Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS



Abstract
1. Introduction
2. Results and Discussion
2.1. CCS Library
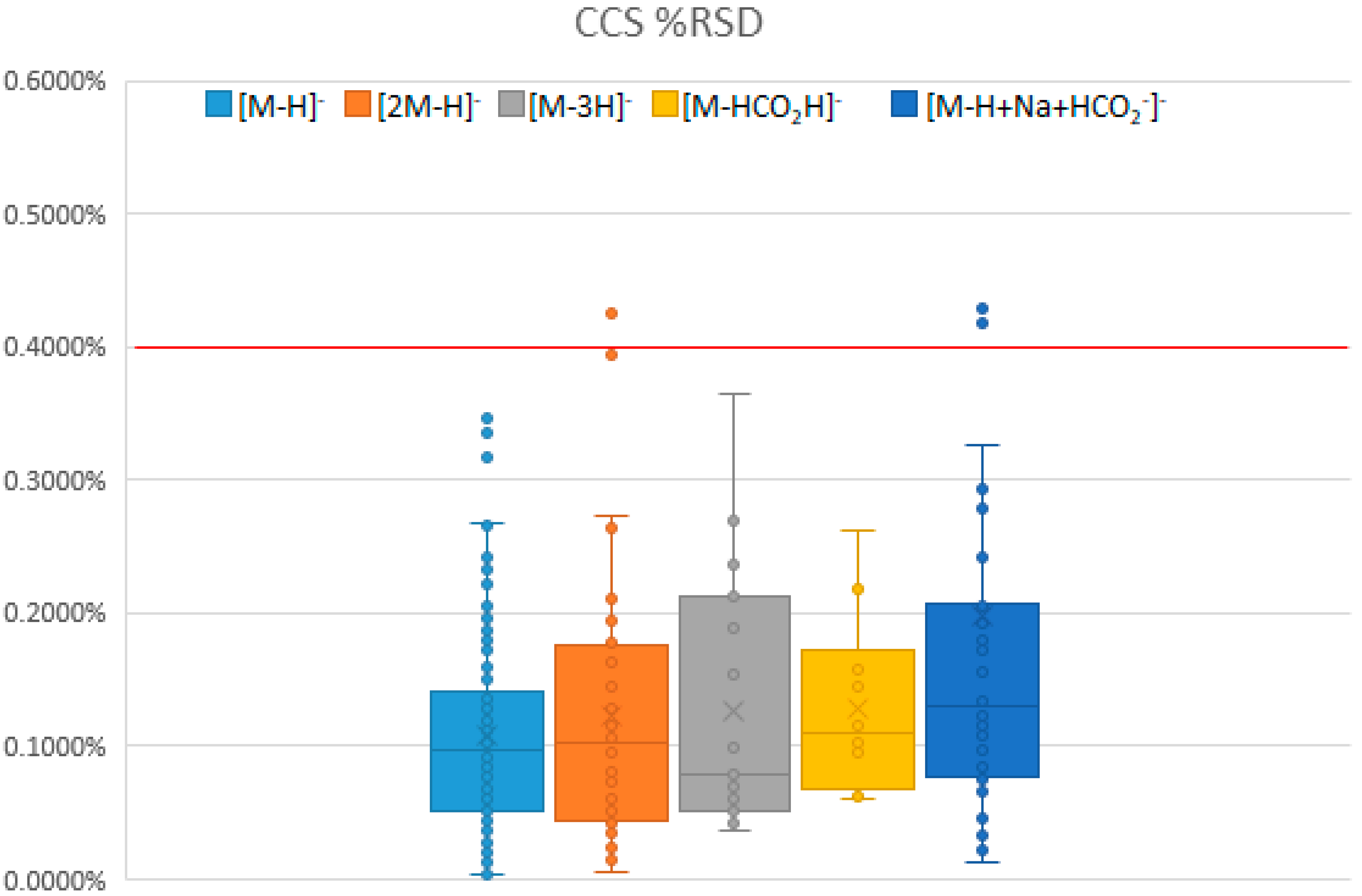
2.1.1. Statistics and Reproducibility
2.1.2. Comparison of CCS Values with Published Data
2.1.3. Isomer Comparisons
2.2. Ion Mobility Spectra Interpretations
2.3. In-TIMS Ion Manipulation
2.4. CCS Matching with Authentic Standards
2.5. CCS Value Matching with Plant Extracts
3. Materials and Methods
3.1. Chemicals
3.2. UHPLC-ESI-TIMS-QTOF-MS
3.2.1. UHPLC
3.2.2. ESI-TIMS-QTOF-MS
3.2.3. Calibration
3.3. Plant Extract Preparation
3.4. Data Processing and CCS Determination
3.5. TIMS Δ6 Adduct Investigations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sumner, L.W.; Lei, Z.; Nikolau, B.J.; Saito, K. Modern plant metabolomics: Advanced natural product gene discoveries, improved technologies, and future prospects. Nat. Prod. Rep. 2015, 32, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Matsuda, F. Metabolomics for Functional Genomics, Systems Biology, and Biotechnology. Annu. Rev. Plant Biol. 2010, 61, 463–489. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Mendes, P.; Dixon, R.A. Plant metabolomics: Large-scale phytochemistry in the functional genomics era. Phytochemistry 2003, 62, 817–836. [Google Scholar] [CrossRef]
- Guy, C.; Kaplan, F.; Kopka, J.; Selbig, J.; Hincha, D.K. Metabolomics of temperature stress. Physiol. Plant. 2008, 132, 220–235. [Google Scholar] [CrossRef]
- Hall, R.D. Plant metabolomics: From holistic hope, to hype, to hot topic. New Phytol. 2006, 169, 453–468. [Google Scholar] [CrossRef]
- Shulaev, V.; Cortes, D.; Miller, G.; Mittler, R. Metabolomics for plant stress response. Physiol. Plant. 2008, 132, 199–208. [Google Scholar] [CrossRef]
- Sumner, L.W.; Styczynski, M.; McLean, J.; Fiehn, O.; Jander, G.; Liao, J.; Sumner, S.; Britz-McKibbin, P.; Welti, R.; Jones, A.D.; et al. Introducing the USA Plant, Algae and Microbial Metabolomics Research Coordination Network (PAMM-NET). Metabolomics 2015, 11, 3–5. [Google Scholar] [CrossRef][Green Version]
- Neue, U.D. Theory of peak capacity in gradient elution. J. Chromatogr. A 2005, 1079, 153–161. [Google Scholar] [CrossRef]
- Plumb, R.; Castro-Perez, J.; Granger, J.; Beattie, I.; Joncour, K.; Wright, A. Ultra-performance liquid chromatography coupled to quadrupole-orthogonal time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 2331–2337. [Google Scholar] [CrossRef]
- Adahchour, M.; Beens, J.; Brinkman, U.A.T. Recent developments in the application of comprehensive two-dimensional gas chromatography. J. Chromatogr. A 2008, 1186, 67–108. [Google Scholar] [CrossRef]
- Stoll, D.R.; Li, X.; Wang, X.; Carr, P.W.; Porter, S.E.G.; Rutan, S.C. Fast, comprehensive two-dimensional liquid chromatography. J. Chromatogr. A 2007, 1168, 3–43. [Google Scholar] [CrossRef] [PubMed]
- Stoll, D.R.; Cohen, J.D.; Carr, P.W. Fast, comprehensive online two-dimensional high performance liquid chromatography through the use of high temperature ultra-fast gradient elution reversed-phase liquid chromatography. J. Chromatogr. A 2006, 1122, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Eiceman, G.A.; Karpas, Z.; Hill, H., Jr. Ion Mobility Spectrometry, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2013; ISBN 1439859973. [Google Scholar]
- May, J.C.; McLean, J.A. Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Anal. Chem. 2015, 87, 1422–1436. [Google Scholar] [CrossRef] [PubMed]
- Michelmann, K.; Silveira, J.A.; Ridgeway, M.E.; Park, M.A. Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 14–24. [Google Scholar] [CrossRef]
- Silveira, J.A.; Michelmann, K.; Ridgeway, M.E.; Park, M.A. Fundamentals of Trapped Ion Mobility Spectrometry Part II: Fluid Dynamics. J. Am. Soc. Mass Spectrom. 2016, 27, 585–595. [Google Scholar] [CrossRef]
- Dixon, R.A.; Pasinetti, G.M. Flavonoids and Isoflavonoids: From Plant Biology to Agriculture and Neuroscience. Plant Physiol. 2010, 154, 453–457. [Google Scholar] [CrossRef]
- Dixon, R.A.; Sumner, L.W. Legume Natural Products: Understanding and Manipulating Complex Pathways for Human and Animal Health. Plant Physiol. 2003, 131, 878–885. [Google Scholar] [CrossRef]
- Cseke, L.; Kirakosyan, A.; Kaufman, P.; Warber, S.; Duke, J.; Brielmann, H. Natural Products from Plants, 2nd ed.; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2006; ISBN 9780849329760. [Google Scholar]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- Creek, D.J.; Dunn, W.B.; Fiehn, O.; Griffin, J.L.; Hall, R.D.; Lei, Z.; Mistrik, R.; Neumann, S.; Schymanski, E.L.; Sumner, L.W.; et al. Metabolite identification: Are you sure? And how do your peers gauge your confidence? Metabolomics 2014, 10, 350–353. [Google Scholar] [CrossRef]
- Lei, Z.; Jing, L.; Qiu, F.; Zhang, H.; Huhman, D.; Zhou, Z.; Sumner, L.W. Construction of an Ultrahigh Pressure Liquid Chromatography-Tandem Mass Spectral Library of Plant Natural Products and Comparative Spectral Analyses. Anal. Chem. 2015, 87, 7373–7381. [Google Scholar] [CrossRef]
- Fine, D.; Wherritt, D.; Barsch, A.; Sumner, L.W. Bruker-Sumner Metabobase Plant Libraries 1.0. 2015. Available online: https://www.bruker.com/service/support-upgrades/software-downloads/mass-spectrometry.html (accessed on 15 July 2016).
- Kanu, A.B.; Dwivedi, P.; Tam, M.; Matz, L.; Hill, H.H. Ion mobility-mass spectrometry. J. Mass Spectrom. 2008, 43, 1–22. [Google Scholar] [CrossRef]
- May, J.C.; Morris, C.B.; McLean, J.A. Ion Mobility Collision Cross Section Compendium. Anal. Chem. 2017, 89, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef]
- Silveira, J.A.; Ridgeway, M.E.; Park, M.A. High Resolution Trapped Ion Mobility Spectrometery of Peptides. Anal. Chem. 2014, 86, 5624–5627. [Google Scholar] [CrossRef]
- Stow, S.M.; Causon, T.J.; Zheng, X.; Kurulugama, R.T.; Mairinger, T.; May, J.C.; Rennie, E.E.; Baker, E.S.; Smith, R.D.; McLean, J.A.; et al. An Interlaboratory Evaluation of Drift Tube Ion Mobility–Mass Spectrometry Collision Cross Section Measurements. Anal. Chem. 2017, 89, 9048–9055. [Google Scholar] [CrossRef]
- Zheng, X.; Aly, N.A.; Zhou, Y.; Dupuis, K.T.; Bilbao, A.; Paurus, V.L.; Orton, D.J.; Wilson, R.; Payne, S.H.; Smith, R.D.; et al. A structural examination and collision cross section database for over 500 metabolites and xenobiotics using drift tube ion mobility spectrometry. Chem. Sci. 2017, 8, 7724–7736. [Google Scholar] [CrossRef]
- Dodds, J.N.; May, J.C.; Mclean, J.A. Correlating Resolving Power, Resolution and Collision Cross Section: Unifying Cross Platform Assessment of Separation Efficiency in Ion Mobility Spectrometry Graphical abstract HHS Public Access. Anal. Chem. 2017, 89, 12176–12184. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UHPLC-TIMS-MS Library CCS 1 | Direct Infusion Mixture CCS | CCS % Difference | |
|---|---|---|---|
| Rutin | 231.05 | 232.37 | 0.56679% |
| Naringin | 215.92 | 215.75 | 0.07876% |
| Naringenin | 163.01 | 162.99 | 0.00818% |
| Chrysin | 156.14 | 156.15 | 0.00854% |
| 6-hydroxyflavone | 155.91 | 155.61 | 0.19688% |
| Average | - | - | 0.16557% |
| Compounds | CCS without Matrix | CCS with Matrix | CCS % Difference |
|---|---|---|---|
| Rutin | 232.56 | 232.64 | 0.03583% |
| Naringin | 215.66 | 215.90 | 0.11431% |
| Naringenin | 162.91 | 162.95 | 0.02455% |
| Chrysin | 156.14 | 156.25 | 0.07042% |
| 6-hydroxyflavone | 156.05 | 155.94 | 0.07265% |
| Average | - | - | 0.06355% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schroeder, M.; Meyer, S.W.; Heyman, H.M.; Barsch, A.; Sumner, L.W. Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS. Metabolites 2020, 10, 13. https://doi.org/10.3390/metabo10010013
Schroeder M, Meyer SW, Heyman HM, Barsch A, Sumner LW. Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS. Metabolites. 2020; 10(1):13. https://doi.org/10.3390/metabo10010013
Chicago/Turabian StyleSchroeder, Mark, Sven W. Meyer, Heino M. Heyman, Aiko Barsch, and Lloyd W. Sumner. 2020. "Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS" Metabolites 10, no. 1: 13. https://doi.org/10.3390/metabo10010013
APA StyleSchroeder, M., Meyer, S. W., Heyman, H. M., Barsch, A., & Sumner, L. W. (2020). Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS. Metabolites, 10(1), 13. https://doi.org/10.3390/metabo10010013