1. Introduction
The main purpose of simulation is to shed light on the underlying mechanisms that control the behavior of a system. It is generally used to understand and predict the behavior of such a system. Predicting how the system will evolve and respond to its environment makes it possible to identify any necessary changes that will help make the system work as expected [
1]. Mechanical simulation is important for multiple applications in different scenarios because of the advantages it offers over performing the real process [
2]. The development of current simulation software is supported by supercomputing and cloud computing environments, which ensure, according to different approaches, the necessary processing, storage, and communication resources [
3].
Molecular dynamics (MD) simulations make it possible to calculate the interaction of a large number of particles at the atomic level. These simulations are analyzed by a broad number of scientists from different fields, such as predicting protein structure, designing new drugs, or guessing binding affinity [
4], to save costs in terms of materials, laboratory costs, and production time.
MD leads to the use of supercomputing environments considering the computing and memory cost of simulating millions of interactions in well-known biological molecules. MD simulators must be concerned with distributing computing and data while avoiding communication problems and maximizing the potential of supercomputers.
In the early 2000s, all the MD algorithms were based on Central Processing Units (CPUs) [
5,
6,
7]. Those were oriented to use multiple CPUs to compute the simulation and usually relied on supercomputers where tens of CPUs performed the tasks. Recently, massively parallel computing has made a significant advance thanks to the introduction of the Graphics Processing Unit (GPUs). These are characterized by multiple parallel operations in the dataset, which is especially suited to the needs of traditional MD. Multi-GPU environments represent the present in MD simulation, but new issues arise due to the difficulty of dividing and sharing the massive molecular constraints between computational resources [
8].
Considering the above, users should face two concerns: (i) spend time and effort implementing those algorithms by installing or developing MD software packages and/or (ii) set up the environment (e.g., supercomputers, cloud services, personal computers) where they run the simulation. Note that end users may not have prior knowledge of MD simulation development or cloud management.
On the one hand, the execution of a MD simulation involves the preparation and processing of the molecule’s data before launching the process. To do this, a molecular structure is usually downloaded from public databases, like [
9]. Users must then perform three relevant tasks:
Establish a physical model, including the details of the atomic interaction.
Add a solution to the molecular system. Normally, the molecule is surrounded by water.
Equilibrate the whole system. Before carrying out the simulation, the molecular system needs to remain in a low-energy state after the introduction in a solution [
10].
All this results in a complex workflow that is often tedious and involves downtime between tasks.
On the other hand, setting up the environment means that users must manage and configure the cloud environment. Cloud computing refers to both applications delivered as services over the Internet and the hardware and software systems in the data centers that provide those services [
11]. This model of computing enables ubiquitous, convenient, on-demand access to a shared pool of configurable computing resources. Cloud computing has gained significant attention for running simulations in both the industrial and academic communities [
12]. Different cloud platforms, such as Amazon Web Services, Google Cloud Platform and Microsoft Azure, allow end users to use all its capacity without the need to manage the system architecture. However, MD software packages require specific configuration in order to take full advantage of the hardware. Users should interact with the remote computer by commands and scripts to set up the simulation.
In this article, we present a web-based tool that allows end users to automate the previously described processes, both the molecular data pipeline and management and execution of the simulation in the cloud environment. Our tool provides an Infrastructure as a Service (IaaS) where users could manage the chosen cloud environment and set it up for MD purposes, and a Software as a Service (SaaS) where the application abstracts the whole workflow to make it transparent to the user. Using web technologies provides a friendly and recognizable UI where users can perform all MD tasks with a higher level of abstraction.
The paper is organized as follows.
Section 2 analyzes different MD simulators and their ability to be used in cloud computing environments.
Section 3 describes the main features of our proposal, from the MD simulation data pipeline to the creation of the infrastructure in the cloud environment.
Section 4 presents how the tool works, carrying out a case study with the complex 4V4L molecular system. Lastly,
Section 5 discusses the results, presents the conclusions drawn, and proposes future work.
2. Related Work
Molecular systems are composed of a large number of atomic particles that interact with each other. MD simulations [
4] recreate these interactions by computational methods to estimate the behavior of a molecular system. Usually, systems are large enough not to be able to solve analytically.
MD considers the atoms’ characteristics, and their force interactions, to accurately reproduce their movement over a time period. Discretization methods divide the simulation into time steps of a magnitude of femtoseconds (fs,
s) and compute interaction forces and new atom positions using numerical integration. These calculations imply high computational and memory costs [
8]. Several parallel computing algorithms have been proposed to accelerate interaction’s calculation [
13,
14], resulting in multiple molecular dynamics simulators [
15,
16,
17,
18,
19].
However, significant conformational changes still take too long when studying the dynamics of complex organic systems [
20], which consist of millions of particles such as virus capsids, or even the entire virus. Usually, a desired biological process requires a high number of interactions that demands considerable simulation time and computing resources.
Even though most supercomputing centers are currently equipped with GPUs to perform massive calculations, this capability is very recent. Until a few years ago, supercomputing environments consisted of a large set of nodes, with multiple CPUs, interconnected among them. Proposed algorithms [
5,
6,
7] were designed to take advantage of the parallelism provided by these types of environments, as the first versions of the well-known NAMD simulator [
17]. Recently most of the current molecular dynamics simulators have been adapted to take advantage of the capabilities offered by modern GPUs to compute the interactions between atoms. NAMD [
21] partitions the simulation of the molecular system into patches, which are distributed among the different computing nodes. GPUs are used as co-processors to perform the electrostatic forces calculations, while bonded forces are still calculated using CPUs [
22]. GROMACS [
18] carries out a spatial partitioning of the system to distribute molecular dynamics in multi-core architectures. The CPU uses the GPU as a co-processor to accelerate force calculations. In a similar way, AmberTools MD software packages (AMBER) [
23] provides MD simulation in different computing environments (single/multi-CPU, single/multi-GPU) to perform a post hoc analysis. Instead, ACEMD [
19] performs the calculation of all atomic forces on GPUs, using a single GPU for each force type. More recent works, such as MDScale [
24], take advantage of massive MultiGPU environments enabling the division and exchange of atoms between GPUs.
If access to specific resources of a supercomputer is requested for MD, extensive bureaucratic procedures and knowledge of how to use it are required before running the simulation. In contrast, in most cases, the availability of cloud computing services for MD is immediate. Unfortunately, users must know how to manage the system and the infrastructure and be able to handle the requested resources, resulting in an additional workload. We found multiple tools that allow users to design and automate these processes. For example, Ref. [
25] allows users to remotely simulate a cloud infrastructure to predict the cost of using specific software. Other software such as Aneka [
26] enables the automatic deployment of a cloud infrastructure. The software provides an API, in which the users through the programming language .NET define the infrastructure and the software that runs on it. Despite the advantages, in both cases specialized knowledge of cloud services is required. Furthermore, the cost of configuring and deploying the cloud environment to run MD simulation software remains high.
The execution of a MD simulation requires a previous process of the molecular system. Typically, molecular data are obtained for general purposes and only provided with the names of the molecules and their positions. Additional data, such as the bonds or the charges of the atoms, are required for MD. It is therefore necessary to transform the existing molecular data, following a data pipeline or workflow, to obtain the necessary parameters to carry out the simulation. For instance, Purawat et al. [
27] present a specific workflow using AMBER. This workflow focuses on the execution and analysis of the MD but leaves the responsibility for the construction of the system to the users. CHARMM-GUI [
28] allows users to build and generate MD input files but does not provide job execution capabilities. VMD (Visual Molecular Dynamics) [
29] complements some simulators (e.g., NAMD) and offers several plug-ins to perform multiple tasks. In particular, QwickMD plug-in [
30] supplies a GUI to use NAMD more easily and simply. Knowing their functionality, users can combine different tools to perform MD simulation.
Table 1 summarizes the software described throughout this section.
We propose a tool in which users can carry out the workflow in such a way that they intervene in the process as little as possible. Our approach is able to deploy all processes using cloud services in any cloud infrastructure. As far as we know, there is no software that fulfills all the necessary characteristics.
3. Method
Generally, users need prior knowledge before performing MD simulation, not only about physical operation but also technical knowledge to be able to run on supercomputers and/or cloud environments. In many cases all this implies a high complexity and a long learning process. To simplify this process, we present a web-based application where users can—through a friendly UI, automatically and transparently—perform a data flow to get a molecular system ready and deal with a whole cloud infrastructure to run MD simulations. We have compiled the different steps needed to easily manage a molecular system and carry out a MD simulation. The application allows users to automate all processes using a remote computer environment.
The following subsections describe in detail the required pre-simulation molecular data processing and the integration mechanisms for running in cloud computing environments.
3.1. Molecular Data Processing
One of the first steps to perform MD is to obtain the data set to be simulated. Open access databases, such as Protein Data Bank (PDB) [
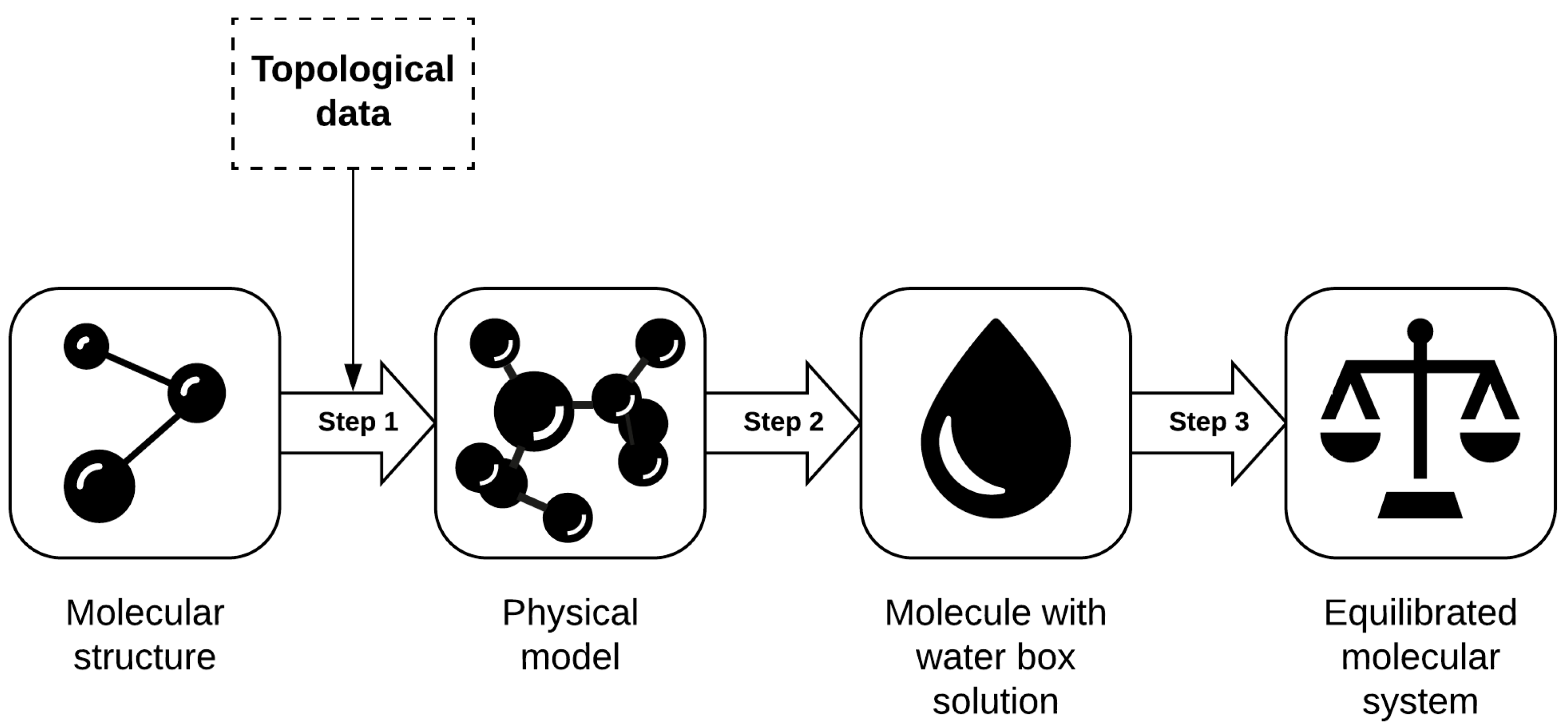
9], offer a worldwide repository of structural data of large biological molecules. The information retrieved from PDB is incomplete because it is only a description of molecular systems, as it only provides the basic molecular data, such as the type of atoms and their positions. However, to carry out MD simulations, a pre-process is required to gather the additional molecule configuration needed. For this purpose we have grouped three relevant tasks that together form a work or data flow to prepare the molecular system and leave it ready for simulation. This data pipeline covers (see
Figure 1) (i) generating a physical model, (ii) adding a solution, and (iii) thermal equilibration.
Generating the physical model is necessary to obtain specific atom’s properties, such as their charges and the types of bonds of which they are part. In the usual molecular archives (e.g., from PBD), available biological structures only define the position and name of the atom, without any additional information. PSFGen plugin [
31] for VMD compares and relates each atom of the molecular system with a series of topological description files. This process allows obtaining their charges and molecular bonds. Although PSFGen automates the task, it is highly demanding process; therefore, it would be advisable to run it in a cloud computing environment.
This result is not yet a realistic representation of the molecule because it is located in a vacuum. Adding a water solution to the molecular system is the next step in the data flow. Solvate plugin [
32] for VMD can perform this operation. It places multiple water molecules surrounding the system, resulting in a box-shaped molecular system.
This water box is inaccurate in physical terms because it has been created independently. This means that, considering the distance between atoms and their properties, the molecular system is in a high-energy state (i.e., unstable). The magnitude of this energy may not exist in the real world and therefore could not be simulated. Because of that, the target system must reach a lower energy state before simulation, which is achieved through a molecular equilibration process [
33]. We have chosen the well-known NAMD molecular dynamics simulator [
17], which offers a molecular system equilibration tool using the Root-Mean-Square Deviation (RMSD) method [
34].
The resulting molecular structure, which can be used in any MD simulator, increases its size by about 40%. Depending on the molecular system, this size can vary from a few megabytes to thousands of gigabytes. For this reason, the described data pipeline implies a high cost of computing resources, and its execution fits perfectly in a cloud computing environment.
3.2. Running MD in the Cloud
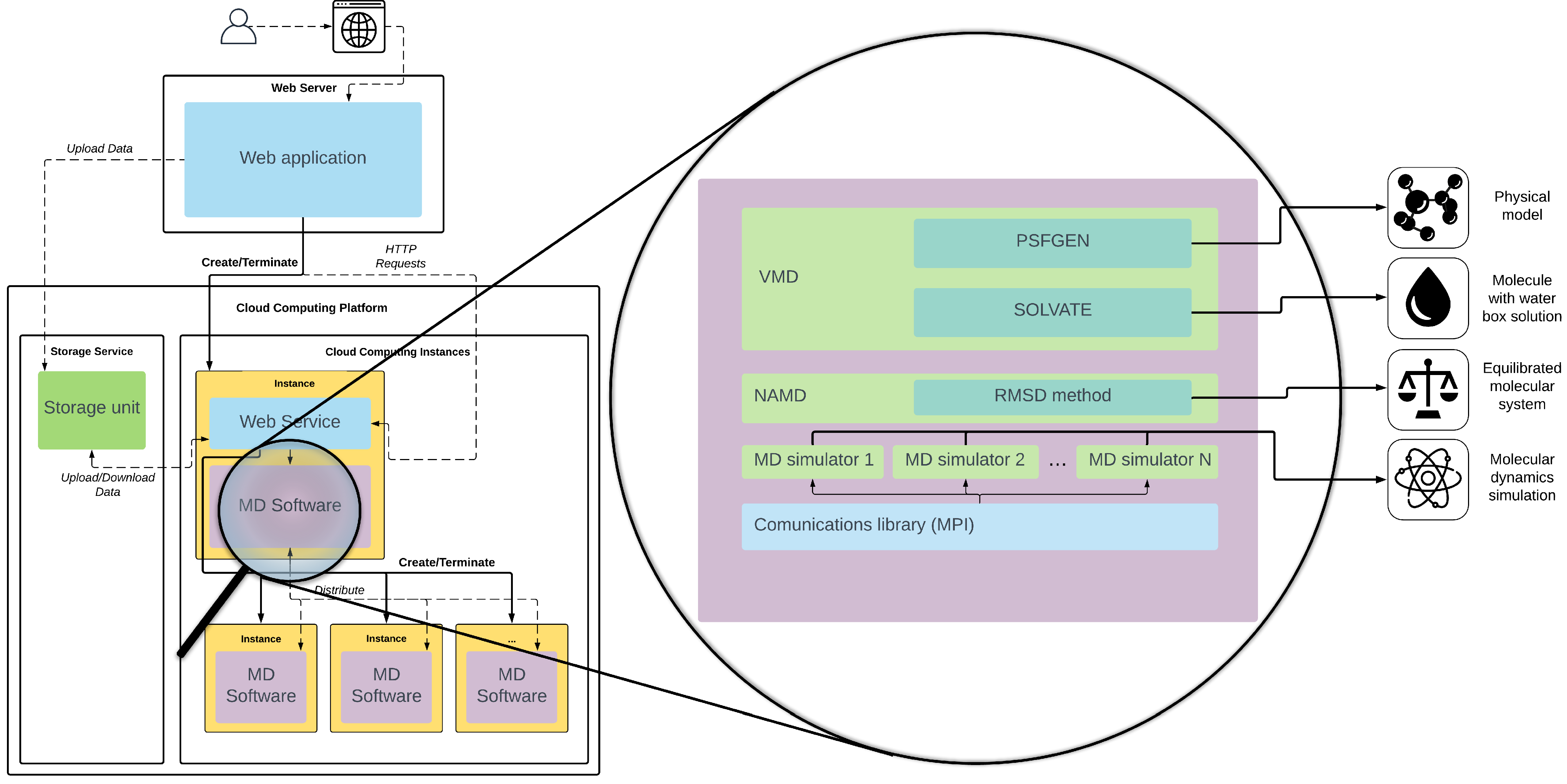
The main objective of our solution is to abstract the user from the resources needed to carry out the simulation. We propose that any process related to MD simulation runs directly in the Cloud. We have developed our tool as a web application, which provides a GPU-accelerated MD service running in cloud computing. Its aim is hiding the complexity of the use of a cloud infrastructure and the MD software.
Figure 2 shows the proposed web application architecture. The main functionality is hosted on a web server that uses remote resources located in the cloud environment to perform the data pipeline shown above and the MD simulation. Since most of the computing workload falls on the cloud service, the web server can be placed internally on the user’s system or on the cloud platform itself. Installation requirements are particularly low so that the web service can be supported by a simple AWS t2 instance [
35], with only 1GB of RAM and a single CPU.
The server has a web service deployed, which controls the entire process. The web service acts as the communication link between the web application and the cloud infrastructure needed for simulation. The web service can be integrated with and manage the most common cloud computing environment, such as Microsoft Azure, Amazon Web Services, Google Cloud Computing, etc. All of these platforms have virtual machines with pre-set hardware (also called instances). They have CPU and GPU optimized instances, the latter even with single, multiple, or fractional GPUs, that can be used to enable a multi-GPU infrastructure for MD. The cloud platforms also provide storage services, which can be remotely accessed from the tool and the instances to store the molecular data and its corresponding metadata as cloud objects.
The web service is implemented as a REST API that allows the main application to make requests through the HTTP protocol, both to start or stop the execution of the software and to query its status. It also collects the data from the storage service and uploads the results obtained. The web service, like all the necessary additional software, is deployed in our own system images designed specifically to take advantage of the Multi-GPU cloud infrastructure. The images are based on a Linux operating system and contain the web service, VMD software with the PSFGen and Solvate plugins, NAMD with the RMSD algorithm, communication libraries, like MPI, and the necessary MD simulation software. The tool starts the instances according to the task to be performed, using only GPU-built instances when necessary. The main objective is that users are not concerned about how the cloud services for MD are started or finished when their run is over.
Our proposal automatically manages the following resources in the cloud platform:
Users. User management allows each user to have their own space, where they can administer their access keys to the cloud platform. Access keys can be obtained directly from the control console of the cloud platform. Each key determines multiple permissions for the user in the cloud infrastructure.
Storage. Since MD simulation requires and generates a large amount of information, we delegate the management of the files to the storage service of the cloud platform. In our tool, for each access key, several workspaces can be configured within the storage service, one for each molecular system to be considered. The tool uses each workspace to store all data related to the data pipe and simulation.
Instances. As seen above, users may want to execute different stages of the data pipeline including simulation. When the execution of a stage is launched, the tool automatically deploys the requested instances into the cloud infrastructure by booting an own pre-configured system image with all the required MD software.
Network communication. We manage the communication between the instances and the storage service in the deployment of the infrastructure. In case of simulation using several GPU-optimized instances at the same time, the tool manages the efficient communication between all of them. Note that the access key must provide the corresponding permissions to be able to raise and communicate instances and services.
Monitoring. Although complete cloud monitoring systems could have been used [
36], we have chosen to provide the user only with information about the execution status in order to simplify their adaptation to the environment. These data are obtained directly from information provided by the cloud infrastructure itself. Each of the instances also executes a web service that communicates with the web server to let it know its execution status, which can be
Preparing: data are being downloaded from the storage service.
Running: the process is being executed.
Finishing: the task has been completed successfully, and data are being uploaded to the storage service.
Terminate: the task has been completed. This status triggers the tool to terminate the launched instances.
Error: there has been an error in the execution of the process. The message is attached to a log. This status also triggers the tool to terminate the launched instances.
The defined architecture allows users to access our tool via a web browser (using smartphones, laptops, etc.) to easily run a MD simulation. As seen, the tool encapsulates the MD simulation functionality and cloud environment configuration, allowing users to perform the entire workflow through an easy-to-use user interface. All the simulation software runs on the cloud infrastructure, and users can leave the task unattended. This behavior approaches the definition of Software as a Service (SaaS).
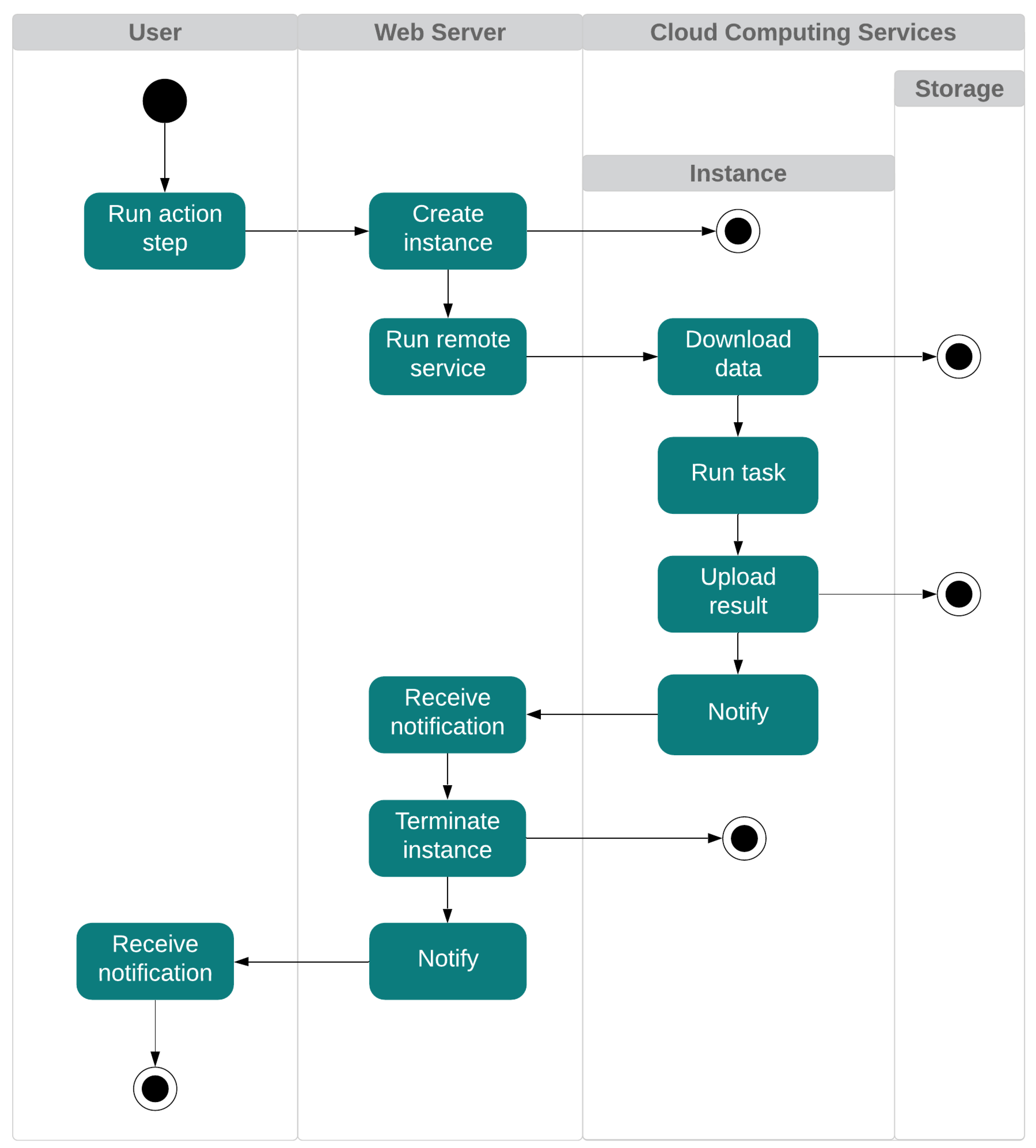
Figure 3 shows the interaction between users and the proposed system. In essence, users interact with the application via a web interface. On the other side, the deployed web server is in charge of running a set of mechanisms related to cloud computing services. The execution process is as follows. First, the user configures the task to be performed in the application. The web server starts the required instance/s according to the configuration selected, using a pre-configured system image. Once the web server checks that communication has been established with the created instance/s, it sends a request with the script to run. The instance/s then download/s from the storage service all the data needed to perform the operation. When the task is completed, each instance uploads the results to the storage service, notifies the web server of its completion, and releases its resources. In case of an error, the instance returns an error message, attaching the log of the executed software. Finally, the web server notifies users via the web interface about the completion of the task.
The tool we propose is designed to use cloud computing services, reducing its cost as much as possible, avoiding unnecessary expenses if the execution fails or ends earlier than expected. Besides, the application allows users to leave it unattended when MD tasks, as usual, take a long time to complete. The tool is fault-tolerant against failures or disconnections of the web server. At each step of the simulation process, the tool stores the execution status and the ID of the instances. This means that if the web server is disconnected, it is possible to recover the execution status without losing progress. Errors that occur during the execution of the software are also managed. In case of failure, the application receives the corresponding status of the instance, which contains the log data of the software that was running. This log is shown to the users via the web interface.
4. Results
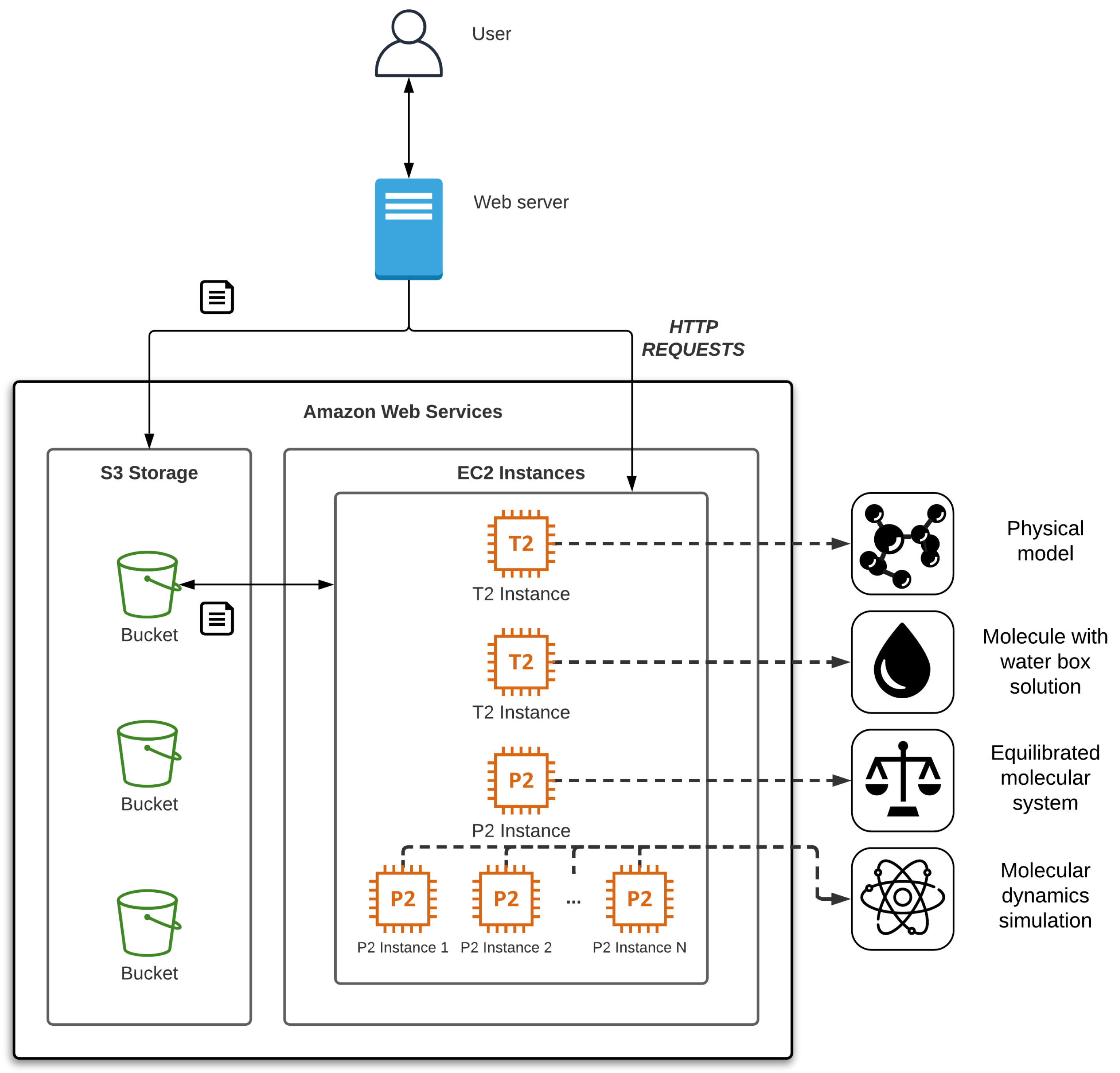
In this section, we present a case study deploying our solution in a local environment and using Amazon Web Services (AWS SDK [
37]) as a cloud platform test. Although we have developed the software for this cloud platform, it can be easily migrated to other environments, such as Microsoft Azure or Google Cloud Platform, using their corresponding SDK. The web server would be adapted to manage the virtual machines using the corresponding SDK. Additionally, new system images should be created with the MD software.
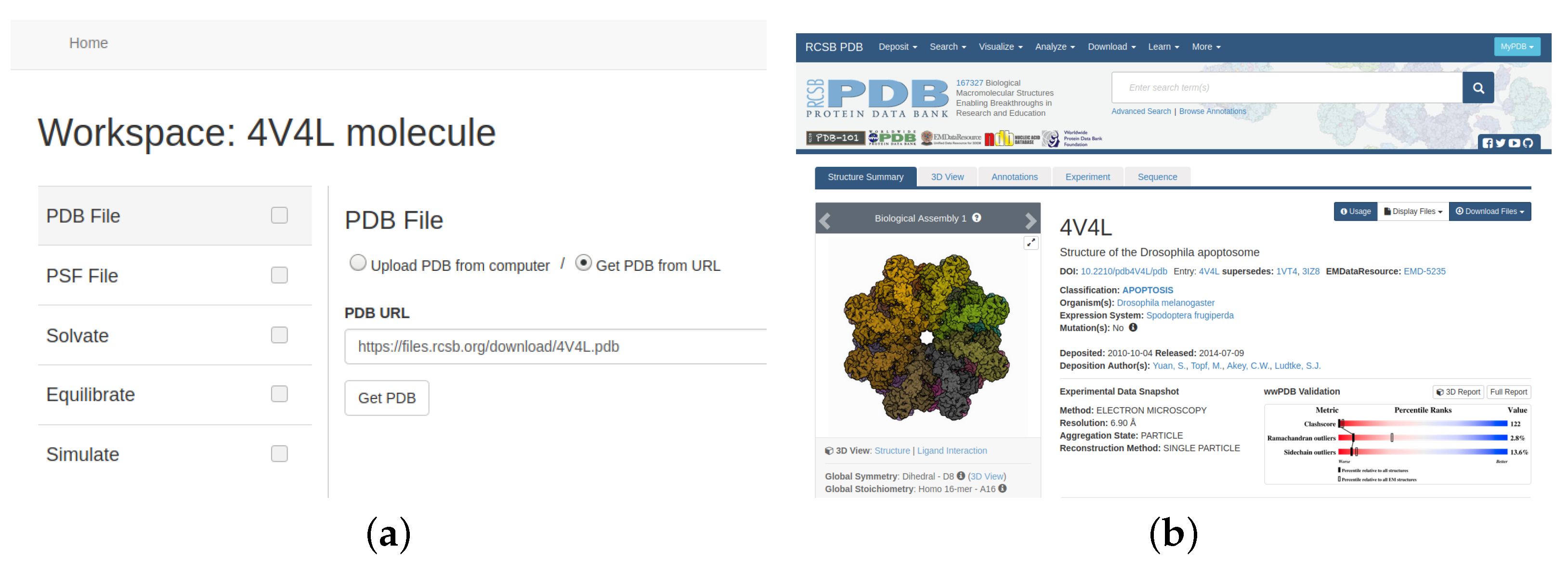
As a molecular system we have chosen the molecule 4V4L as an example of a complex system downloaded from PDB (see
Figure 4). The 4V4L molecular system obtained from PDB [
38] is composed of about 128,000 atoms reaching up to 500,000 atoms at the end of the process. The development of the data pipeline to build the molecular system to be simulated implies a RAM memory cost of 15 gigabytes. In this case, this amount can be easily achieved with a basic AWS Elastic Compute Cloud (EC2) CPU-based instance, like t2.2xlarge [
35]). Larger molecular systems, such as 3J3Q [
39], can reach up to 65 million atoms, requiring about 3 terabytes of RAM and necessarily involving the need to use supercomputers or powerful cloud computing environments. For example, x1e.32xlarge AWS EC2 CPU-based instance [
40] provides 3.9 terabytes of RAM, enough to solve this problem, but it would also be possible to use t2.2xlarge EC2 instances with an additional 3 terabyte hard disk used as swap. The latter configuration is much cheaper but involves a much longer runtime.
For equilibration and simulation processes it is strongly recommended to use AWS EC2 instances that provide GPUs due to the high computational workload of these operations. For instance, p2.16xlarge AWS EC2 GPU-optimized instance [
41] enables up to 16 Nvidia Tesla K80 GPUs. Note that the simulation of larger molecular systems may require even more GPUs. In a regular environment it would be necessary to properly configure the network communication between the different instances to have a functional Multi-GPU environment, besides installing and managing the necessary software. Our web-based approach initializes, deploys instances with the required number of GPUs, and enables communication among them without user intervention.
As a cloud object storage service, we used AWS Simple Cloud Storage Service (S3), which provides scalability and data availability. In the AWS nomenclature, a bucket is similar to a folder in the storage service that stores objects, which consist of data, in this case molecular systems, and its descriptive metadata.
Figure 5 shows the complete architecture of the test bed running the AWS infrastructure required for the deployment and use of the application. In the next paragraphs, we describe the entire procedure from the user management to the execution of MD simulation going through the workflow using our tool. Note that it is not necessary to carry out all the steps in the process. It is possible to load the result of some of them, and users can also rerun each step without having to restart the whole process.
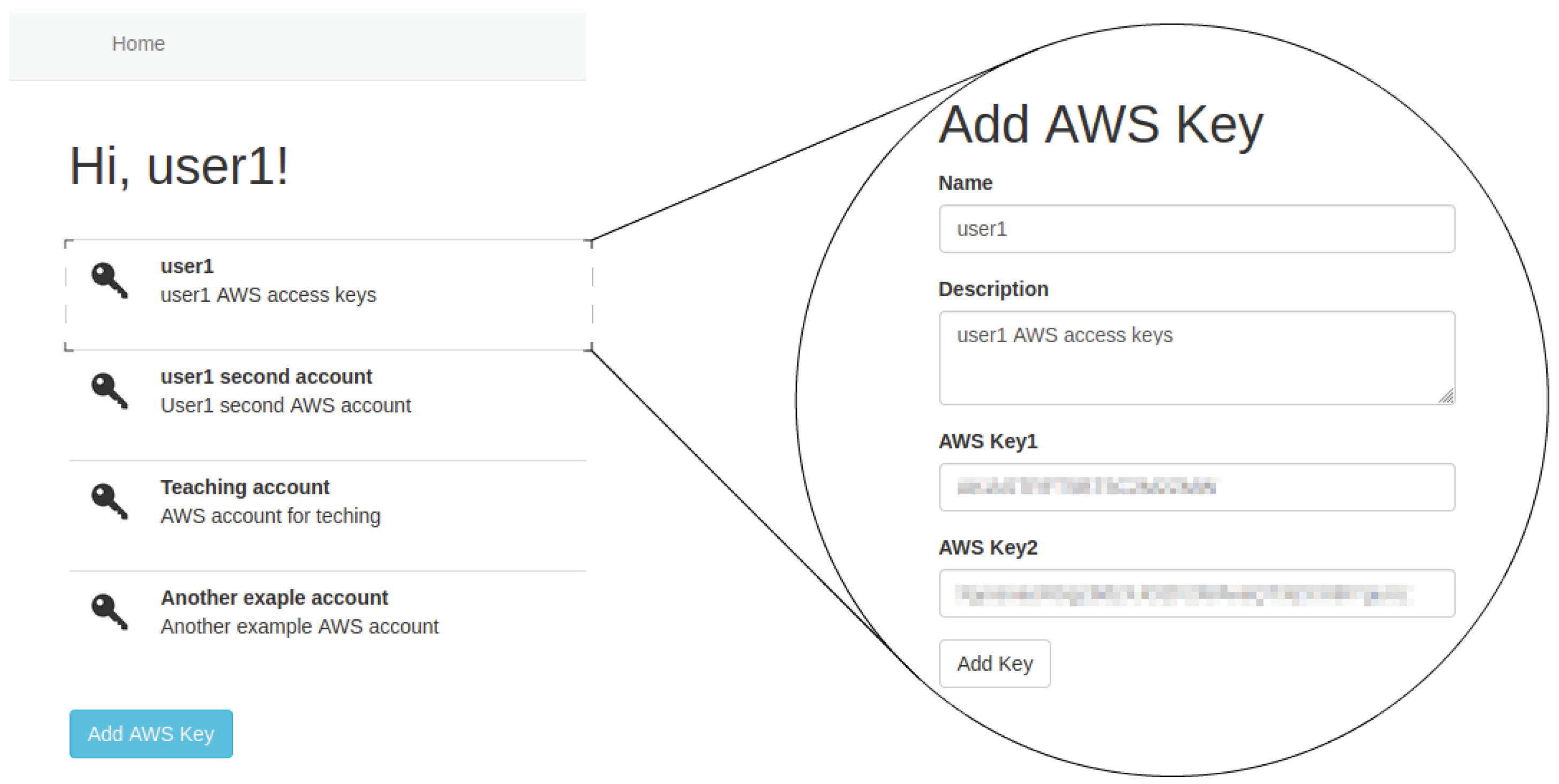
The first step is to register access to the cloud platform. User data are encrypted and stored in a SQL database on the web server. Specifically, users will be asked for the credentials they want to use on the specific cloud platform, in this case AWS. Users must have an AWS account and have generated the external access keys in the AWS Identity and Access Management (IAM) service. Note that the cloud AWS user must have permissions to remotely manage EC2 instances and store data in S3. Access keys consist of two parts: an access key ID and a secret access key. Each access key is stored and assigned to the logged-in user as shown in
Figure 6. Once users log in the tool, they can start managing their work environment.
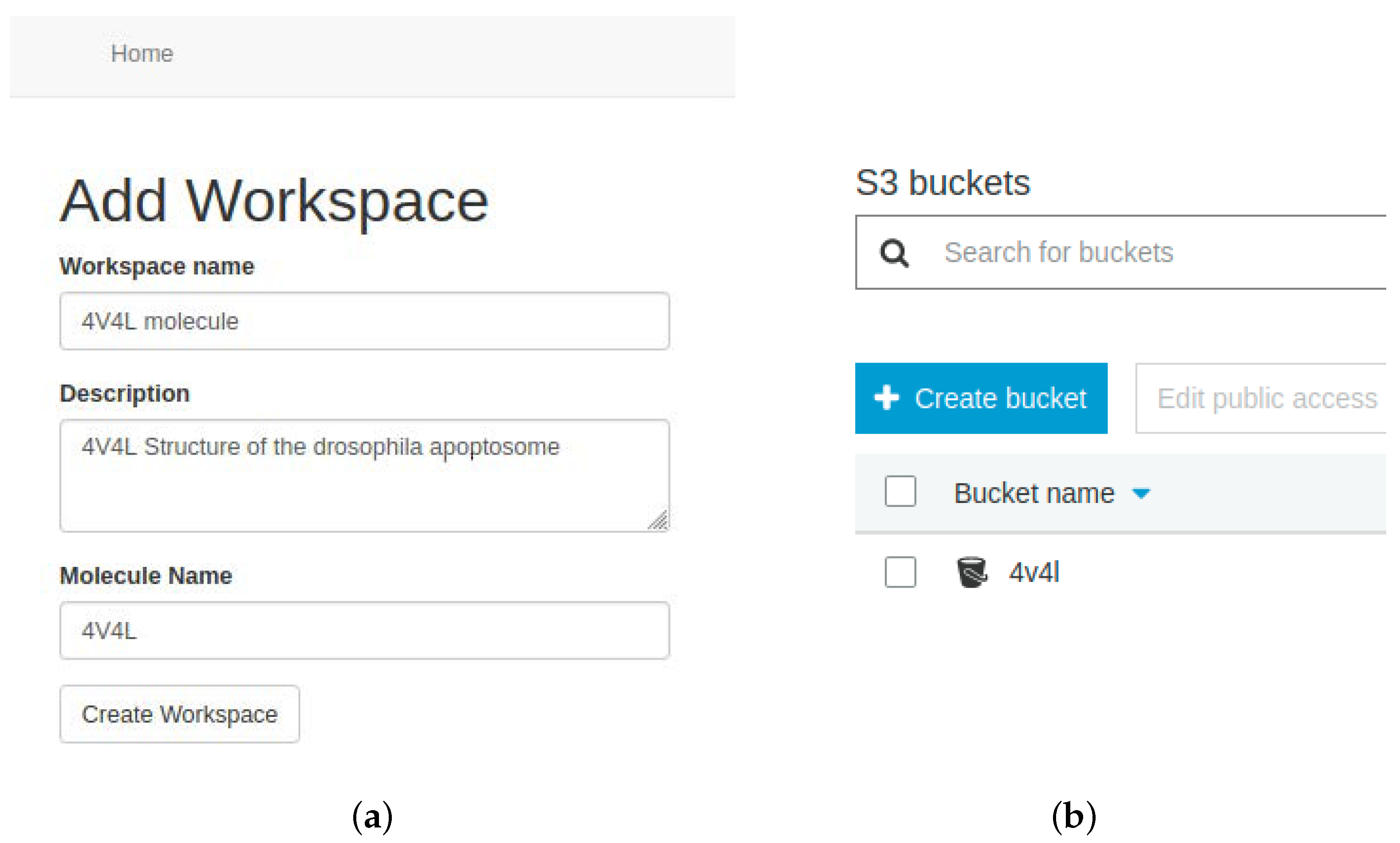
At this point, users must configure a workspace associated with their account, which represents a bucket within the AWS S3 storage service. It is recommended to create a single workspace per molecular system to be simulated. The bucket stores all data related to the selected molecular system. EC2 instances will access this bucket to retrieve the data and upload the results of the MD process.
Figure 7 shows a workspace created in the tool, along with its representation in S3.
The next step is to select the data of the molecular system to work with. To simplify the process, our tool offers two options to upload a file to the cloud storage service: choose a local file with the information or provide a link to gather it directly from PDB, as shown in
Figure 8.
Once molecular data have been uploaded, users can start the data pipeline depending on their needs. The first stage they can run consists of relating the atoms to the molecule to which they belong, assigning their bonds and electrical charges. Users can upload and use a specific topology file, but if not, they can carry out that calculation in a single AWS EC2 t2 instance using the VMD software [
29], which compares molecular data with different existing topologies. The tool performs everything related to the management of this instance. Users thus only have to configure the execution and the startup request. The status of the stage execution can be consulted in the application, which is retrieved directly from the own EC2 instance.
Figure 9 depicts how the tool is configured to create a Protein Structure File (PSF). PSF files contain all the molecular information that is not included in the previously obtained PDB data, such as bonds or atom charges. They should be used together with the PDB file to simulate MD.
As described in
Section 3.1, a molecular system cannot be located in the vacuum and needs to be inside of a water solution. The second stage consists of creating a molecular structure that forms a cubic system composed of the original data surrounded by multiple water molecules (see
Figure 10). The size of the structure depends on the magnitude of the original system, but users can set how large the margin will be. VMD software also performs this process on a CPU-based t2 instance of the cloud infrastructure. Just like the previous stage, users select some parameters, and the tool manages the whole process.
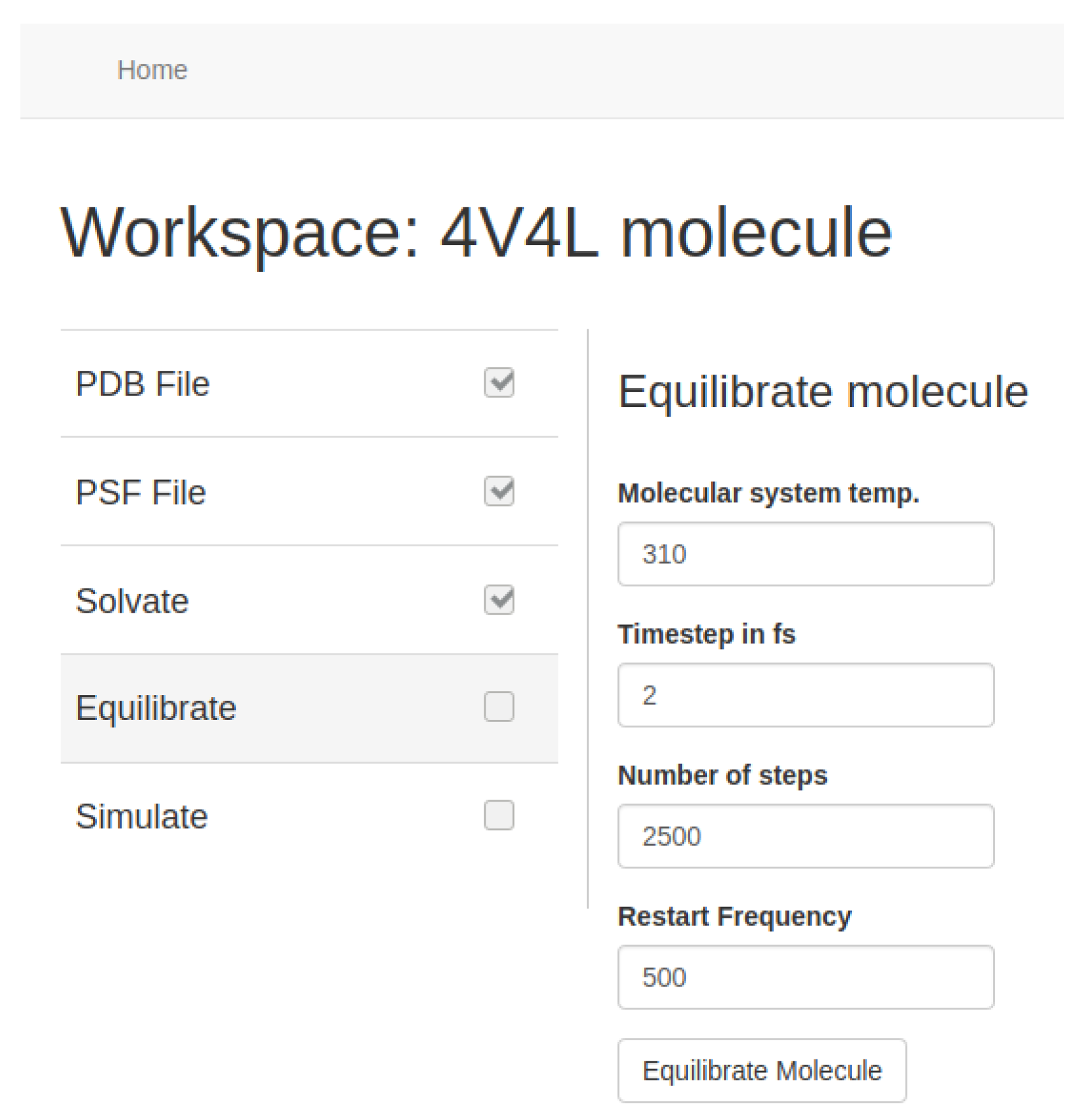
Introducing water into the system makes it unstable for MD simulation. To solve this we use NAMD software, which implements an option to balance molecular systems. As in the previous stages, the tool manages the execution of this process in a single EC2 GPU-optimized p2 instance. Users only have to select the relevant parameters to perform a small simulation to look for the low-energy state. Our tool allows users to choose multiple settings like simulation temperature, time step (in fs), simulation steps, and step reset speed values.
Figure 11 shows an example of the configuration of this stage.
With the previous stages the molecule would be ready to be simulated. Finally, our tool offers to run the simulation using different MD simulators. We have configured an AWS EC2 instance, which can be replicated in a cloud environment by connecting between its copies, by installing NAMD and our own implemented Multi-GPU version of [
8], named as MDScale [
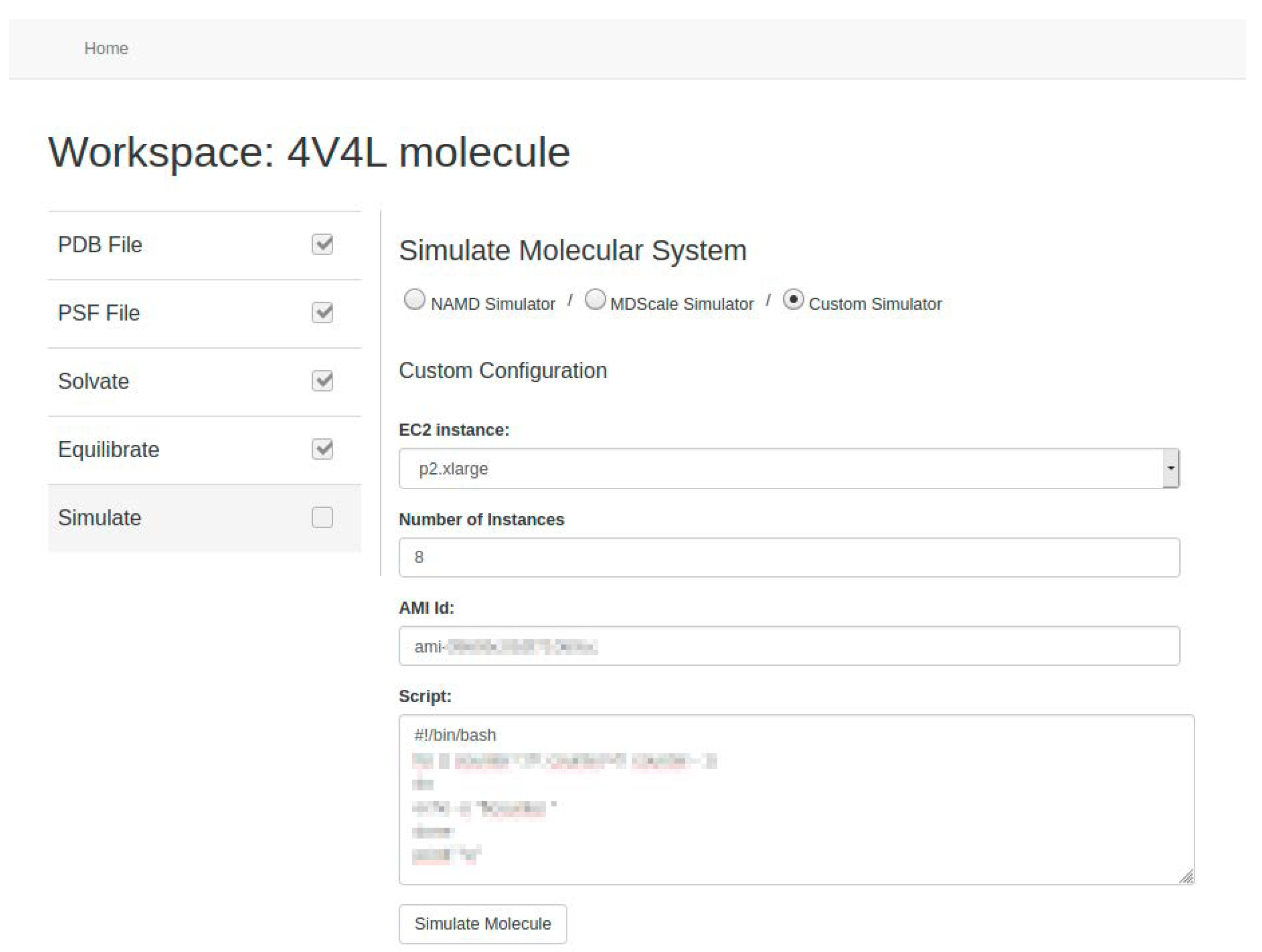
24]. As future work we propose to include other MD simulators, such as GROMACS or AMBER. In any case, we allow users, if they wish, to use their own system image (called Amazon Machine Image in AWS) to carry out the simulation process using custom software. To do this, users must also provide the script needed to run the simulator (see
Figure 12).
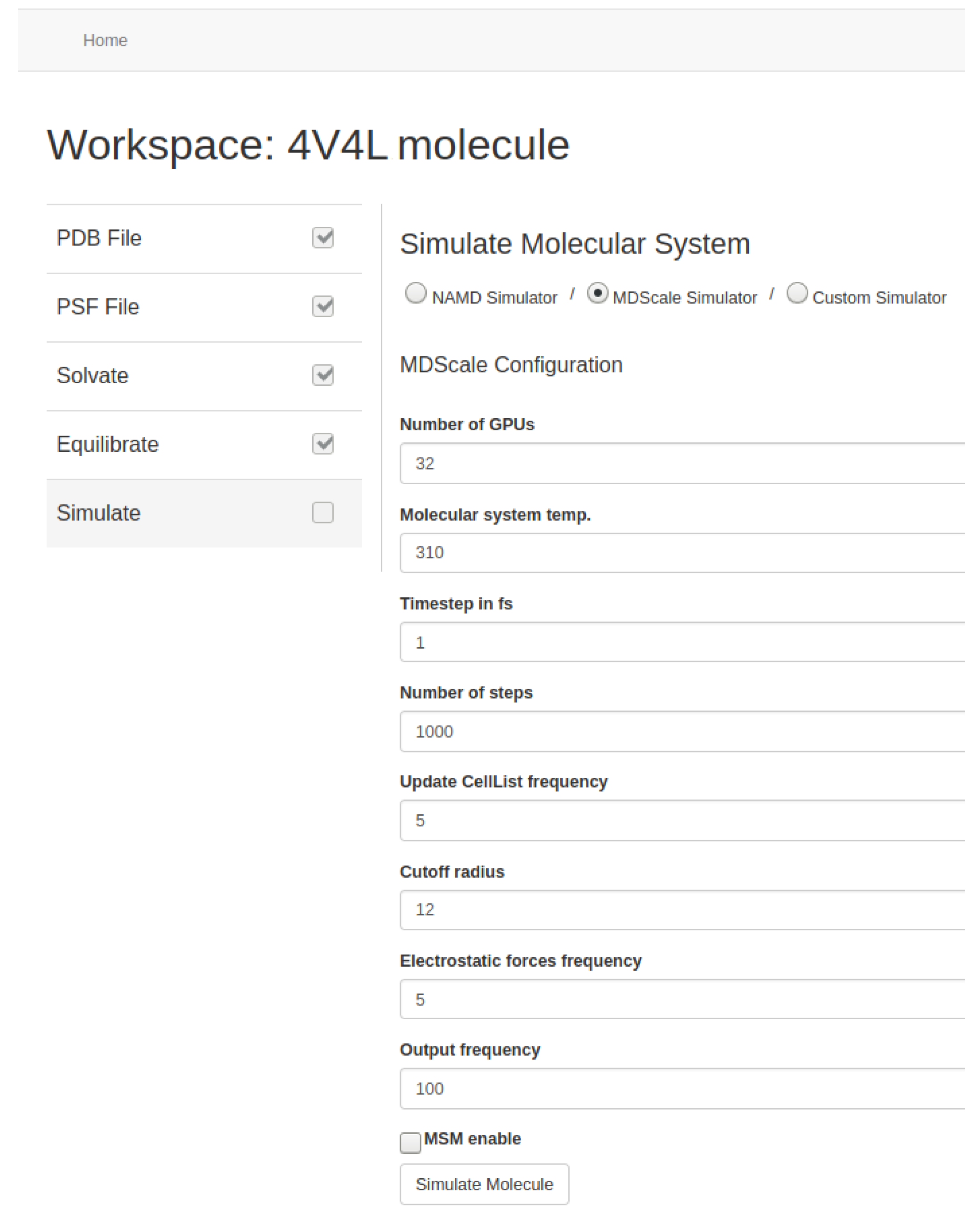
Users can set the basic parameters related to the simulation such as temperature, time-step, or output writing frequency. Depending on the MD simulator used, they can also set other optional parameters such as steps per cycle, cutoff radius and frequency for the calculation of the short-range electrostatic forces, cell-list update frequency, and/or the use of specific algorithms such as PME [
42] or MSM [
43] to calculate the long-range electrostatic forces (see
Figure 13 and
Figure 14). In addition, users can choose the number of GPUs, specifically Nvidia Tesla K80 in AWS, deployed for simulation. Our tool automates the initialization of the required EC2 p2 instances based on the number of GPUs requested and manages the network communication among them. At the end, the resulting data from the molecular system are stored in the AWS S3 storage service.
Finally, we carried out the entire simulation process using the 4V4L molecular system in NAMD and MDScale to demonstrate the possibilities of our tool.
Table 2 shows the execution times for each step of the workflow, from the initial data processing to the simulation of the molecular system. The deployment and execution time for cloud environments depend on the resources required and their availability at any given time; therefore, there is not much emphasis on this matter. Thanks to the possibility of configuring the simulation, users are able to carry out balanced tests with the different types of atomic forces.
5. Conclusions
In this paper, we have presented a web-based application that facilitates MD simulation using cloud computing services and automates related tasks. This application allows users to manage both data preparation and simulation of molecular systems in a Multi-GPU cloud computing environment. Through a friendly user interface, our system provides an easy way to perform GPU-accelerated MD simulation even for non-specialist users.
We designed the tool in order to save time in setting up a cloud environment and cost managing the cloud resources efficiently to prepare molecular systems and run MD simulators. The main target of this paper was to discuss the functionality of the tool in a cloud computing environment.
Our solution relies on a typical web application structure, which consists of a web server and a web service following the REST API architecture. Users interact with the simulation through a web browser, run any task, and leave it unattended. The application can stop any cloud resource to avoid unnecessary cost.
Our tool has been designed to facilitate the use of different MD software. We installed and compiled tools like VMD, NAMD, etc., in a cloud instance to perform the MD workflow without the need to understand how to configure and execute them. Furthermore, this design is open to changes. Any software or algorithm can be updated, added, or replaced in order to perform any task of MD simulation process. This would enable users to generate comparisons between new algorithms and techniques that will be developed in the future.
The code of the tool is freely available for use (
http://monkey.etsii.urjc.es/md/easymd). We expect our tool can be used in the future for anybody interested in MD simulation. This application could also be extended to create other solutions used a cloud environment. Just by changing the instances and slightly modifying the web service, this tool can be easily adjusted to perform different tasks that require cloud computing.
Our tool has some limitations in supporting cloud environments. We designed the application with Amazon Web Services in mind, due to the possibility of using GPU instances. However, we have intended to offer support for other cloud environments with different configurations and SDK, such as Google Cloud Platform or Microsoft Azure, or even for use in custom local clusters or supercomputing centers.
As future work, we propose to implement security mechanisms to establish communication between the web service and the infrastructure, providing also protection against DoS, SQL injection, and similar attacks. In addition to the simulation process, rendering of molecular dynamic images is also a lengthy and costly process. Rendering can also be performed within the cloud computing environment, and this feature will be explored in future work.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}