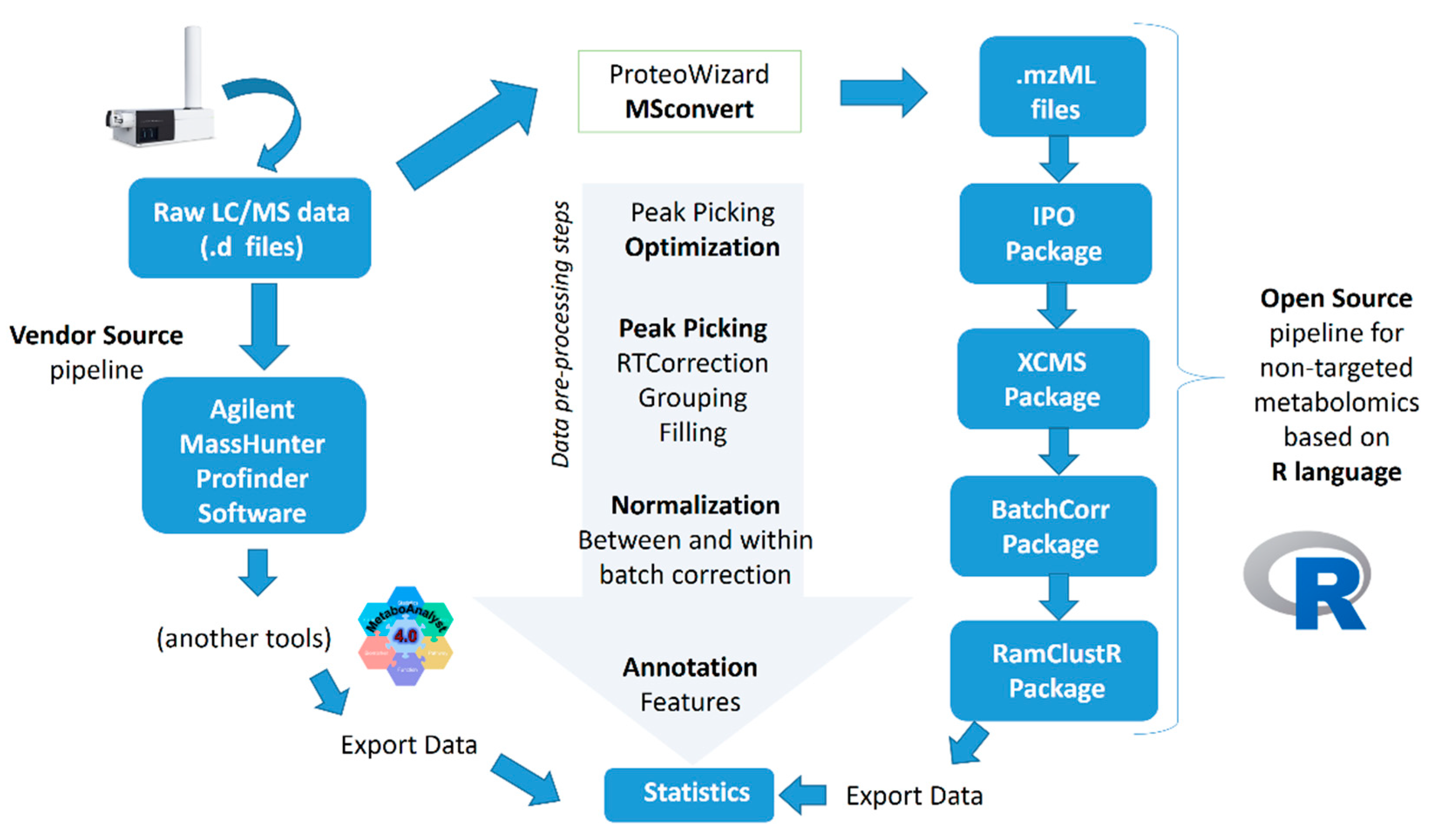
2.1. Peak Picking
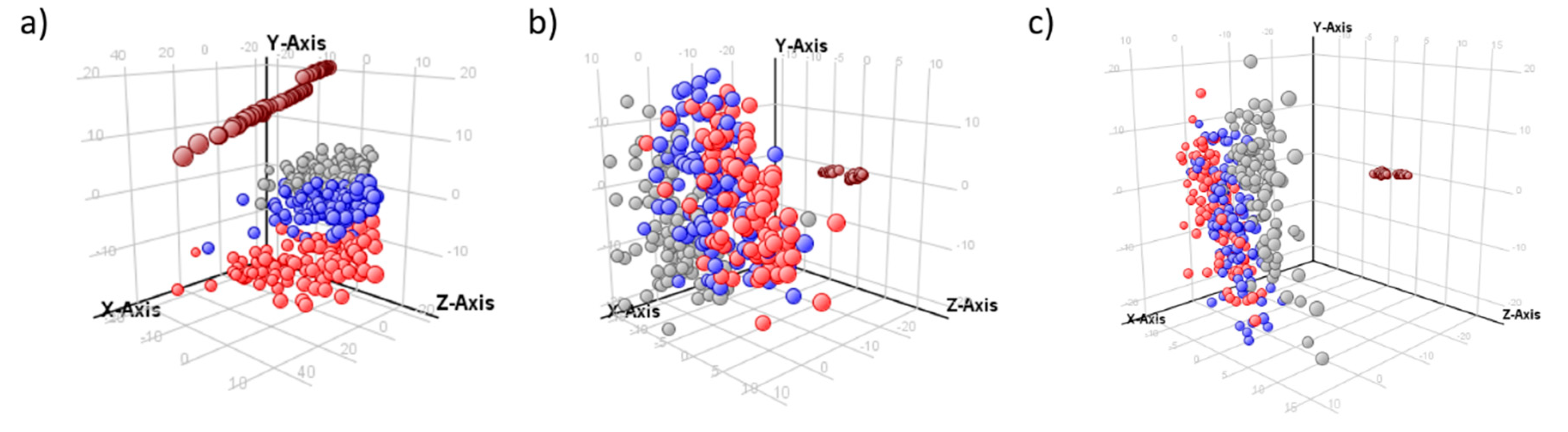
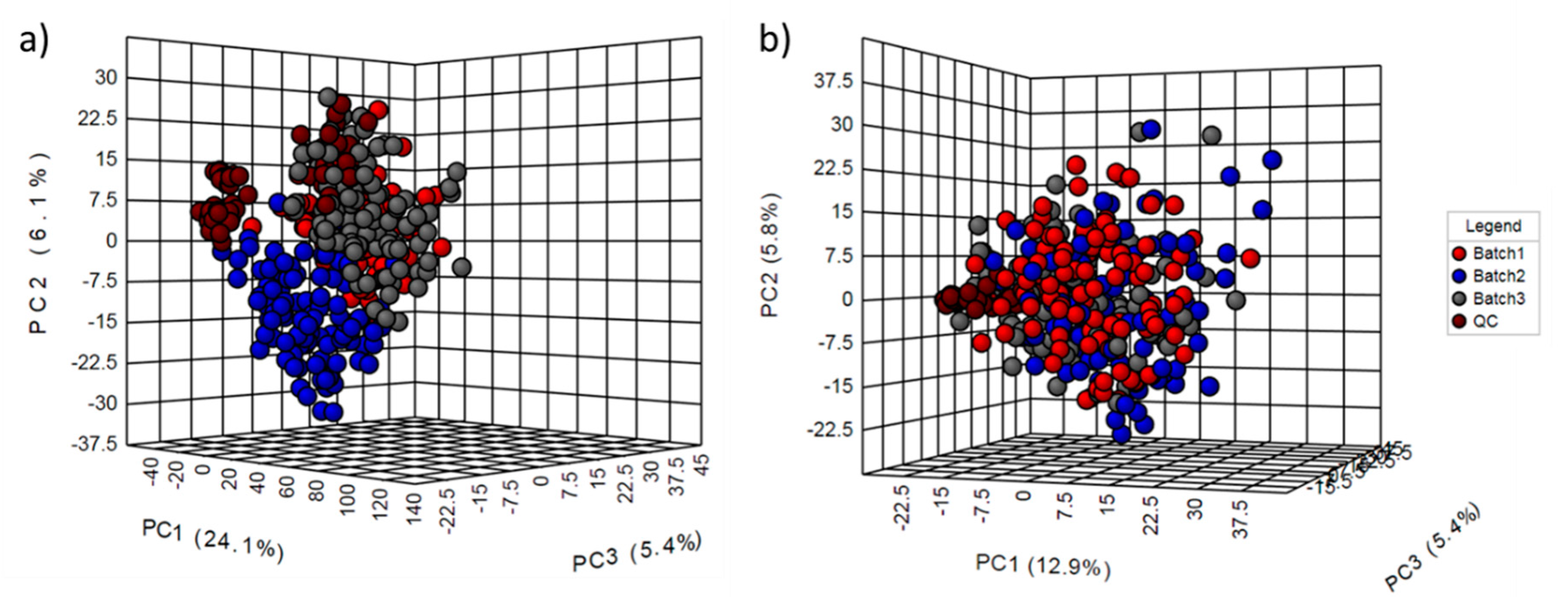
2.2. Normalization Results
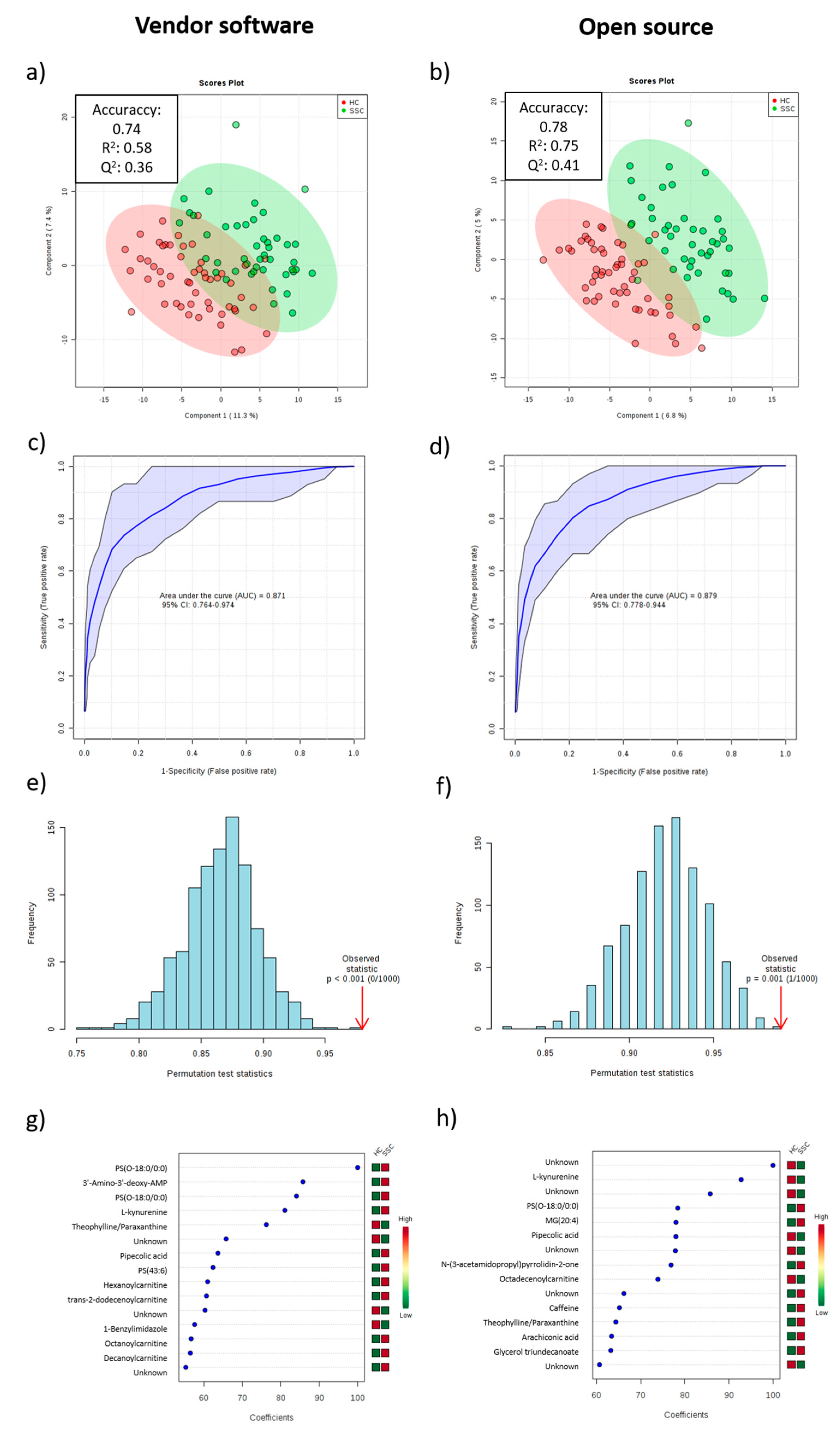
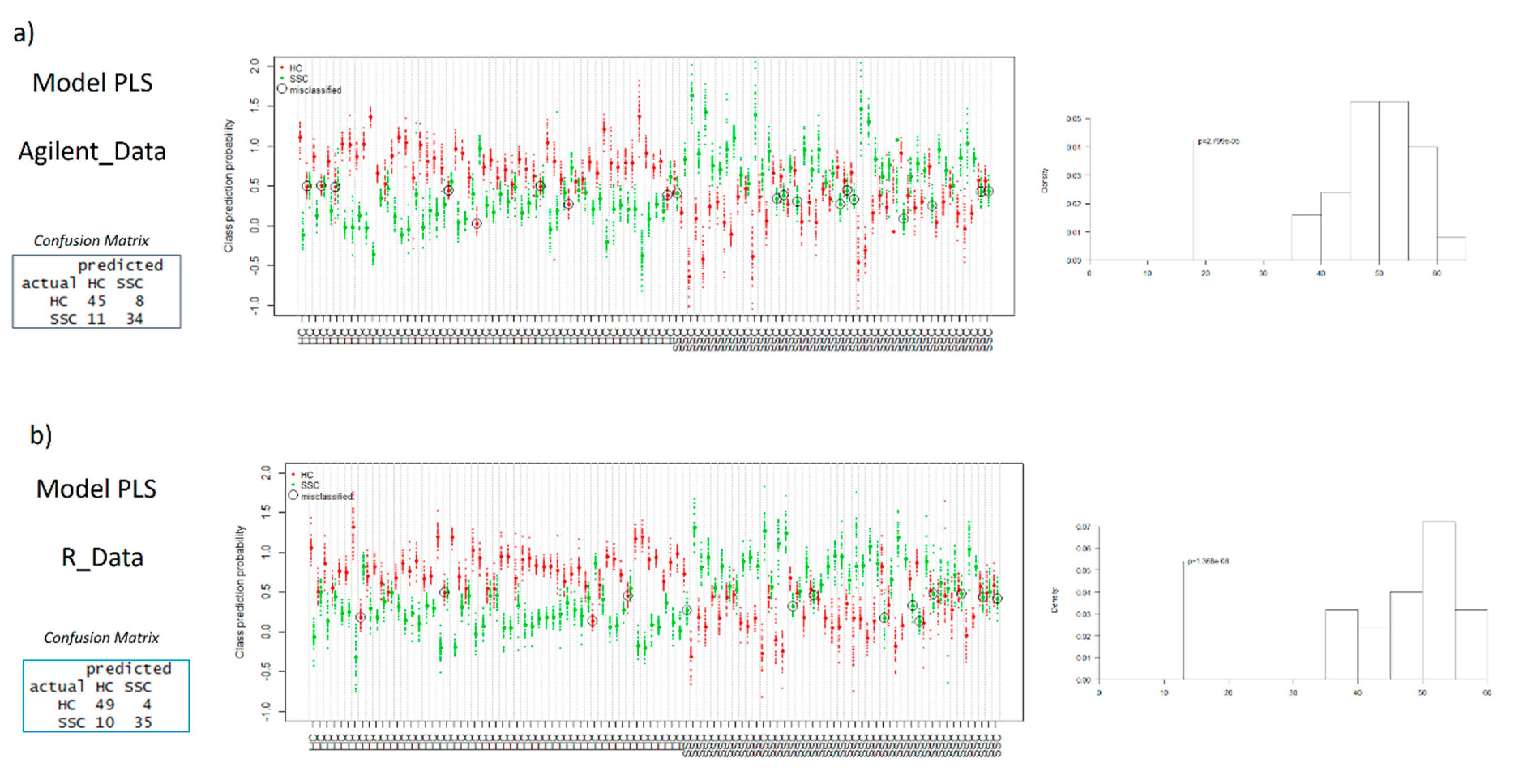
2.3. Multivariate Models
2.4. Global Comparison of Both Methodologies
3. Materials and Methods
3.1. Dataset
3.2. Data Pre-Processing
3.2.1. Agilent MassHunter Profinder Software Approach
3.2.2. R-Based Approach
3.3. Statistics and Metabolite Annotation
4. Conclusions and Future Research
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Agin, A.; Heintz, D.; Ruhland, E.; Chao de la Barca, J.M.; Zumsteg, J.; Moal, V.; Gauchez, A.S.; Namer, I.J. Metabolomics—An overview. From basic principles to potential biomarkers (part 1). Med. Nucl. 2016, 40, 4–10. [Google Scholar] [CrossRef]
- Parfieniuk, E.; Zbucka-Kretowska, M.; Ciborowski, M.; Kretowski, A.; Barbas, C. Untargeted metabolomics: An overview of its usefulness and future potential in prenatal diagnosis. Expert Rev. Proteom. 2018, 15, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Marsal, S.; Julià, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Ulaszewska, M.M.; Weinert, C.H.; Trimigno, A.; Portmann, R.; Andres Lacueva, C.; Badertscher, R.; Brennan, L.; Brunius, C.; Bub, A.; Capozzi, F.; et al. Nutrimetabolomics: An Integrative Action for Metabolomic Analyses in Human Nutritional Studies. Mol. Nutr. Food Res. 2019, 63, 1800384. [Google Scholar] [CrossRef]
- Wu, Y.; Li, L. Sample normalization methods in quantitative metabolomics. J. Chromatogr. A 2016, 1430, 80–95. [Google Scholar] [CrossRef]
- Emwas, A.H.M. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 2015, 1277, 161–193. [Google Scholar]
- Spicer, R.; Salek, R.M.; Moreno, P.; Cañueto, D.; Steinbeck, C. Navigating freely-available software tools for metabolomics analysis. Metabolomics 2017, 13, 106. [Google Scholar] [CrossRef]
- Castillo, S.; Gopalacharyulu, P.; Yetukuri, L.; Orešič, M. Algorithms and tools for the preprocessing of LC–MS metabolomics data. Chemom. Intell. Lab. Syst. 2011, 108, 23–32. [Google Scholar] [CrossRef]
- Sugimoto, M.; Kawakami, M.; Robert, M.; Soga, T.; Tomita, M. Bioinformatics Tools for Mass Spectroscopy-Based Metabolomic Data Processing and Analysis. Curr. Bioinform. 2012, 7, 96–108. [Google Scholar] [CrossRef]
- Katajamaa, M.; Orešič, M. Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A 2007, 1158, 318–328. [Google Scholar] [CrossRef]
- Hao, L.; Wang, J.; Page, D.; Asthana, S.; Zetterberg, H.; Carlsson, C.; Okonkwo, O.C.; Li, L. Comparative Evaluation of MS-based Metabolomics Software and Its Application to Preclinical Alzheimer’s Disease. Sci. Rep. 2018, 8, 9291. [Google Scholar] [CrossRef]
- Vettukattil, R. Preprocessing of Raw Metabonomic Data; Humana Press: New York, NY, USA, 2015; pp. 123–136. [Google Scholar]
- Vaclavik, L.; Lacina, O.; Hajslova, J.; Zweigenbaum, J. The use of high performance liquid chromatography-quadrupole time-of-flight mass spectrometry coupled to advanced data mining and chemometric tools for discrimination and classification of red wines according to their variety. Anal. Chim. Acta 2011, 685, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Sánchez De Medina, V.; Priego-Capote, F.; Luque De Castro, M.D. Characterization of refined edible oils enriched with phenolic extracts from olive leaves and pomace. J. Agric. Food Chem. 2012, 60, 5866–5873. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Giacomoni, F.; Le Corguille, G.; Monsoor, M.; Landi, M.; Pericard, P.; Petera, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef]
- Lommen, A. MetAlign: Interface-Driven, Versatile Metabolomics Tool for Hyphenated Full-Scan Mass Spectrometry Data Preprocessing. Anal. Chem. 2009, 81, 3079–3086. [Google Scholar] [CrossRef]
- Bertsch, A.; Gröpl, C.; Reinert, K.; Kohlbacher, O. OpenMS and TOPP: Open Source Software for LC-MS Data Analysis. Methods Mol. Biol.. 2011, 696, 353–367. [Google Scholar]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Weber, R.J.M.; Lawson, T.N.; Salek, R.M.; Ebbels, T.M.D.; Glen, R.C.; Goodacre, R.; Griffin, J.L.; Haug, K.; Koulman, A.; Moreno, P.; et al. Computational tools and workflows in metabolomics: An international survey highlights the opportunity for harmonisation through Galaxy. Metabolomics 2017, 13, 12. [Google Scholar] [CrossRef]
- Li, Z.; Lu, Y.; Guo, Y.; Cao, H.; Wang, Q.; Shui, W. Comprehensive evaluation of untargeted metabolomics data processing software in feature detection, quantification and discriminating marker selection. Anal. Chim. Acta 2018, 1029, 50–57. [Google Scholar] [CrossRef]
- Libiseller, G.; Dvorzak, M.; Kleb, U.; Gander, E.; Eisenberg, T.; Madeo, F.; Neumann, S.; Trausinger, G.; Sinner, F.; Pieber, T.; et al. IPO: A tool for automated optimization of XCMS parameters. BMC Bioinform. 2015, 16, 118. [Google Scholar] [CrossRef]
- Brunius, C.; Shi, L.; Landberg, R. Large-scale untargeted LC-MS metabolomics data correction using between-batch feature alignment and cluster-based within-batch signal intensity drift correction. Metabolomics 2016, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Broeckling, C.D.; Afsar, F.A.; Neumann, S.; Ben-Hur, A.; Prenni, J.E. RAMClust: A novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal. Chem. 2014, 86, 6812–6817. [Google Scholar] [CrossRef] [PubMed]
- Ejigu, B.A.; Valkenborg, D.; Baggerman, G.; Vanaerschot, M.; Witters, E.; Dujardin, J.-C.; Burzykowski, T.; Berg, M. Evaluation of normalization methods to pave the way towards large-scale LC-MS-based metabolomics profiling experiments. OMICS 2013, 17, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Ueda, K.; Kobayashi, Y.; Tsuyama, N.; Todoroki, K.; Min, J.Z.; Toyo’oka, T. The great importance of normalization of LC-MS data for highly-accurate non-targeted metabolomics. Biomed. Chromatogr. 2017, 31, 1–7. [Google Scholar] [CrossRef]
- Shen, X.; Gong, X.; Cai, Y.; Guo, Y.; Tu, J.; Li, H.; Zhang, T.; Wang, J.; Xue, F.; Zhu, Z.-J. Normalization and integration of large-scale metabolomics data using support vector regression. Metabolomics 2016, 12, 89. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.; Lim, M.; Seong, S.J.; Seo, J.J.; Park, S.M.; Lee, H.W.; Yoon, Y.-R. Quantile normalization approach for liquid chromatography-mass spectrometry-based metabolomic data from healthy human volunteers. Anal. Sci. 2012, 28, 801–805. [Google Scholar] [CrossRef]
- Nodzenski, M.; Muehlbauer, M.J.; Bain, J.R.; Reisetter, A.C.; Lowe, W.L.; Scholtens, D.M. Metabomxtr: An R package for mixture-model analysis of non-targeted metabolomics data. Bioinformatics 2014, 30, 3287–3288. [Google Scholar] [CrossRef]
- Chawade, A.; Alexandersson, E.; Levander, F. Normalyzer: A Tool for Rapid Evaluation of Normalization Methods for Omics Data Sets. J. Proteome Res. 2014, 13, 3114–3120. [Google Scholar] [CrossRef]
- De Livera, A.M.; Olshansky, G.; Simpson, J.A.; Creek, D.J. NormalizeMets: Assessing, selecting and implementing statistical methods for normalizing metabolomics data. Metabolomics 2018, 14, 54. [Google Scholar] [CrossRef]
- Shi, L.; Westerhuis, J.A.; Rosén, J.; Landberg, R.; Brunius, C. Variable selection and validation in multivariate modelling. Bioinformatics 2018, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.; Liu, A. What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Rev. Neurother 2015, 15, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, A.A.; Trygg, J.; Wuttge, D.M.; Sturfelt, G.; Theander, E.; Donten, M.; Moritz, T.; Sennbro, C.J.; Torell, F.; Lood, C.; et al. Metabolic profiling of systemic lupus erythematosus and comparison with primary Sjögren’s syndrome and systemic sclerosis. PLoS ONE 2016, 11, e0159384. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ochoa, Á.; Quirantes-Piné, R.; Borrás-Linares, I.; Gemperline, D.; Alarcón Riquelme, M.E.; Beretta, L.; Segura-Carretero, A. Urinary and plasma metabolite differences detected by HPLC-ESI-QTOF-MS in systemic sclerosis patients. J. Pharm. Biomed. Anal. 2019, 162, 82–90. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2016, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Adusumilli, R.; Mallick, P. Data Conversion with ProteoWizard msConvert; Humana Press: New York, NY, USA, 2017; pp. 339–368. [Google Scholar]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef]
- Lindgren, F.; Hansen, B.; Karcher, W.; Sjöström, M.; Eriksson, L. Model validation by permutation tests: Applications to variable selection. J. Chemom. 1996, 10, 521–532. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Gil de la Fuente, A.; Grace Armitage, E.; Otero, A.; Barbas, C.; Godzien, J. Differentiating signals to make biological sense—A guide through databases for MS-based non-targeted metabolomics. Electrophoresis 2017, 38, 2242–2256. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS-MUVR Models | nVar | Class (%) | AUC | nComp | p-Value |
|---|---|---|---|---|---|
| R Data | 15 | 86.8 | 0.931 | 2 | 1.38 × 10−6 |
| Profinder software Data | 67 | 81.7 | 0.893 | 3 | 2.80 × 10−5 |
| Profinder Software Methodology | R-Based Methodology | ||
|---|---|---|---|
 |  | | |
| Easy to use, user-friendly interface | License fee | Open source | Steep learning curve |
| High quality of the plots | Limited capacity to process a high number of samples | Greater number of packages, functions, and methods (e.g., normalization) | Low plot quality (plots obtained with the specific R packages used) |
| No need to transform the format of the data | Few normalization techniques. Difficulties to normalize large between-batch effects | High capacity for faster processing of a high number of samples | Data format transformation |
| Easy to inspect features, integration results, and MS spectra. Easy to predict molecular formula | Errors in peak integration | Possibility of carrying out all the steps of pre-processing and statistical analysis in the same environment | More cumbersome to show integration results, MS spectra, and to predict molecular formula |
| Easy to manually correct areas | Low control of the processing (only some parameters can be modified) | Flexibility and versatility | Some level of coding skills is required |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Ochoa, Á.; Quirantes-Piné, R.; Borrás-Linares, I.; Cádiz-Gurrea, M.d.l.L.; PRECISESADS Clinical Consortium; Alarcón Riquelme, M.E.; Brunius, C.; Segura-Carretero, A. A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics. Metabolites 2020, 10, 28. https://doi.org/10.3390/metabo10010028
Fernández-Ochoa Á, Quirantes-Piné R, Borrás-Linares I, Cádiz-Gurrea MdlL, PRECISESADS Clinical Consortium, Alarcón Riquelme ME, Brunius C, Segura-Carretero A. A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics. Metabolites. 2020; 10(1):28. https://doi.org/10.3390/metabo10010028
Chicago/Turabian StyleFernández-Ochoa, Álvaro, Rosa Quirantes-Piné, Isabel Borrás-Linares, María de la Luz Cádiz-Gurrea, PRECISESADS Clinical Consortium, Marta E. Alarcón Riquelme, Carl Brunius, and Antonio Segura-Carretero. 2020. "A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics" Metabolites 10, no. 1: 28. https://doi.org/10.3390/metabo10010028
APA StyleFernández-Ochoa, Á., Quirantes-Piné, R., Borrás-Linares, I., Cádiz-Gurrea, M. d. l. L., PRECISESADS Clinical Consortium, Alarcón Riquelme, M. E., Brunius, C., & Segura-Carretero, A. (2020). A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics. Metabolites, 10(1), 28. https://doi.org/10.3390/metabo10010028