Interplay among ATP-Dependent Chromatin Remodelers Determines Chromatin Organisation in Yeast




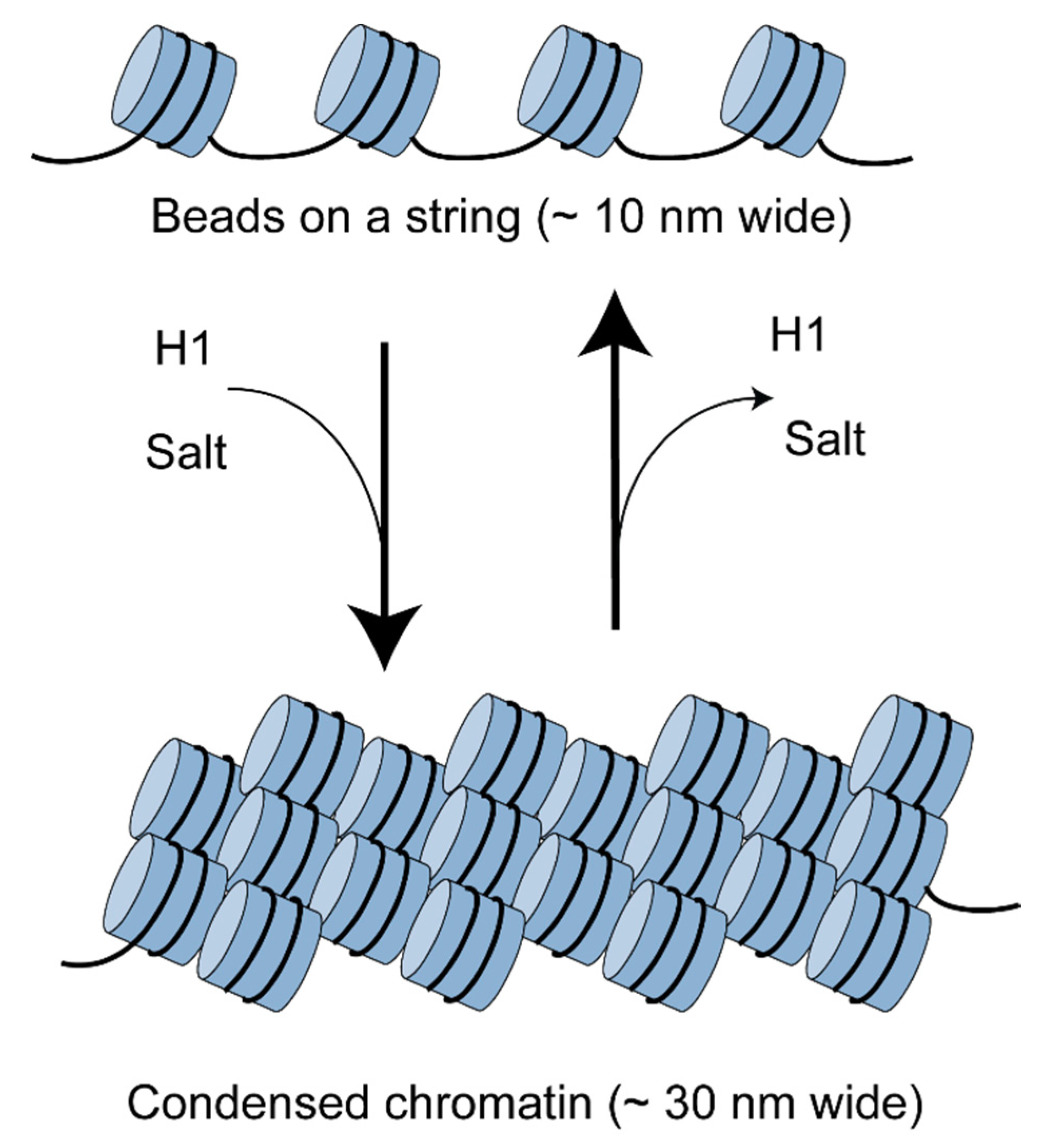{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
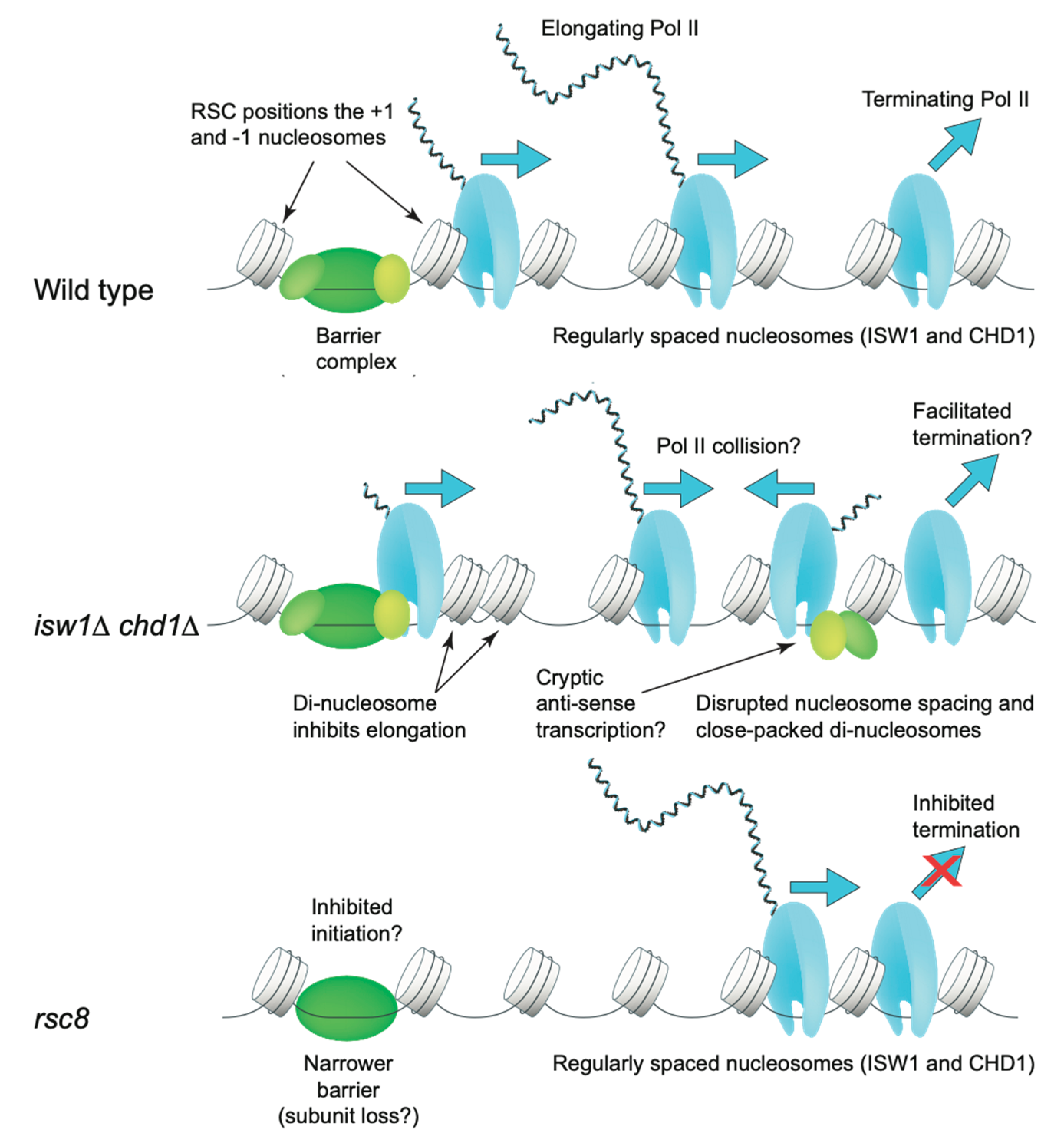{kind=link}
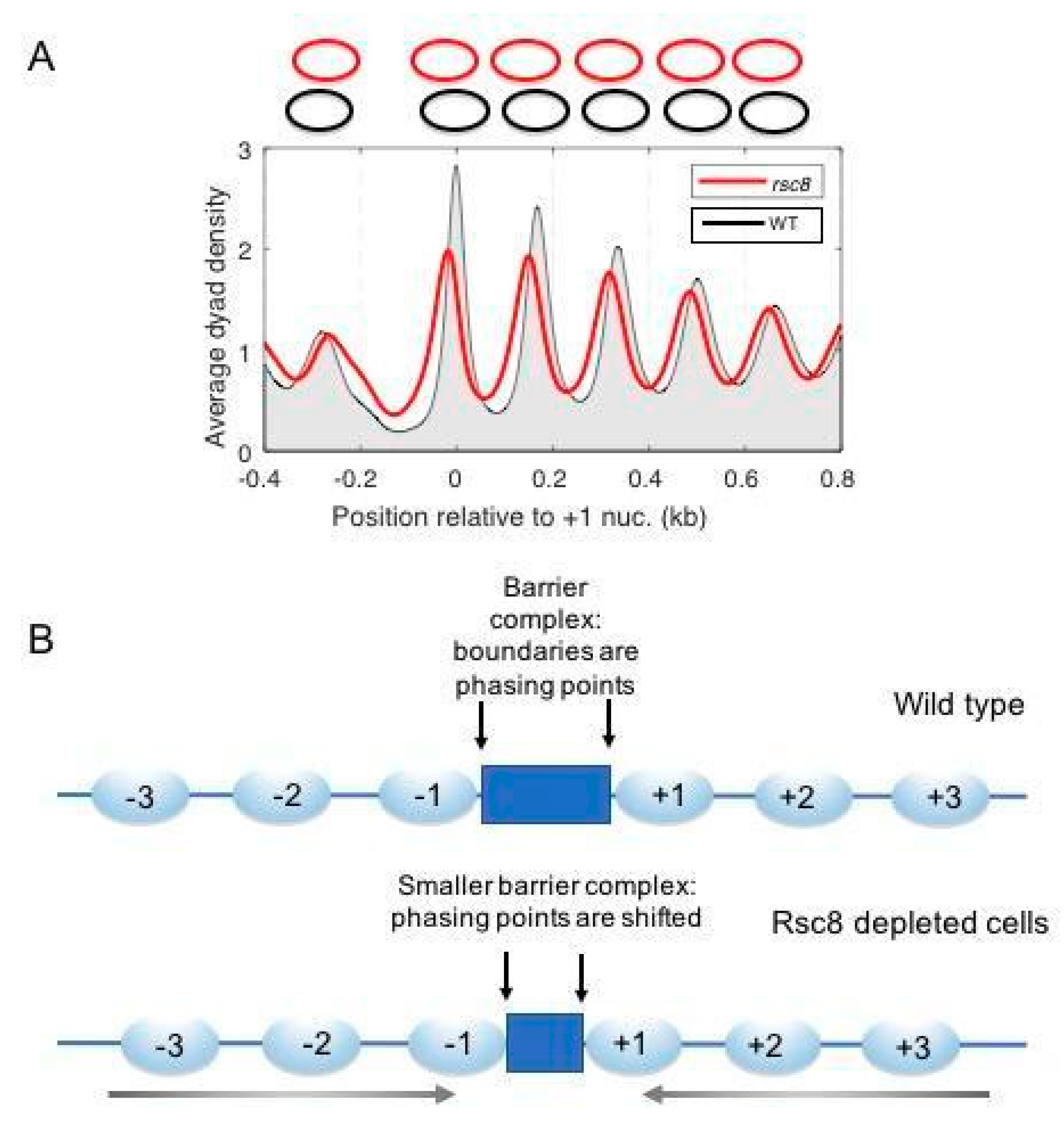{kind=link}
Abstract
1. Introduction
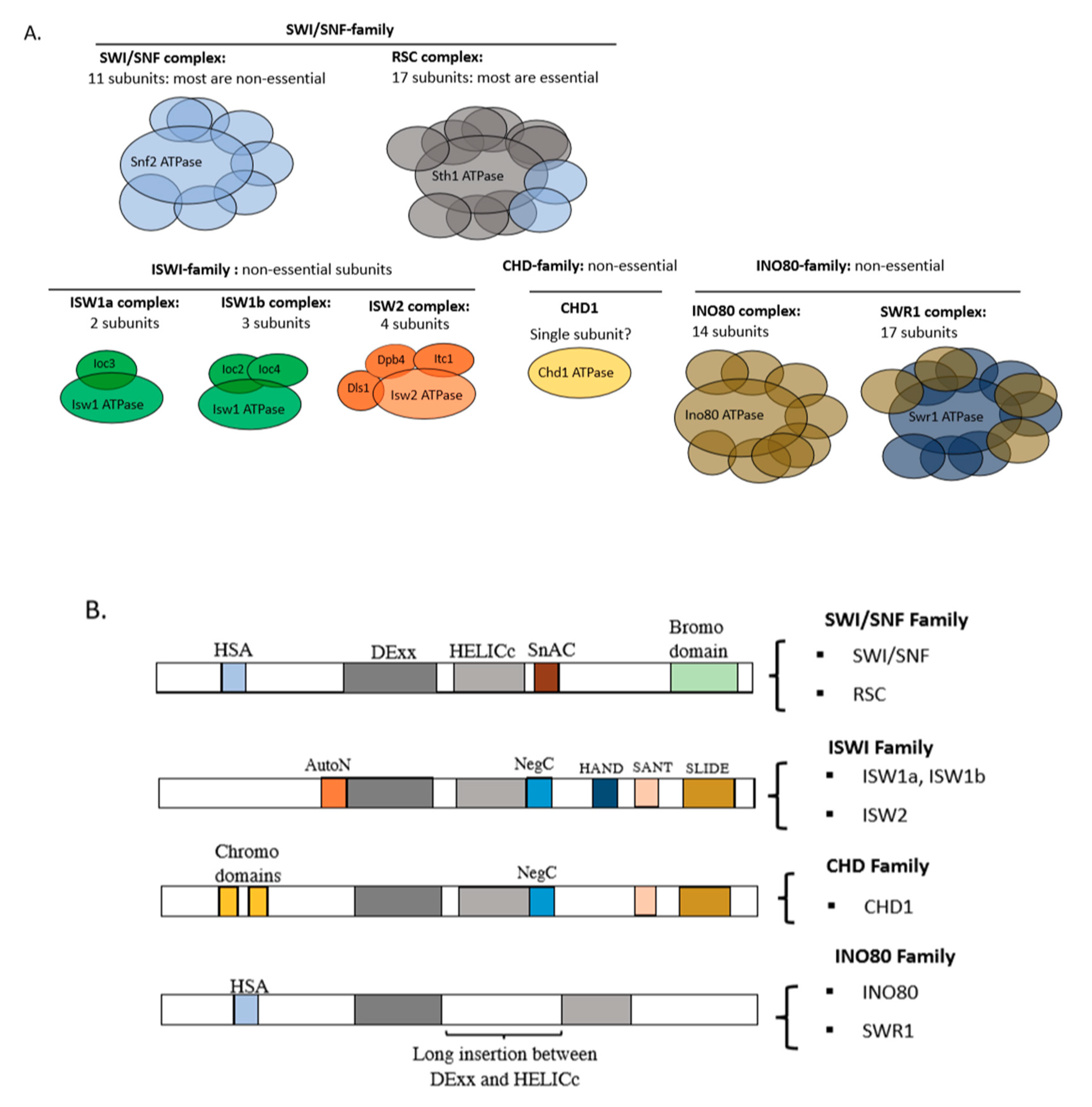
2. The ATP-Dependent Chromatin Remodelers
3. Nucleosome Mapping: MNase-Seq and Chemical Mapping
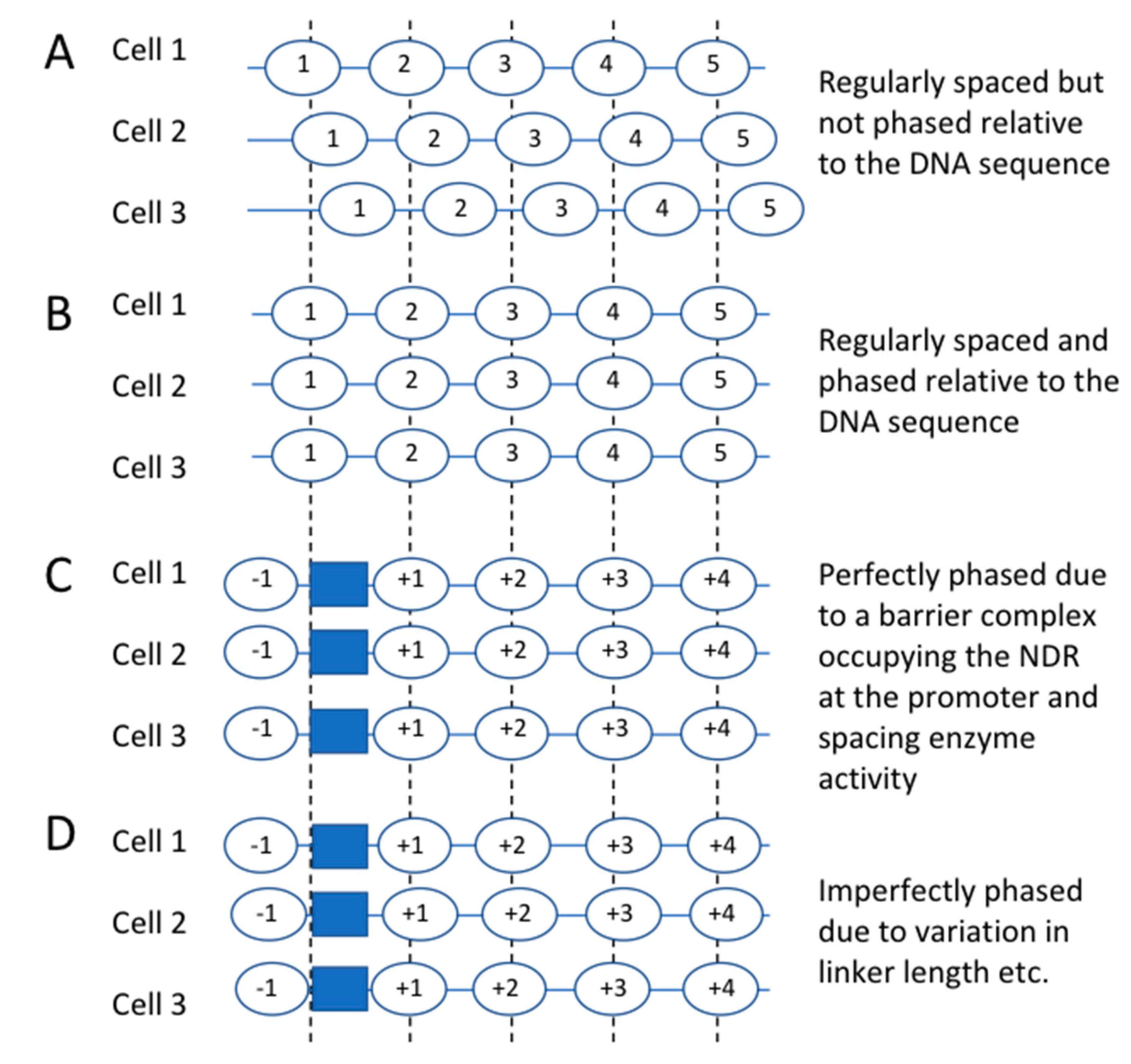
4. Nucleosome-Depleted Regions, Nucleosome Spacing and Nucleosome Phasing
5. The NDR May Contain a “Barrier” Complex
6. The Yeast Nucleosome-Spacing Enzymes: ISW1a, ISW1b, ISW2, CHD1 and INO80
6.1. Nucleosome-Spacing Enzymes in Yeast Are Not Functionally Redundant
6.2. ISW1 and CHD1 Compete to Set Nucleosome Spacing on Most Genes
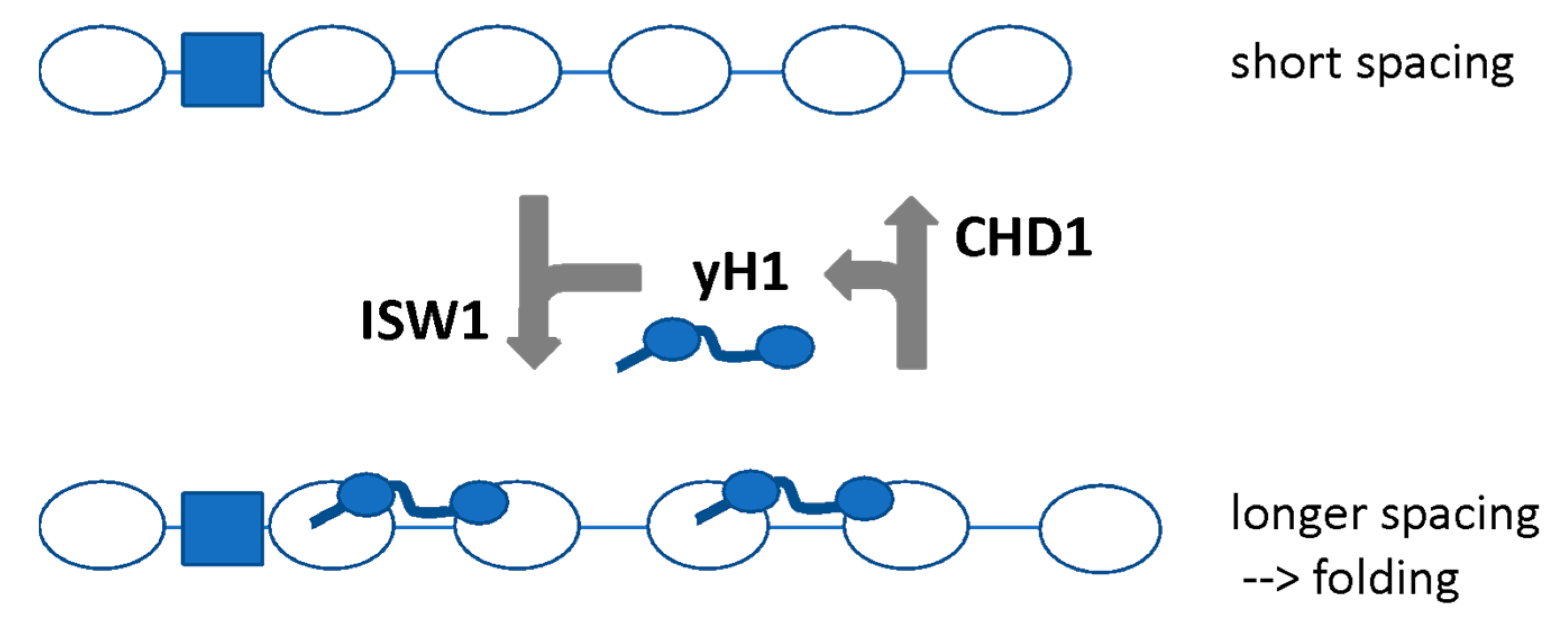
6.3. Competing Remodelers May Control H1 Binding and Chromatin Folding by Regulating Spacing
6.4. Nucleosome-Spacing Enzymes and Transcription
7. Nucleosome Remodeling Enzymes: RSC and SWI/SNF
7.1. Remodeling Activities of the Yeast SWI/SNF-Family Complexes, RSC and SWI/SNF
7.2. Roles of RSC and SWI/SNF in Determining NDRs In Vivo
7.3. RSC, SWI/SNF and Transcription
7.4. Dynamic Nucleosome Remodeling by RSC In Vivo
8. Important Issues for Future Study
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Yadon, A.N.; Tsukiyama, T. SnapShot: Chromatin remodeling: ISWI. Cell 2011, 144, 453. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.M.; Clapier, C.R.; Cairns, B.R. SnapShot: Chromatin remodeling: SWI/SNF. Cell 2011, 144, 310. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, G.J.; Sundaramoorthy, R.; Owen-Hughes, T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 2013, 154, 490–503. [Google Scholar] [CrossRef]
- Thoma, F.; Koller, T.; Klug, A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J. Cell Biol. 1979, 83, 403–427. [Google Scholar] [CrossRef]
- Widom, J. Physicochemical studies of the folding of the 100 A nucleosome filament into the 300 A filament. Cation dependence. J. Mol. Biol. 1986, 190, 411–424. [Google Scholar] [CrossRef]
- Clark, D.J.; Kimura, T. Electrostatic mechanism of chromatin folding. J. Mol. Biol. 1990, 211, 883–896. [Google Scholar] [CrossRef]
- Felsenfeld, G. Chromatin unfolds. Cell 1996, 86, 13–19. [Google Scholar] [CrossRef]
- Van Holde, K.E. Chromatin. Series in Molecular Biology; Springer: New York, NY, USA, 1989. [Google Scholar]
- Pearson, E.C.; Bates, D.L.; Prospero, T.D.; Thomas, J.O. Neuronal nuclei and glial nuclei from mammalian cerebral cortex. Nucleosome repeat lengths, DNA contents and H1 contents. Eur. J. Biochem. 1984, 144, 353–360. [Google Scholar] [CrossRef]
- Clark, S.C.; Chereji, R.V.; Lee, P.R.; Fields, R.D.; Clark, D.J. Differential nucleosome spacing in neurons and glia. Neurosci. Lett. 2020, 714, 134559. [Google Scholar] [CrossRef] [PubMed]
- Lohr, D.; Van Holde, K.E. Organization of spacer DNA in chromatin. Proc. Natl. Acad. Sci. USA 1979, 76, 6326–6330. [Google Scholar] [CrossRef] [PubMed]
- Brogaard, K.; Xi, L.; Wang, J.-P.; Widom, J. A map of nucleosome positions in yeast at base-pair resolution. Nature 2012, 486, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Bulger, M.; Pazin, M.J.; Kobayashi, R.; Kadonaga, J.T. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell 1997, 90, 145–155. [Google Scholar] [CrossRef]
- Tsukiyama, T.; Wu, C. Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 1995, 83, 1011–1020. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Ryan, D.P.; Sundaramoorthy, R.; Martin, D.; Singh, V.; Owen-Hughes, T. The DNA-binding domain of the Chd1 chromatin-remodelling enzyme contains SANT and SLIDE domains. EMBO J. 2011, 30, 2596–2609. [Google Scholar] [CrossRef]
- Brehm, A.; Tufteland, K.R.; Aasland, R.; Becker, P.B. The many colours of chromodomains. Bioessays 2004, 26, 133–140. [Google Scholar] [CrossRef]
- Flanagan, J.F.; Mi, L.-Z.; Chruszcz, M.; Cymborowski, M.; Clines, K.L.; Kim, Y.; Minor, W.; Rastinejad, F.; Khorasanizadeh, S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 2005, 438, 1181–1185. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Paul, S.; Bartholomew, B. Regulation of ATP-dependent chromatin remodelers: Accelerators/brakes, anchors and sensors. Biochem. Soc. Trans. 2018, 46, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Luk, E.; Ranjan, A.; Fitzgerald, P.C.; Mizuguchi, G.; Huang, Y.; Wei, D.; Wu, C. Stepwise histone replacement by SWR1 requires dual activation with histone H2A.Z and canonical nucleosome. Cell 2010, 143, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, G.; Shen, X.; Landry, J.; Wu, W.-H.; Sen, S.; Wu, C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 2004, 303, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ranjan, A.; Wei, D.; Wu, C. Comment on “A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme”. Science 2016, 353, 358. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Radman-Livaja, M.; Rando, O.J.; Peterson, C.L. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science 2013, 340, 195–199. [Google Scholar] [CrossRef]
- Watanabe, S.; Peterson, C.L. Response to Comment on “A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme”. Science 2016, 353, 358. [Google Scholar] [CrossRef]
- Qiu, H.; Biernat, E.; Govind, C.K.; Rawal, Y.; Chereji, R.V.; Clark, D.J.; Hinnebusch, A.G. Chromatin remodeler Ino80C acts independently of H2A.Z to evict promoter nucleosomes and stimulate transcription of highly expressed genes in yeast. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Badis, G.; Chan, E.T.; van Bakel, H.; Pena-Castillo, L.; Tillo, D.; Tsui, K.; Carlson, C.D.; Gossett, A.J.; Hasinoff, M.J.; Warren, C.L.; et al. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol. Cell 2008, 32, 878–887. [Google Scholar] [CrossRef]
- Lorch, Y.; Maier-Davis, B.; Kornberg, R.D. Histone Acetylation Inhibits RSC and Stabilizes the +1 Nucleosome. Mol. Cell 2018, 72, 594–600. [Google Scholar] [CrossRef]
- Kasten, M.; Szerlong, H.; Erdjument-Bromage, H.; Tempst, P.; Werner, M.; Cairns, B.R. Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys14. EMBO J. 2004, 23, 1348–1359. [Google Scholar] [CrossRef]
- VanDemark, A.P.; Kasten, M.M.; Ferris, E.; Heroux, A.; Hill, C.P.; Cairns, B.R. Autoregulation of the rsc4 tandem bromodomain by gcn5 acetylation. Mol. Cell 2007, 27, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Cole, H.A.; Howard, B.H.; Clark, D.J. Genome-wide mapping of nucleosomes in yeast using paired-end sequencing. Methods Enzymol. 2012, 513, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Chereji, R.V.; Ocampo, J.; Clark, D.J. MNase-Sensitive Complexes in Yeast: Nucleosomes and Non-histone Barriers. Mol. Cell 2017, 65, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Flaus, A.; Luger, K.; Tan, S.; Richmond, T.J. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proc. Natl. Acad. Sci. USA 1996, 93, 1370–1375. [Google Scholar] [CrossRef]
- Voong, L.N.; Xi, L.; Sebeson, A.C.; Xiong, B.; Wang, J.-P.; Wang, X. Insights into Nucleosome Organization in Mouse Embryonic Stem Cells through Chemical Mapping. Cell 2016, 167, 1555–1570. [Google Scholar] [CrossRef]
- Chereji, R.V.; Ramachandran, S.; Bryson, T.D.; Henikoff, S. Precise genome-wide mapping of single nucleosomes and linkers in vivo. Genome Biol. 2018, 19, 1–20. [Google Scholar] [CrossRef]
- Lai, B.; Gao, W.; Cui, K.; Xie, W.; Tang, Q.; Jin, W.; Hu, G.; Ni, B.; Zhao, K. Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing. Nature 2018, 562, 281–285. [Google Scholar] [CrossRef]
- Shipony, Z.; Marinov, G.K.; Swaffer, M.P.; Sinnott-Armstrong, N.A.; Skotheim, J.M.; Kundaje, A.; Greenleaf, W.J. Long-range single-molecule mapping of chromatin accessibility in eukaryotes. Nat. Methods 2020, 17, 319–327. [Google Scholar] [CrossRef]
- Lee, C.-K.; Shibata, Y.; Rao, B.; Strahl, B.D.; Lieb, J.D. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat. Genet. 2004, 36, 900–905. [Google Scholar] [CrossRef]
- Yuan, G.-C.; Liu, Y.-J.; Dion, M.F.; Slack, M.D.; Wu, L.F.; Altschuler, S.J.; Rando, O.J. Genome-scale identification of nucleosome positions in S. cerevisiae. Science 2005, 309, 626–630. [Google Scholar] [CrossRef]
- Nagarajavel, V.; Iben, J.R.; Howard, B.H.; Maraia, R.J.; Clark, D.J. Global “bootprinting” reveals the elastic architecture of the yeast TFIIIB-TFIIIC transcription complex in vivo. Nucleic Acids Res. 2013, 41, 8135–8143. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Ioshikhes, I.P.; Venters, B.J.; Jiang, C.; Tomsho, L.P.; Qi, J.; Schuster, S.C.; Albert, I.; Pugh, B.F. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008, 18, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Tillo, D.; Bray, N.; Morse, R.H.; Davis, R.W.; Hughes, T.R.; Nislow, C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet. 2007, 39, 1235–1244. [Google Scholar] [CrossRef]
- Shen, C.H.; Leblanc, B.P.; Alfieri, J.A.; Clark, D.J. Remodeling of yeast CUP1 chromatin involves activator-dependent repositioning of nucleosomes over the entire gene and flanking sequences. Mol. Cell. Biol. 2001, 21, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; McLaughlin, N.; Lindstrom, K.; Tsukiyama, T.; Clark, D.J. Activation of Saccharomyces cerevisiae HIS3 results in Gcn4p-dependent, SWI/SNF-dependent mobilization of nucleosomes over the entire gene. Mol. Cell. Biol. 2006, 26, 8607–8622. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, G.; John, S.; Roberts, M.S.; Hager, G.L. Nucleosome positioning on the MMTV LTR results from the frequency-biased occupancy of multiple frames. Genes Dev. 1995, 9, 1933–1947. [Google Scholar] [CrossRef] [PubMed]
- Cole, H.A.; Howard, B.H.; Clark, D.J. Activation-induced disruption of nucleosome position clusters on the coding regions of Gcn4-dependent genes extends into neighbouring genes. Nucleic Acids Res. 2011, 39, 9521–9535. [Google Scholar] [CrossRef]
- Chereji, R.V.; Eriksson, P.R.; Ocampo, J.; Prajapati, H.K.; Clark, D.J. Accessibility of promoter DNA is not the primary determinant of chromatin-mediated gene regulation. Genome Res. 2019, 29, 1985–1995. [Google Scholar] [CrossRef]
- Oberbeckmann, E.; Wolff, M.; Krietenstein, N.; Heron, M.; Ellins, J.L.; Schmid, A.; Krebs, S.; Blum, H.; Gerland, U.; Korber, P.; et al. Absolute nucleosome occupancy map for the Saccharomyces cerevisiae genome. Genome Res. 2019, 29, 1996–2009. [Google Scholar] [CrossRef]
- Krietenstein, N.; Wal, M.; Watanabe, S.; Park, B.; Peterson, C.L.; Pugh, B.F.; Korber, P. Genomic Nucleosome Organization Reconstituted with Pure Proteins. Cell 2016, 167, 709–721. [Google Scholar] [CrossRef]
- Chereji, R.V.; Clark, D.J. Major determinants of nucleosome positioning. Biophys. J. 2018, 114, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.; Hughes, A.; Yassour, M.; Rando, O.J.; Friedman, N. High-resolution nucleosome mapping reveals transcription-dependent promoter packaging. Genome Res. 2010, 20, 90–100. [Google Scholar] [CrossRef]
- Kubik, S.; Bruzzone, M.J.; Albert, B.; Shore, D. A Reply to “MNase-Sensitive Complexes in Yeast: Nucleosomes and Non-histone Barriers”, by Chereji et al. Mol. Cell 2017, 65, 578–580. [Google Scholar] [CrossRef][Green Version]
- Kubik, S.; Bruzzone, M.J.; Jacquet, P.; Falcone, J.-L.; Rougemont, J.; Shore, D. Nucleosome Stability Distinguishes Two Different Promoter Types at All Protein-Coding Genes in Yeast. Mol. Cell 2015, 60, 422–434. [Google Scholar] [CrossRef]
- Xi, Y.; Yao, J.; Chen, R.; Li, W.; He, X. Nucleosome fragility reveals novel functional states of chromatin and poises genes for activation. Genome Res. 2011, 21, 718–724. [Google Scholar] [CrossRef]
- Brahma, S.; Henikoff, S. RSC-Associated Subnucleosomes Define MNase-Sensitive Promoters in Yeast. Mol. Cell 2019, 73, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Mivelaz, M.; Cao, A.-M.; Kubik, S.; Zencir, S.; Hovius, R.; Boichenko, I.; Stachowicz, A.M.; Kurat, C.F.; Shore, D.; Fierz, B. Chromatin fiber invasion and nucleosome displacement by the rap1 transcription factor. Mol. Cell 2020, 77, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Hartley, P.D.; Madhani, H.D. Mechanisms that specify promoter nucleosome location and identity. Cell 2009, 137, 445–458. [Google Scholar] [CrossRef]
- Ganapathi, M.; Palumbo, M.J.; Ansari, S.A.; He, Q.; Tsui, K.; Nislow, C.; Morse, R.H. Extensive role of the general regulatory factors, Abf1 and Rap1, in determining genome-wide chromatin structure in budding yeast. Nucleic Acids Res. 2011, 39, 2032–2044. [Google Scholar] [CrossRef]
- Yan, C.; Chen, H.; Bai, L. Systematic Study of Nucleosome-Displacing Factors in Budding Yeast. Mol. Cell 2018, 71, 294–305. [Google Scholar] [CrossRef]
- Kubik, S.; O’Duibhir, E.; de Jonge, W.J.; Mattarocci, S.; Albert, B.; Falcone, J.-L.; Bruzzone, M.J.; Holstege, F.C.P.; Shore, D. Sequence-Directed Action of RSC Remodeler and General Regulatory Factors Modulates +1 Nucleosome Position to Facilitate Transcription. Mol. Cell 2018, 71, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Chereji, R.V.; Morozov, A.V. Ubiquitous nucleosome crowding in the yeast genome. Proc. Natl. Acad. Sci. USA 2014, 111, 5236–5241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wippo, C.J.; Wal, M.; Ward, E.; Korber, P.; Pugh, B.F. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science 2011, 332, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Vary, J.C.; Fazzio, T.G.; Tsukiyama, T. Assembly of yeast chromatin using ISWI complexes. Methods Enzymol. 2004, 375, 88–102. [Google Scholar] [PubMed]
- Tsukiyama, T.; Palmer, J.; Landel, C.C.; Shiloach, J.; Wu, C. Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev. 1999, 13, 686–697. [Google Scholar] [CrossRef]
- Lusser, A.; Urwin, D.L.; Kadonaga, J.T. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat. Struct. Mol. Biol. 2005, 12, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, C.; Flaus, A.; Ferreira, H.; Owen-Hughes, T. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J. Biol. Chem. 2006, 281, 16279–16288. [Google Scholar] [CrossRef]
- Udugama, M.; Sabri, A.; Bartholomew, B. The INO80 ATP-dependent chromatin remodeling complex is a nucleosome spacing factor. Mol. Cell. Biol. 2011, 31, 662–673. [Google Scholar] [CrossRef]
- Lieleg, C.; Ketterer, P.; Nuebler, J.; Ludwigsen, J.; Gerland, U.; Dietz, H.; Mueller-Planitz, F.; Korber, P. Nucleosome spacing generated by ISWI and CHD1 remodelers is constant regardless of nucleosome density. Mol. Cell. Biol. 2015, 35, 1588–1605. [Google Scholar] [CrossRef]
- Pointner, J.; Persson, J.; Prasad, P.; Norman-Axelsson, U.; Strålfors, A.; Khorosjutina, O.; Krietenstein, N.; Svensson, J.P.; Ekwall, K.; Korber, P.; et al. CHD1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in S. pombe. EMBO J. 2012, 31, 4388–4403. [Google Scholar] [CrossRef]
- Torigoe, S.E.; Patel, A.; Khuong, M.T.; Bowman, G.D.; Kadonaga, J.T. ATP-dependent chromatin assembly is functionally distinct from chromatin remodeling. eLife 2013, 2, e00863. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, J.; Chereji, R.V.; Eriksson, P.R.; Clark, D.J. The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacing in vivo. Nucleic Acids Res. 2016, 44, 4625–4635. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Sigal, N.; Barkai, N. Widespread remodeling of mid-coding sequence nucleosomes by Isw1. Genome Biol. 2010, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Vinayachandran, V.; Batta, K.; Koerber, R.T.; Pugh, B.F. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 2012, 149, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Gkikopoulos, T.; Schofield, P.; Singh, V.; Pinskaya, M.; Mellor, J.; Smolle, M.; Workman, J.L.; Barton, G.J.; Owen-Hughes, T. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science 2011, 333, 1758–1760. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, J.; Chereji, R.V.; Eriksson, P.R.; Clark, D.J. Contrasting roles of the RSC and ISW1/CHD1 chromatin remodelers in RNA polymerase II elongation and termination. Genome Res. 2019, 29, 407–417. [Google Scholar] [CrossRef]
- Kato, D.; Osakabe, A.; Arimura, Y.; Mizukami, Y.; Horikoshi, N.; Saikusa, K.; Akashi, S.; Nishimura, Y.; Park, S.-Y.; Nogami, J.; et al. Crystal structure of the overlapping dinucleosome composed of hexasome and octasome. Science 2017, 356, 205–208. [Google Scholar] [CrossRef]
- Dechassa, M.L.; Sabri, A.; Pondugula, S.; Kassabov, S.R.; Chatterjee, N.; Kladde, M.P.; Bartholomew, B. SWI/SNF has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol. Cell 2010, 38, 590–602. [Google Scholar] [CrossRef]
- Engeholm, M.; de Jager, M.; Flaus, A.; Brenk, R.; van Noort, J.; Owen-Hughes, T. Nucleosomes can invade DNA territories occupied by their neighbors. Nat. Struct. Mol. Biol. 2009, 16, 151–158. [Google Scholar] [CrossRef]
- Ulyanova, N.P.; Schnitzler, G.R. Human SWI/SNF generates abundant, structurally altered dinucleosomes on polynucleosomal templates. Mol. Cell. Biol. 2005, 25, 11156–11170. [Google Scholar] [CrossRef]
- Fazzio, T.G.; Tsukiyama, T. Chromatin remodeling in vivo: Evidence for a nucleosome sliding mechanism. Mol. Cell 2003, 12, 1333–1340. [Google Scholar] [CrossRef]
- Whitehouse, I.; Rando, O.J.; Delrow, J.; Tsukiyama, T. Chromatin remodelling at promoters suppresses antisense transcription. Nature 2007, 450, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Van Bakel, H.; Tsui, K.; Gebbia, M.; Mnaimneh, S.; Hughes, T.R.; Nislow, C. A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet. 2013, 9, e1003479. [Google Scholar] [CrossRef] [PubMed]
- Simic, R.; Lindstrom, D.L.; Tran, H.G.; Roinick, K.L.; Costa, P.J.; Johnson, A.D.; Hartzog, G.A.; Arndt, K.M. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J. 2003, 22, 1846–1856. [Google Scholar] [CrossRef]
- Rhee, H.S.; Bataille, A.R.; Zhang, L.; Pugh, B.F. Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 2014, 159, 1377–1388. [Google Scholar] [CrossRef]
- Landsman, D. Histone H1 in Saccharomyces cerevisiae: A double mystery solved? Trends Biochem. Sci. 1996, 21, 287–288. [Google Scholar]
- Cutter, A.R.; Hayes, J.J. A brief review of nucleosome structure. FEBS Lett. 2015, 589, 2914–2922. [Google Scholar] [CrossRef]
- Patterton, H.G.; Landel, C.C.; Landsman, D.; Peterson, C.L.; Simpson, R.T. The biochemical and phenotypic characterization of Hho1p, the putative linker histone H1 of Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 7268–7276. [Google Scholar] [CrossRef]
- Ali, T.; Coles, P.; Stevens, T.J.; Stott, K.; Thomas, J.O. Two homologous domains of similar structure but different stability in the yeast linker histone, Hho1p. J. Mol. Biol. 2004, 338, 139–148. [Google Scholar] [CrossRef]
- Ali, T.; Thomas, J.O. Distinct properties of the two putative “globular domains” of the yeast linker histone, Hho1p. J. Mol. Biol. 2004, 337, 1123–1135. [Google Scholar] [CrossRef]
- Cole, H.A.; Cui, F.; Ocampo, J.; Burke, T.L.; Nikitina, T.; Nagarajavel, V.; Kotomura, N.; Zhurkin, V.B.; Clark, D.J. Novel nucleosomal particles containing core histones and linker DNA but no histone H1. Nucleic Acids Res. 2016, 44, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Rando, O.J. Comparative genomics reveals chd1 as a determinant of nucleosome spacing in vivo. G3 Genes Genomes Genet. 2015, 5, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Freidkin, I.; Katcoff, D.J. Specific distribution of the Saccharomyces cerevisiae linker histone homolog HHO1p in the chromatin. Nucleic Acids Res. 2001, 29, 4043–4051. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.L.; Thomas, J.O. Histones H1 and H5: One or two molecules per nucleosome? Nucleic Acids Res. 1981, 9, 5883–5894. [Google Scholar] [CrossRef]
- Lindstrom, K.C.; Vary, J.C.; Parthun, M.R.; Delrow, J.; Tsukiyama, T. Isw1 functions in parallel with the NuA4 and Swr1 complexes in stress-induced gene repression. Mol. Cell. Biol. 2006, 26, 6117–6129. [Google Scholar] [CrossRef]
- Cole, H.A.; Ocampo, J.; Iben, J.R.; Chereji, R.V.; Clark, D.J. Heavy transcription of yeast genes correlates with differential loss of histone H2B relative to H4 and queued RNA polymerases. Nucleic Acids Res. 2014, 42, 12512–12522. [Google Scholar] [CrossRef]
- Elfving, N.; Chereji, R.V.; Bharatula, V.; Björklund, S.; Morozov, A.V.; Broach, J.R. A dynamic interplay of nucleosome and Msn2 binding regulates kinetics of gene activation and repression following stress. Nucleic Acids Res. 2014, 42, 5468–5482. [Google Scholar] [CrossRef]
- Smolle, M.; Venkatesh, S.; Gogol, M.M.; Li, H.; Zhang, Y.; Florens, L.; Washburn, M.P.; Workman, J.L. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat. Struct. Mol. Biol. 2012, 19, 884–892. [Google Scholar] [CrossRef]
- Schwabish, M.A.; Struhl, K. Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol. Cell. Biol. 2004, 24, 10111–10117. [Google Scholar] [CrossRef]
- Schwabish, M.A.; Struhl, K. The Swi/Snf complex is important for histone eviction during transcriptional activation and RNA polymerase II elongation in vivo. Mol. Cell. Biol. 2007, 27, 6987–6995. [Google Scholar] [CrossRef]
- Cui, F.; Cole, H.A.; Clark, D.J.; Zhurkin, V.B. Transcriptional activation of yeast genes disrupts intragenic nucleosome phasing. Nucleic Acids Res. 2012, 40, 10753–10764. [Google Scholar] [CrossRef] [PubMed]
- Zawadzki, K.A.; Morozov, A.V.; Broach, J.R. Chromatin-dependent transcription factor accessibility rather than nucleosome remodeling predominates during global transcriptional restructuring in Saccharomyces cerevisiae. Mol. Biol. Cell 2009, 20, 3503–3513. [Google Scholar] [CrossRef] [PubMed]
- Field, Y.; Kaplan, N.; Fondufe-Mittendorf, Y.; Moore, I.K.; Sharon, E.; Lubling, Y.; Widom, J.; Segal, E. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput. Biol. 2008, 4, e1000216. [Google Scholar] [CrossRef]
- Shivaswamy, S.; Bhinge, A.; Zhao, Y.; Jones, S.; Hirst, M.; Iyer, V.R. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008, 6, e65. [Google Scholar] [CrossRef]
- Levendosky, R.F.; Sabantsev, A.; Deindl, S.; Bowman, G.D. The Chd1 chromatin remodeler shifts hexasomes unidirectionally. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Morillon, A.; Karabetsou, N.; O’Sullivan, J.; Kent, N.; Proudfoot, N.; Mellor, J. Isw1 chromatin remodeling ATPase coordinates transcription elongation and termination by RNA polymerase II. Cell 2003, 115, 425–435. [Google Scholar] [CrossRef]
- Alén, C.; Kent, N.A.; Jones, H.S.; O’Sullivan, J.; Aranda, A.; Proudfoot, N.J. A role for chromatin remodeling in transcriptional termination by RNA polymerase II. Mol. Cell 2002, 10, 1441–1452. [Google Scholar] [CrossRef]
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodeling by RSC involves ATP-dependent DNA translocation. Genes Dev. 2002, 16, 2120–2134. [Google Scholar] [CrossRef]
- Harada, B.T.; Hwang, W.L.; Deindl, S.; Chatterjee, N.; Bartholomew, B.; Zhuang, X. Stepwise nucleosome translocation by RSC remodeling complexes. eLife 2016, 5. [Google Scholar] [CrossRef]
- Lorch, Y.; Kornberg, R.D. Chromatin-remodeling for transcription. Q. Rev. Biophys. 2017, 50, e5. [Google Scholar] [CrossRef]
- Lorch, Y.; Zhang, M.; Kornberg, R.D. RSC unravels the nucleosome. Mol. Cell 2001, 7, 89–95. [Google Scholar] [CrossRef]
- Shukla, M.S.; Syed, S.H.; Montel, F.; Faivre-Moskalenko, C.; Bednar, J.; Travers, A.; Angelov, D.; Dimitrov, S. Remosomes: RSC generated non-mobilized particles with approximately 180 bp DNA loosely associated with the histone octamer. Proc. Natl. Acad. Sci. USA 2010, 107, 1936–1941. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.S.; Syed, S.H.; Boopathi, R.; Simon, E.B.; Nahata, S.; Ramos, L.; Dalkara, D.; Moskalenko, C.; Travers, A.; Angelov, D.; et al. Generation of remosomes by the SWI/SNF chromatin remodeler family. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cairns, B.R.; Lorch, Y.; Li, Y.; Zhang, M.; Lacomis, L.; Erdjument-Bromage, H.; Tempst, P.; Du, J.; Laurent, B.; Kornberg, R.D.; et al. RSC, an essential, abundant chromatin-remodeling complex. Cell 1996, 87, 1249–1260. [Google Scholar] [CrossRef]
- Ganguli, D.; Chereji, R.V.; Iben, J.R.; Cole, H.A.; Clark, D.J. RSC-dependent constructive and destructive interference between opposing arrays of phased nucleosomes in yeast. Genome Res. 2014, 24, 1637–1649. [Google Scholar] [CrossRef] [PubMed]
- Rawal, Y.; Chereji, R.V.; Qiu, H.; Ananthakrishnan, S.; Govind, C.K.; Clark, D.J.; Hinnebusch, A.G. SWI/SNF and RSC cooperate to reposition and evict promoter nucleosomes at highly expressed genes in yeast. Genes Dev. 2018, 32, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Parnell, T.J.; Schlichter, A.; Wilson, B.G.; Cairns, B.R. The chromatin remodelers RSC and ISW1 display functional and chromatin-based promoter antagonism. eLife 2015, 4, e06073. [Google Scholar] [CrossRef]
- Qiu, H.; Chereji, R.V.; Hu, C.; Cole, H.A.; Rawal, Y.; Clark, D.J.; Hinnebusch, A.G. Genome-wide cooperation by HAT Gcn5, remodeler SWI/SNF, and chaperone Ydj1 in promoter nucleosome eviction and transcriptional activation. Genome Res. 2016, 26, 211–225. [Google Scholar] [CrossRef]
- Ramachandran, S.; Zentner, G.E.; Henikoff, S. Asymmetric nucleosomes flank promoters in the budding yeast genome. Genome Res. 2015, 25, 381–390. [Google Scholar] [CrossRef]
- Ng, H.H.; Robert, F.; Young, R.A.; Struhl, K. Genome-wide location and regulated recruitment of the RSC nucleosome-remodeling complex. Genes Dev. 2002, 16, 806–819. [Google Scholar] [CrossRef]
- Floer, M.; Wang, X.; Prabhu, V.; Berrozpe, G.; Narayan, S.; Spagna, D.; Alvarez, D.; Kendall, J.; Krasnitz, A.; Stepansky, A.; et al. A RSC/nucleosome complex determines chromatin architecture and facilitates activator binding. Cell 2010, 141, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Spain, M.M.; Ansari, S.A.; Pathak, R.; Palumbo, M.J.; Morse, R.H.; Govind, C.K. The RSC complex localizes to coding sequences to regulate Pol II and histone occupancy. Mol. Cell 2014, 56, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Qiu, H.; Swanson, M.J.; Hinnebusch, A.G. Recruitment of SWI/SNF by Gcn4p does not require Snf2p or Gcn5p but depends strongly on SWI/SNF integrity, SRB mediator, and SAGA. Mol. Cell. Biol. 2003, 23, 8829–8845. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Jackson, B.M.; Zhou, H.; Winston, F.; Hinnebusch, A.G. Transcriptional activation by Gcn4p involves independent interactions with the SWI/SNF complex and the SRB/mediator. Mol. Cell 1999, 4, 657–664. [Google Scholar] [CrossRef]
- Neely, K.E.; Hassan, A.H.; Wallberg, A.E.; Steger, D.J.; Cairns, B.R.; Wright, A.P.; Workman, J.L. Activation domain-mediated targeting of the SWI/SNF complex to promoters stimulates transcription from nucleosome arrays. Mol. Cell 1999, 4, 649–655. [Google Scholar] [CrossRef]
- Kim, Y.; Clark, D.J. SWI/SNF-dependent long-range remodeling of yeast HIS3 chromatin. Proc. Natl. Acad. Sci. USA 2002, 99, 15381–15386. [Google Scholar] [CrossRef]
- Parnell, T.J.; Huff, J.T.; Cairns, B.R. RSC regulates nucleosome positioning at Pol II genes and density at Pol III genes. EMBO J. 2008, 27, 100–110. [Google Scholar] [CrossRef]
- Shandilya, J.; Roberts, S.G.E. The transcription cycle in eukaryotes: From productive initiation to RNA polymerase II recycling. Biochim. Biophys. Acta 2012, 1819, 391–400. [Google Scholar] [CrossRef]
- Klein-Brill, A.; Joseph-Strauss, D.; Appleboim, A.; Friedman, N. Dynamics of Chromatin and Transcription during Transient Depletion of the RSC Chromatin Remodeling Complex. Cell Rep. 2019, 26, 279–292. [Google Scholar] [CrossRef]
- Sudarsanam, P.; Iyer, V.R.; Brown, P.O.; Winston, F. Whole-genome expression analysis of snf/swi mutants of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2000, 97, 3364–3369. [Google Scholar] [CrossRef]
- Wilson, C.J.; Chao, D.M.; Imbalzano, A.N.; Schnitzler, G.R.; Kingston, R.E.; Young, R.A. RNA polymerase II holoenzyme contains SWI/SNF regulators involved in chromatin remodeling. Cell 1996, 84, 235–244. [Google Scholar] [CrossRef]
- Goldstein, I.; Hager, G.L. Dynamic enhancer function in the chromatin context. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1390. [Google Scholar] [CrossRef] [PubMed]
- Swinstead, E.E.; Paakinaho, V.; Presman, D.M.; Hager, G.L. Pioneer factors and ATP-dependent chromatin remodeling factors interact dynamically: A new perspective: Multiple transcription factors can effect chromatin pioneer functions through dynamic interactions with ATP-dependent chromatin remodeling factors. Bioessays 2016, 38, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Stavreva, D.A.; Garcia, D.A.; Fettweis, G.; Gudla, P.R.; Zaki, G.F.; Soni, V.; McGowan, A.; Williams, G.; Huynh, A.; Palangat, M.; et al. Transcriptional Bursting and Co-bursting Regulation by Steroid Hormone Release Pattern and Transcription Factor Mobility. Mol. Cell 2019, 75, 1161–1177. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Z.; Li, L.; Chen, B.-C.; Revyakin, A.; Hajj, B.; Legant, W.; Dahan, M.; Lionnet, T.; Betzig, E.; et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 2014, 156, 1274–1285. [Google Scholar] [CrossRef]
- Zhen, C.Y.; Tatavosian, R.; Huynh, T.N.; Duc, H.N.; Das, R.; Kokotovic, M.; Grimm, J.B.; Lavis, L.D.; Lee, J.; Mejia, F.J.; et al. Live-cell single-molecule tracking reveals co-recognition of H3K27me3 and DNA targets polycomb Cbx7-PRC1 to chromatin. eLife 2016, 5, e17667. [Google Scholar] [CrossRef]
- Paakinaho, V.; Presman, D.M.; Ball, D.A.; Johnson, T.A.; Schiltz, R.L.; Levitt, P.; Mazza, D.; Morisaki, T.; Karpova, T.S.; Hager, G.L.; et al. Single-molecule analysis of steroid receptor and cofactor action in living cells. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Ball, D.A.; Mehta, G.D.; Salomon-Kent, R.; Mazza, D.; Morisaki, T.; Mueller, F.; McNally, J.G.; Karpova, T.S. Single molecule tracking of Ace1p in Saccharomyces cerevisiae defines a characteristic residence time for non-specific interactions of transcription factors with chromatin. Nucleic Acids Res. 2016, 44, e160. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.D.; Ball, D.A.; Eriksson, P.R.; Chereji, R.V.; Clark, D.J.; McNally, J.G.; Karpova, T.S. Single-Molecule Analysis Reveals Linked Cycles of RSC Chromatin Remodeling and Ace1p Transcription Factor Binding in Yeast. Mol. Cell 2018, 72, 875–887. [Google Scholar] [CrossRef]
- Babour, A.; Shen, Q.; Dos-Santos, J.; Murray, S.; Gay, A.; Challal, D.; Fasken, M.; Palancade, B.; Corbett, A.; Libri, D.; et al. The Chromatin Remodeler ISW1 Is a Quality Control Factor that Surveys Nuclear mRNP Biogenesis. Cell 2016, 167, 1201–1214. [Google Scholar] [CrossRef][Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prajapati, H.K.; Ocampo, J.; Clark, D.J. Interplay among ATP-Dependent Chromatin Remodelers Determines Chromatin Organisation in Yeast. Biology 2020, 9, 190. https://doi.org/10.3390/biology9080190
Prajapati HK, Ocampo J, Clark DJ. Interplay among ATP-Dependent Chromatin Remodelers Determines Chromatin Organisation in Yeast. Biology. 2020; 9(8):190. https://doi.org/10.3390/biology9080190
Chicago/Turabian StylePrajapati, Hemant K., Josefina Ocampo, and David J. Clark. 2020. "Interplay among ATP-Dependent Chromatin Remodelers Determines Chromatin Organisation in Yeast" Biology 9, no. 8: 190. https://doi.org/10.3390/biology9080190
APA StylePrajapati, H. K., Ocampo, J., & Clark, D. J. (2020). Interplay among ATP-Dependent Chromatin Remodelers Determines Chromatin Organisation in Yeast. Biology, 9(8), 190. https://doi.org/10.3390/biology9080190