


Multimodal Gene Expression and Methylation Profiling Reveals Misclassified Tumors Beyond Histological Diagnosis


Abstract
1. Introduction
2. Materials and Methods
2.1. Overview of This Study
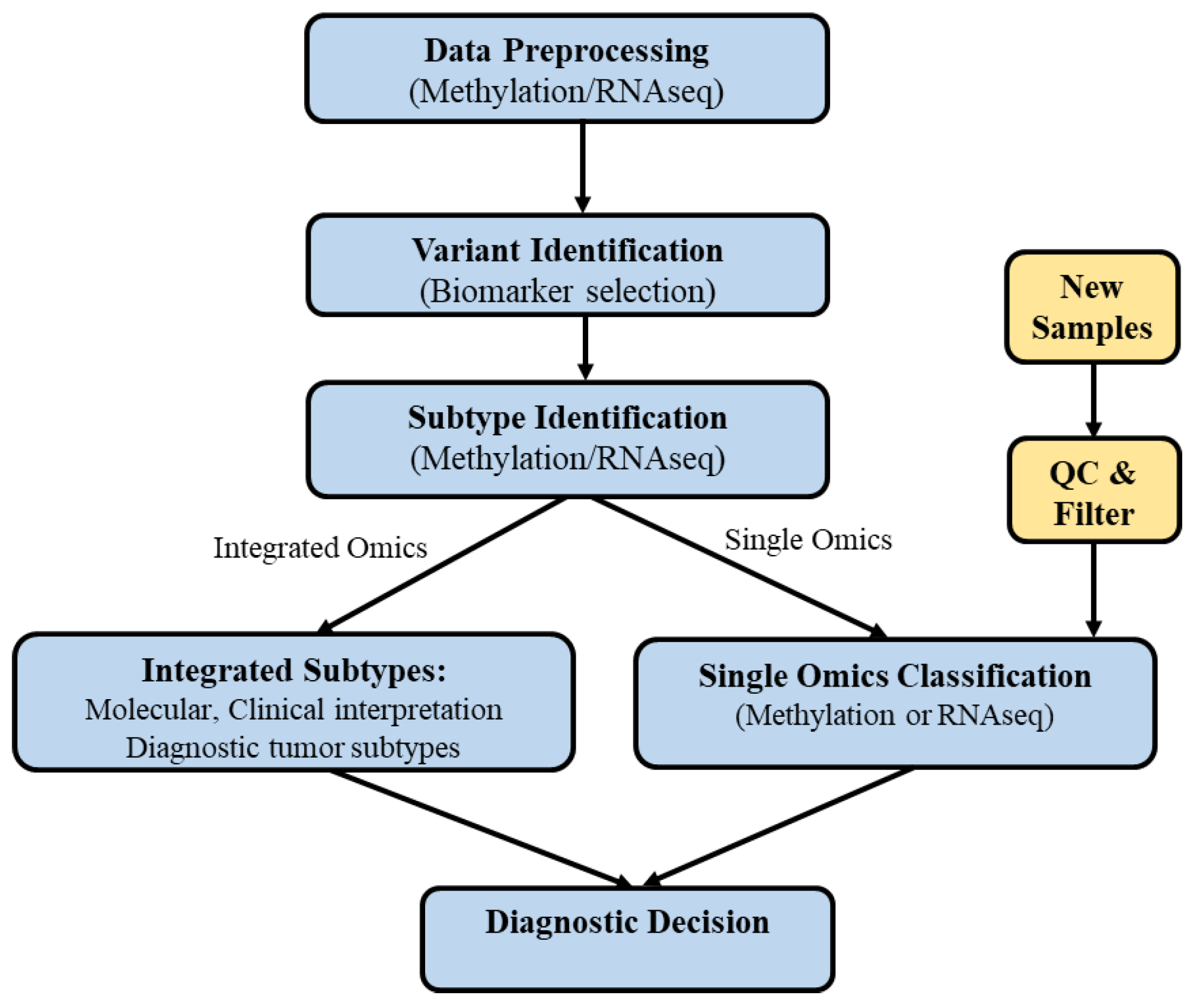
2.2. Developed Pipeline
2.3. Data Collection and Preprocessing
2.4. Feature Selection and Clustering
2.5. Classification Models
2.6. Limitations of Existing Approaches and Rationale for Our Strategy
3. Results
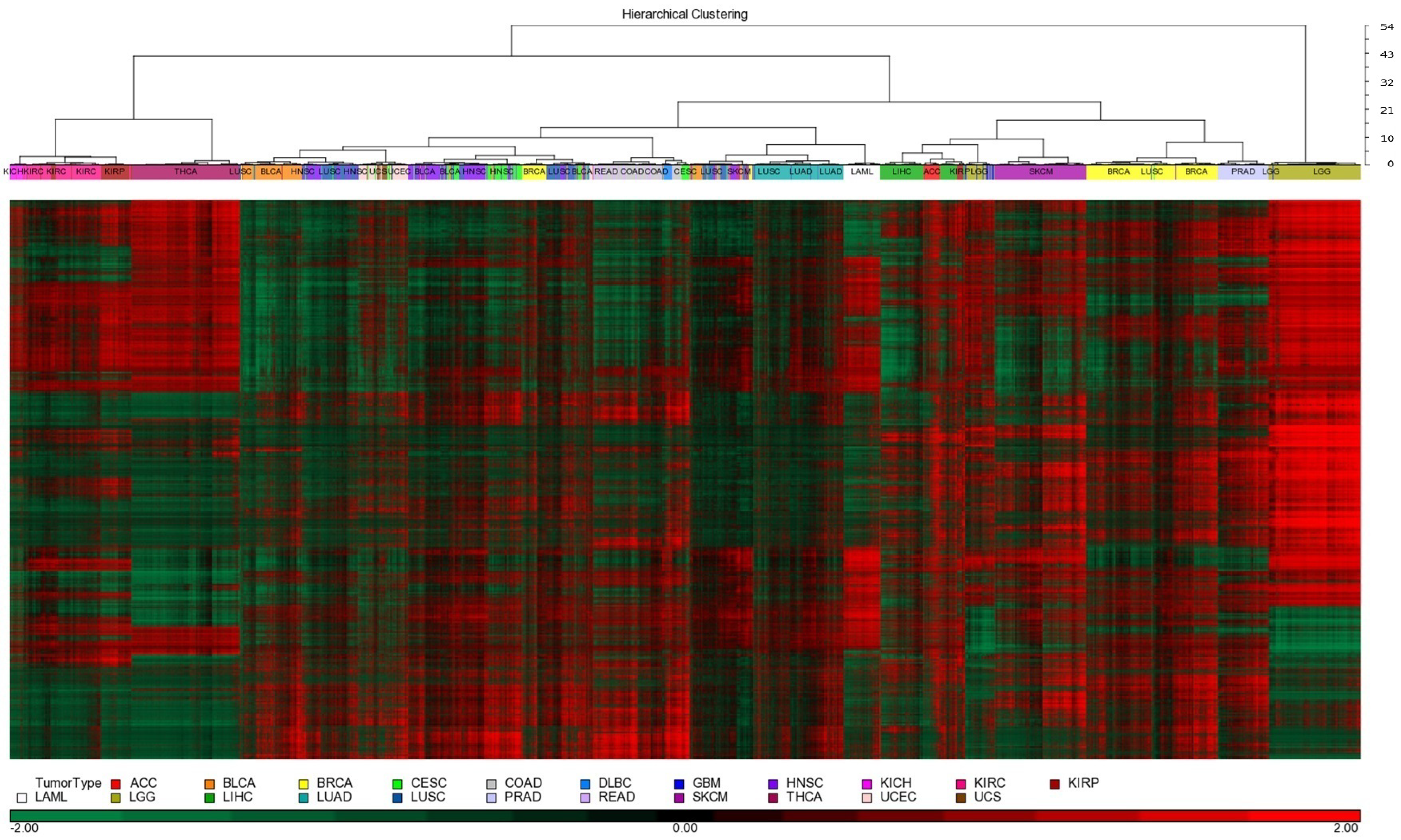
3.1. Patient Samples, Molecular Platforms, and Clustering
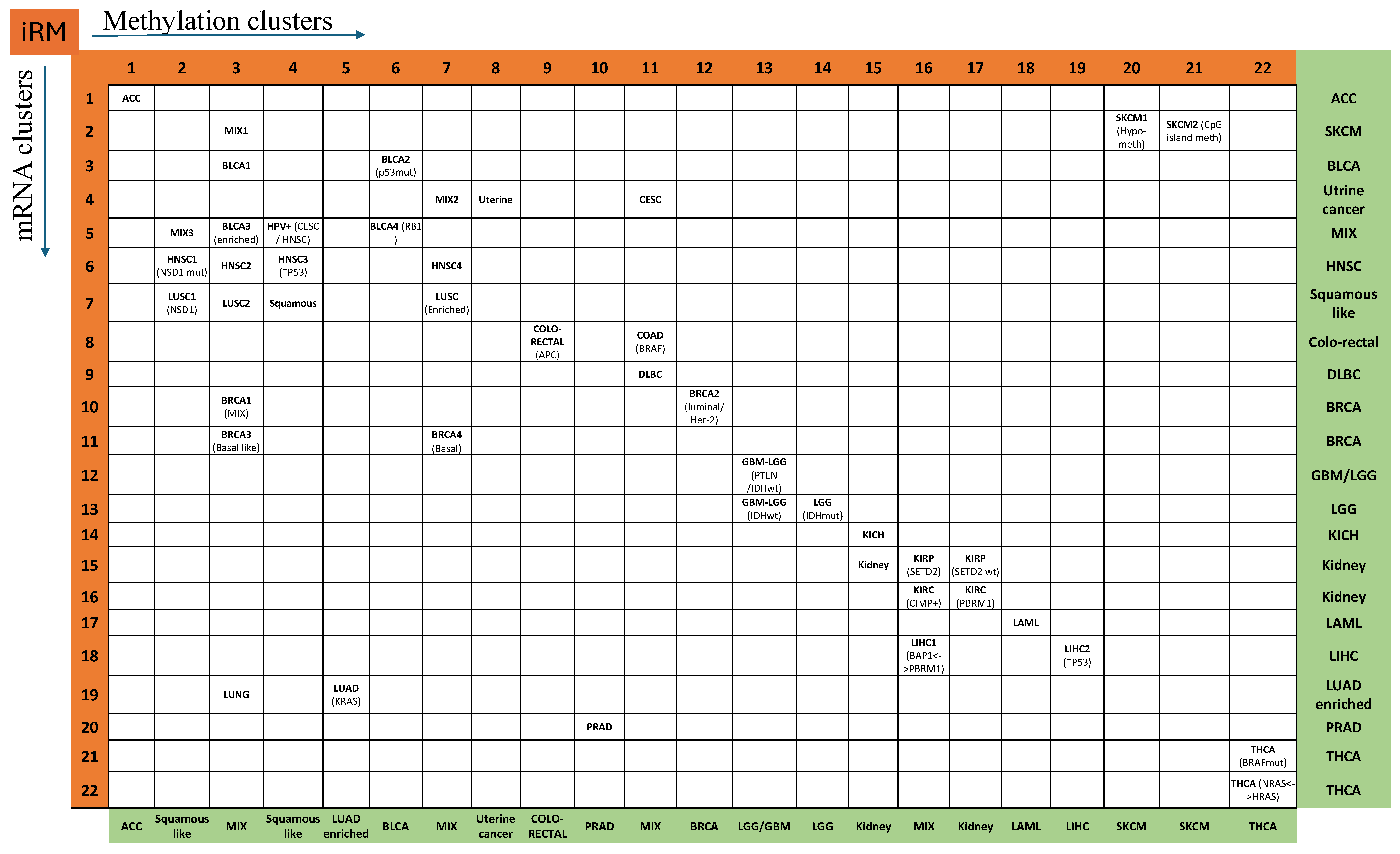
3.2. Integrated RNA and Methylation (iRM) Molecular Taxonomy
3.3. Clinically Significant iRM Signatures
3.4. Histologic Features of Outlier Samples Based on iRM Signatures
3.5. Classification to Identify Misclassified Samples
3.6. mRNA-Based Misclassification Identification
3.7. Methylation-Based Misclassification Identification
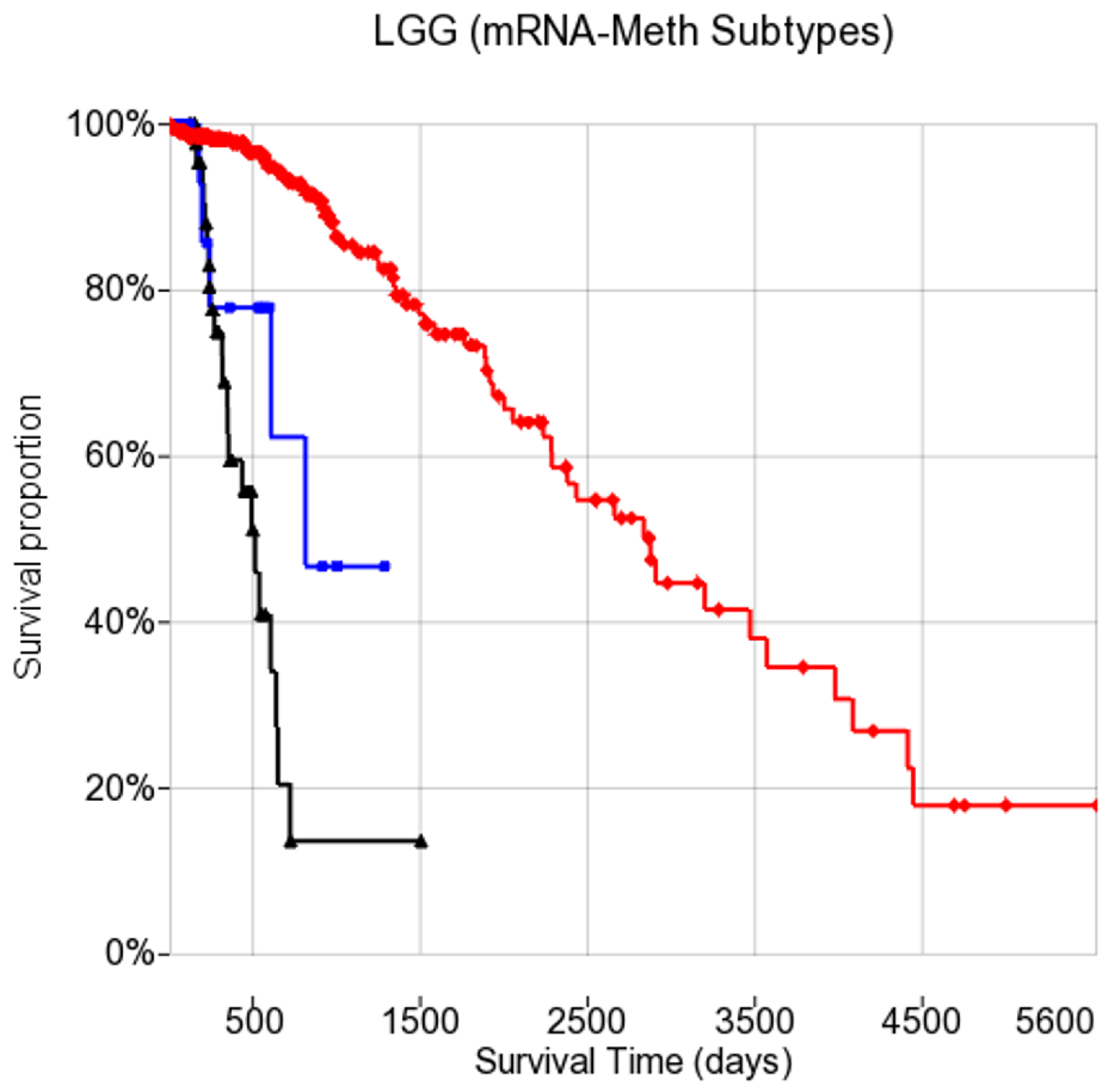
3.8. Subtype Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.; Miller, K.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cibas, E.; Ducatman, B. Cibas and Ducatman’s Cytology: Diagnostic Principles and Clinical Correlates, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2025. [Google Scholar]
- Wang, W.; Zhao, Y.; Teng, L.; Yan, J.; Guo, Y.; Qiu, Y.; Ji, Y.; Yu, B.; Pei, D.; Duan, W.; et al. Neuropathologist-level integrated classification of adult-type diffuse gliomas using deep learning from whole-slide pathological images. Nat. Commun. 2023, 14, 6359. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Wang, Z.; Cheng, Z.; Wang, W.; Lu, S.; Zhang, Z.; Anene, C.; Khan, F.; Chen, Y.; Bailey, E.; et al. The integrated molecular and histological analysis defines subtypes of esophageal squamous cell carcinoma. Nat. Commun. 2024, 15, 8988. [Google Scholar] [CrossRef] [PubMed]
- Moran, S.; Martínez-Cardús, A.; Sayols, S.; Musulén, E.; Balañá, C.; Estival-Gonzalez, A.; Moutinho, C.; Heyn, H.; Diaz-Lagares, A.; de Moura, M.; et al. Epigenetic profiling to classify cancer of unknown primary: A multicentre, retrospective analysis. Lancet Oncol. 2016, 17, 1386–1395. [Google Scholar] [CrossRef]
- Elmore, J.; Longton, G.; Carney, P.; Geller, B.; Onega, T.; Tosteson, A.; Nelson, H.; Pepe, M.; Allison, K.; Schnitt, S.; et al. Diagnostic concordance among pathologists interpreting breast biopsy specimens. JAMA 2015, 313, 1122–1132. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Berger, A.; Korkut, A.; Kanchi, R.; Hegde, A.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef]
- Sturm, D.; Capper, D.; Andreiuolo, F.; Gessi, M.; Kölsche, C.; Reinhardt, A.; Sievers, P.; Wefers, A.; Ebrahimi, A.; Suwala, A.; et al. Author Correction: Multiomic neuropathology improves diagnostic accuracy in pediatric neuro-oncology. Nat. Med. 2024, 30, 306. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Q.; Zhang, Z.; Long, L.; Ezhilarasan, R.; Karp, J.; Tsirigos, A.; Snuderl, M.; Wiestler, B.; Wick, W.; et al. DNA methylation-based epigenetic signatures predict somatic genomic alterations in gliomas. Nat. Commun. 2022, 13, 4410. [Google Scholar] [CrossRef]
- Weinstein, J.; Collisson, E.; Mills, G.; Shaw, K.; Ozenberger, B.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.; The Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Hoadley, K.; Yau, C.; Wolf, D.; Cherniack, A.; Tamborero, D.; Ng, S.; Leiserson, M.; Niu, B.; McLellan, M.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Overby, C.; Tarczy-Hornoch, P. Personalized Medicine: Challenges and Opportunities for Translational Bioinformatics. Per. Med. 2013, 10, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Reuss, D.; Mamatjan, Y.; Schrimpf, D.; Capper, D.; Hovestadt, V.; Kratz, A.; Sahm, F.; Koelsche, C.; Korshunov, A.; Olar, A.; et al. IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: A grading problem for WHO. Acta Neuropathol. 2015, 129, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Mamatjan, Y.; Agnihotri, S.; Goldenberg, A.; Tonge, P.; Mansouri, S.; Zadeh, G.; Aldape, K. Molecular Signatures for Tumor Classification: An Analysis of The Cancer Genome Atlas Data. J. Mol. Diagn. 2017, 19, 881–891. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Taskesen, E.; Babaei, S.; Reinders, M.; de Ridder, J. Integration of gene expression and DNA-methylation profiles improves molecular subtype classification in acute myeloid leukemia. BMC Bioinform. 2015, 16, S5. [Google Scholar] [CrossRef]
- Tanvir, R.; Islam, M.; Sobhan, M.; Luo, D.; Mondal, A. MOGAT: A Multi-Omics Integration Framework Using Graph Attention Networks for Cancer Subtype Prediction. Int. J. Mol. Sci. 2024, 25, 2788. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.; Xiao, S.; Liu, G.; Wu, W.; Wang, S. DeepMoIC: Multi-Omics Data Integration via Deep Graph Convolutional Networks for Cancer Subtype Classification. BMC Genom. 2024, 25, 1209. [Google Scholar] [CrossRef]
- Waqas, A.; Tripathi, A.; Ahmed, S.; Mukund, A.; Farooq, H.; Schabath, M.; Stewart, P.; Naeini, M.; Rasool, G. SeNMo: A self-normalizing deep learning model for enhanced multi-omics data analysis in oncology. arXiv 2024, arXiv:2405.08226. Available online: https://arxiv.org/abs/2405.08226 (accessed on 10 June 2025). [Google Scholar] [CrossRef]
- Hoadley, K.; Yau, C.; Hinoue, T.; Wolf, D.; Lazar, A.; Drill, E.; Shen, R.; Taylor, A.; Cherniack, A.; Thorsson, V.; et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef]
- Haibe-Kains, B.; Adam, G.; Hosny, A.; Khodakarami, F.; Massive Analysis Quality Control (MAQC) Society Board of Directors; Waldron, L.; Wang, B.; McIntosh, C.; Goldenberg, A.; Kundaje, A.; et al. Transparency and reproducibility in artificial intelligence. Nature 2020, 586, E14–E16. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. The Cancer Genome Atlas Program (TCGA) Homepage. 2025. Available online: https://portal.gdc.cancer.gov/ (accessed on 20 April 2025).
- Brown, M.; Grundy, W.; Lin, D.; Cristianini, N.; Sugnet, C.; Ares, M.; Haussler, D. Support Vector Machine Classification of Microarray Gene Expression Data; Technical Report UCSC-CRL-99-09; University of California: Santa Cruz, CA, USA, 1999. [Google Scholar]
- Chu, F.; Wang, L. Gene expression data analysis using support vector machines. In Proceedings of the International Joint Conference on Neural Networks, Portland, OR, USA, 20–24 July 2003. [Google Scholar] [CrossRef]
- Berger, T.; Wen, P.; Lang-Orsini, M.; Chukwueke, U. World Health Organization 2021 Classification of Central Nervous System Tumors and Implications for Therapy for Adult-Type Gliomas: A Review. JAMA Oncol. 2022, 8, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.; Perry, A.; Wesseling, P.; Brat, D.; Cree, I.; Figarella-Branger, D.; Hawkins, C.; Ng, H.; Pfister, S.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.; Verhaak, R.; Aldape, K.; Yung, W.; Salama, S.; Cooper, L.; Rheinbay, E.; Miller, C.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef]
- Tran, D.; Nguyen, H.; Pham, V.; Nguyen, P.; Nguyen Luu, H.; Minh Phan, L.; DeStefano, C.; Yeung, S.; Nguyen, T. A comprehensive review of cancer survival prediction using multi-omics integration and clinical variables. Brief Bioinform. 2025, 26, bbaf150. [Google Scholar] [CrossRef]
- Zhou, J.; Theesfeld, C.; Yao, K.; Chen, K.; Wong, A.; Troyanskaya, O. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 2018, 50, 1171–1179. [Google Scholar] [CrossRef]
- Whalen, S.; Schreiber, J.; Noble, W.; Pollard, K. Navigating the pitfalls of applying machine learning in genomics. Nat. Rev. Genet. 2022, 23, 169–181. [Google Scholar] [CrossRef]
- Mamatjan, Y. Pan-Cancer Classification System with Explainable AI Interpretation: A Feasibility Study. In Proceedings of the IEEE Congress on Evolutionary Computation (CEC), Yokohama, Japan, 30 June–5 July 2024; pp. 1–6. [Google Scholar] [CrossRef]
- Simon, M.; Kuschel, L.; von Hoff, K.; Yuan, D.; Hernáiz Driever, P.; Hain, E.; Koch, A.; Capper, D.; Schulz, M.; Thomale, U.; et al. Rapid DNA methylation-based classification of pediatric brain tumors from ultrasonic aspirate specimens. J. Neurooncol. 2024, 169, 73–83. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef]
- Chaudhary, K.; Poirion, O.; Lu, L.; Garmire, L. Deep Learning-Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef]
- Campanella, G.; Hanna, M.; Geneslaw, L.; Miraflor, A.; Werneck Krauss Silva, V.; Busam, K.; Brogi, E.; Reuter, V.; Klimstra, D.; Fuchs, T. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 2019, 25, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Valous, N.; Popp, F.; Zörnig, I.; Jäger, D.; Charoentong, P. Graph machine learning for integrated multi-omics analysis. Br. J. Cancer 2024, 131, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Liang, J.; Li, Z.; Wang, J.; Wang, J.; Yu, G. Cancer molecular subtyping using limited multi-omics data with missingness. PLoS Comput. Biol. 2024, 20, e1012710. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | BLCA (N: 366) | KIRP (N: 225) | Misclassified Sample ID | |
|---|---|---|---|---|
| 7130 | 7287 | |||
| KDM6A | 76 | 6 | 1 | – |
| FGFR3 | 53 | 3 | 1 | – |
| STAG2 | 38 | 5 | 1 | 1 |
| BAP1 | 14 | 4 | – | – |
| PBRM1 | 18 | 6 | – | – |
| VHL | – | 1 | – | – |
| CDH1 | 7 | 1 | – | – |
| NSD1 | 21 | 4 | – | – |
| TP53 | 151 | 3 | – | – |
| PIK3CA | 83 | 2 | – | – |
| # of sequenced samples | 366 | 153 | ||
| Gene Name | R12–M13 | R13–M13 | R13–M14 | p-Value |
|---|---|---|---|---|
| IDH1 | 0 (0%) | 0 (0%) | 401 (96%) | |
| EGFR | 25 (40%) | 6 (24%) | 2 (1%) | |
| PTEN | 20 (32%) | 1 (4%) | 4 (1%) | |
| TP53 | 7 (11%) | 5 (20%) | 238 (57%) | |
| NF1 | 9 (15%) | 9 (35%) | 12 (3%) | |
| ATRX | 2 (4%) | 5 (20%) | 190 (46%) | |
| BRAF | 1 (2%) | 3 (12%) | 0 (0%) | |
| CIC | 0 (0%) | 1 (4%) | 111 (27%) | |
| ATM | 0 (0%) | 2 (8%) | 3 (1%) | 0.002 |
| ARID2 | 0 (0%) | 3 (12%) | 8 (2%) | 0.002 |
| NOTCH1 | 0 (0%) | 1 (4%) | 43 (11%) | 0.02 |
| # of sequenced samples | 64 | 26 | 422 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamatjan, Y.; Abulizi, N. Multimodal Gene Expression and Methylation Profiling Reveals Misclassified Tumors Beyond Histological Diagnosis. Electronics 2025, 14, 2442. https://doi.org/10.3390/electronics14122442
Mamatjan Y, Abulizi N. Multimodal Gene Expression and Methylation Profiling Reveals Misclassified Tumors Beyond Histological Diagnosis. Electronics. 2025; 14(12):2442. https://doi.org/10.3390/electronics14122442
Chicago/Turabian StyleMamatjan, Yasin, and Nijiati Abulizi. 2025. "Multimodal Gene Expression and Methylation Profiling Reveals Misclassified Tumors Beyond Histological Diagnosis" Electronics 14, no. 12: 2442. https://doi.org/10.3390/electronics14122442
APA StyleMamatjan, Y., & Abulizi, N. (2025). Multimodal Gene Expression and Methylation Profiling Reveals Misclassified Tumors Beyond Histological Diagnosis. Electronics, 14(12), 2442. https://doi.org/10.3390/electronics14122442