Organismal Fructose Metabolism in Health and Non-Alcoholic Fatty Liver Disease



{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
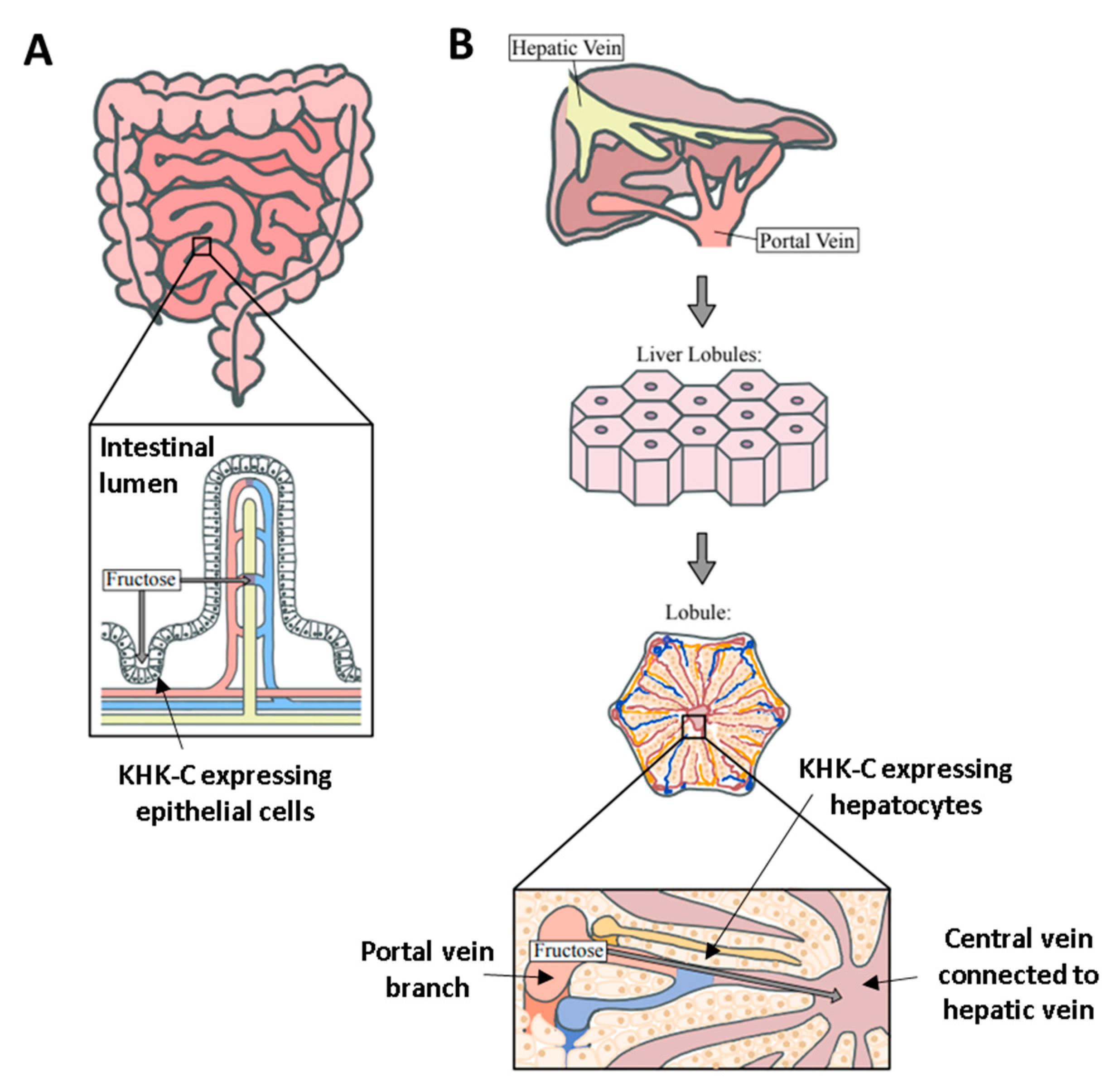
2. Intestinal Fructose Absorption and Metabolism
3. Hepatic Fructose Metabolism and NAFLD
4. Relationship between Intestinal and Hepatic Fructose Metabolism in NAFLD
5. Fructose Metabolism by Other Host Organs
6. Microbial Fructose Metabolism in NAFLD
7. Future Perspectives: Developments in the Prevention of Fructose-Induced NAFLD
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Andres-Hernando, A.; Johnson, R.J.; Lanaspa, M.A. Endogenous fructose production: What do we know and how relevant is it? Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 289–294. [Google Scholar] [CrossRef]
- Patel, C.; Douard, V.; Yu, S.; Tharabenjasin, P.; Gao, N.; Ferraris, R.P. Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R499–R509. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 2018, 27, 351–361.e3. [Google Scholar] [CrossRef] [PubMed]
- Gouyon, F.; Caillaud, L.; Carriere, V.; Klein, C.; Dalet, V.; Citadelle, D.; Kellett, G.L.; Thorens, B.; Leturque, A.; Brot-Laroche, E. Simple-sugar meals target GLUT2 at enterocyte apical membranes to improve sugar absorption: A study in GLUT2-null mice. J. Physiol. 2003, 552, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, C.I. GLUT2 is the transporter for fructose across the rat intestinal basolateral membrane. Gastroenterology 1993, 105, 1050–1056. [Google Scholar] [CrossRef]
- Wasserman, D.; Hoekstra, J.H.; Tolia, V.; Tayor, C.J.; Kirschner, B.S.; Takeda, J.; Bell, G.I.; Taub, R.; Rand, E.B. Molecular analysis of the fructose transporter gene (GLUT5) in isolated fructose malabsorption. J. Clin. Investig. 1996, 98, 2398–2402. [Google Scholar] [CrossRef]
- Sullivan, J.S.; Le, M.T.; Pan, Z.; Rivard, C.; Love-Osborne, K.; Robbins, K.; Johnson, R.J.; Sokol, R.J.; Sundaram, S.S. Oral fructose absorption in obese children with non-alcoholic fatty liver disease. Pediatr. Obes. 2005, 10, 188–195. [Google Scholar] [CrossRef]
- Fukuzawa, T.; Fukazawa, M.; Ueda, O.; Shimada, H.; Kito, A.; Kakefuda, M.; Kawase, Y.; Wada, N.A.; Goto, C.; Fukushima, N.; et al. SGLT5 reabsorbs fructose in the kidney but its deficiency paradoxically exacerbates hepatic steatosis induced by fructose. PLoS ONE 2013, 8, e56681. [Google Scholar] [CrossRef]
- Todoric, J.; di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Heinz, F.; Lamprecht, W.; Kirsch, J. Enzymes of fructose metabolism in human liver. J. Clin. Investig. 1968, 47, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and localization of the principal fructose-metabolizing enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Wada, S.; Yang, S.; Gosis, B.; Zeng, X.; Zhang, Z.; Shen, Y.; Lee, G.; Arany, Z.; Rabinowitz, J.D. The small intestine shields the liver from fructose-induced steatosis. Nat. Metab. 2020, 2, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Andres-Hernando, A.; Orlicky, D.J.; Kuwabara, M.; Ishimoto, T.; Nakagawa, T.; Johnson, R.J.; Lanaspa, M.A. Deletion of fructokinase in the liver or in the intestine reveals differential effects on sugar-induced metabolic dysfunction. Cell Metab. 2020, 32, 117–127.e3. [Google Scholar] [CrossRef] [PubMed]
- Mavrias, D.A.; Mayer, R.J. Metabolism of fructose in the small intestine. The effect of fructose feeding on fructose transport and metabolism in rat small intestine. Biochim. Biophys. Acta 1973, 291, 531–537. [Google Scholar] [CrossRef]
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; Maclean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325. [Google Scholar] [CrossRef]
- Kim, J.; Kang, J.; Kang, Y.L.; Woo, J.; Kim, Y.; Huh, J.; Park, J.W. Ketohexokinase-A acts as a nuclear protein kinase that mediates fructose-induced metastasis in breast cancer. Nat. Commun. 2020, 11, 5436. [Google Scholar] [CrossRef]
- Kim, M.; Astapova, I.I.; Flier, S.N.; Hannou, S.A.; Doridot, L.; Sargsyan, A.; Kou, H.H.; Fowler, A.J.; Liang, G.; Herman, M.A. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight 2017, 2, e96703. [Google Scholar] [CrossRef]
- Blanco, A.; Blanco, G. Carbohydrate metabolism. In Medical Biochemistry; Academic Press: London, UK; San Diego, CA, USA, 2017; pp. 283–323. [Google Scholar]
- Liu, L.; Li, T.; Liao, Y.; Wang, Y.; Gao, Y.; Hu, H.; Huang, H.; Wu, F.; Chen, Y.G.; Xu, S.; et al. Triose kinase controls the lipogenic potential of fructose and dietary tolerance. Cell Metab. 2020, 32, 605–618. [Google Scholar] [CrossRef]
- Opelt, S.A.; Sennott, E.M.; Tolan, D.R. Aldolase-B knockout in mice phenocopies hereditary fructose intolerance in humans. Mol. Genet. Metab. 2015, 114, 445–450. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Andres-Hernando, A.; Orlicky, D.J.; Cicerchi, C.; Jang, C.; Li, N.; Milagres, T.; Kuwabara, M.; Wempe, M.F.; Rabinowitz, J.D.; et al. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J. Clin. Investig. 2018, 128, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- van Norman, G.A. Limitations of animal studies for predicting toxicity in clinical trials: Is it time to rethink our current approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Te Morenga, L.; Mallard, S.; Mann, J. Dietary sugars and body weight: Systematic review and meta-analyses of randomised controlled trials and cohort studies. BMJ 2012, 346, e7492. [Google Scholar] [CrossRef]
- Chung, M.; Ma, J.; Patel, K.; Berger, S.; Lau, J.; Lichtenstein, A.H. Fructose, high-fructose corn syrup, sucrose, and nonalcoholic fatty liver disease or indexes of liver health: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2014, 100, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.; Sievenpiper, J.L.; de Souza, R.J.; Cozma, A.I.; Mirrahimi, A.; Carleton, A.J.; Ha, V.; di Buono, M.; Jenkins, A.L.; Leiter, L.A.; et al. Effect of fructose on markers of non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of controlled feeding trials. Eur. J. Clin. Nutr. 2014, 68, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Tsilas, C.S.; Tsilas, C.S.; de Souza, R.J.; Mejia, S.B.; Mirrahimi, A.; Cozma, A.I.; Jayalath, V.H.; Ha, V.; Tawfik, R.; di Buono, M.; et al. Relation of total sugars, fructose and sucrose with incident type 2 diabetes: A systematic review and meta-analysis of prospective cohort studies. CMAJ 2017, 189, E711–E720. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.H.S.; Wong, S.H. Microbiota, obesity and NAFLD. In Obesity, Fatty Liver and Liver Cancer; Advances in Experimental Medicine and Biology; Yu, J., Ed.; Springer: Singapore, 2018; pp. 111–125. [Google Scholar]
- Zhang, Y.H.; An, T.; Zhang, R.C.; Zhou, Q.; Huang, Y.; Zhang, J. Very high fructose intake increases serum LDL-cholesterol and total cholesterol: A meta-analysis of controlled feeding trials. J. Nutr. 2013, 143, 1391–1398. [Google Scholar] [CrossRef]
- Sievenpiper, J.L.; de Souza, R.J.; Mirrahimi, A.; Yu, M.E.; Carleton, A.J.; Beyene, J.; Chiavaroli, L.; di Buono, M.; Jenkins, A.L.; Leiter, L.A.; et al. Effect of fructose on body weight in controlled feeding trials: A systematic review and meta-analysis. Ann. Intern. Med. 2012, 156, 291–304. [Google Scholar] [CrossRef]
- Tappy, L. Fructose-containing caloric sweeteners as a cause of obesity and metabolic disorders. J. Exp. Biol. 2018, 221 (Suppl. 1), jeb164202. [Google Scholar] [CrossRef]
- Tajima, R.; Kimura, T.; Enomoto, A.; Saito, A.; Kobayashi, S.; Masuda, K.; Iida, K. No association between fruits or vegetables and non-alcoholic fatty liver disease in middle-aged men and women. Nutrition 2019, 61, 119–124. [Google Scholar] [CrossRef]
- Mai, B.H.; Yan, L.J. The negative and detrimental effects of high fructose on the liver, with special reference to metabolic disorders. Diabetes Metab. Syndr. Obes. 2019, 12, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.D.; Stephenson, M.C.; Crossland, H.; Cordon, S.M.; Palcidi, E.; Cox, E.F.; Taylor, M.A.; Aithal, G.P.; Macdonald, I.A. No difference between high-fructose and high-glucose diets on liver triacylglycerol or biochemistry in healthy overweight men. Gastroenterology 2013, 145, 1016–1025.e2. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab. 2019, 30, 735–753.e4. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Lozada, L.G.; Mu, W.; Roncal, C.; Sautin, Y.Y.; Abdelmalek, M.; Reungjui, S.; Le, M.; Nakagawa, T.; Lan, H.Y.; Yu, X.; et al. Comparison of free fructose and glucose to sucrose in the ability to cause fatty liver. Eur. J. Nutr. 2010, 49, 1–9. [Google Scholar] [CrossRef]
- Softic, S.; Gupta, M.K.; Wang, G.X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling [published correction appears in J Clin Invest. 2018 Mar 1;128(3):1199]. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef]
- Patel, C.; Douard, V.; Yu, S.; Gao, N.; Ferraris, R.P. Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. FASEB J. 2015, 29, 4046–4058. [Google Scholar] [CrossRef]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef]
- Merino, B.; Fernández-Díaz, C.M.; Cózar-Castellano, I.; Perdomo, G. Intestinal fructose and glucose metabolism in health and disease. Nutrients 2019, 12, 94. [Google Scholar] [CrossRef]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef]
- Mayes, P.A. Intermediary metabolism of fructose. Am. J. Clin. Nutr. 1993, 58 (Suppl. 5), 754S–765S. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Lanaspa, M.A.; Johnson, R.J. The effects of fruit consumption in patients with hyperuricaemia or gout. Rheumatology 2019, 58, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Dalbeth, N.; Yin, H.; Li, C.; Merriman, T.R.; Wei, W.H. Mouse models for human hyperuricaemia: A critical review. Nat. Rev. Rheumatol. 2019, 15, 413–426. [Google Scholar] [CrossRef]
- Lu, J.; Hou, X.; Yuan, X.; Cui, L.; Liu, Z.; Li, X.; Ma, L.; Cheng, X.; Xin, Y.; Wang, C.; et al. Knockout of the urate oxidase gene provides a stable mouse model of hyperuricemia associated with metabolic disorders. Kidney Int. 2018, 93, 69–80. [Google Scholar] [CrossRef]
- Malhotra, P.; Gill, R.K.; Saksena, S.; Alrefai, W.A. Disturbances in cholesterol homeostasis and non-alcoholic fatty liver diseases. Front. Med. 2020, 7, 467. [Google Scholar] [CrossRef]
- Silbernagel, G.; Lütjohann, D.; Machann, J.; Meichsner, S.; Kantartzis, K.; Schick, F.; Häring, H.U.; Stefan, N.; Fritsche, A. Cholesterol synthesis is associated with hepatic lipid content and dependent on fructose/glucose intake in healthy humans. Exp. Diabetes Res. 2012, 361863. [Google Scholar] [CrossRef]
- Feingold, K.R.; Moser, A.H. Effect of glucose or fructose feeding on cholesterol synthesis in diabetic animals. Am. J. Physiol. 1985, 249, G634–G641. [Google Scholar] [CrossRef]
- Jegatheesan, P.; de Bandt, J.P. Fructose and NAFLD: The multifaceted aspects of fructose metabolism. Nutrients 2017, 9, 230. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M.; Nonalcoholic steatohepatitis clinical research network. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef]
- Koopman, K.E.; Caan, M.W.; Nederveen, A.J.; Pels, A.; Ackermans, M.T.; Fliers, E.; la Fleur, S.E.; Serlie, M.J. Hypercaloric diets with increased meal frequency, but not meal size, increase intrahepatic triglycerides: A randomized controlled trial. Hepatology 2014, 60, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Haidari, M.; Leung, N.; Mahbub, F.; Uffelman, K.D.; Kohen-Avramoglu, R.; Lewis, G.F.; Adeli, K. Fasting and postprandial overproduction of intestinally derived lipoproteins in an animal model of insulin resistance. J. Biol. Chem. 2002, 277, 31646–31655. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, R.P. Dietary and developmental regulation of intestinal sugar transport. Biochem. J. 2001, 360, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M. Microscopic anatomy of the liver. Clin. Liver Dis. 2013, 2 (Suppl. 1), S4–S7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.M.; Jiao, R.Q.; Kong, L.D. High dietary fructose: Direct or indirect dangerous factors disturbing tissue and organ functions. Nutrients 2017, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Mirtschink, P.; Krishnan, J.; Grimm, F.; Sarre, A.; Hörl, M.; Kayikci, M.; Fankhauser, N.; Christinat, Y.; Cortijo, C.; Feehan, O.; et al. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature 2015, 522, 444–449. [Google Scholar] [CrossRef]
- Gersch, M.; Cirillo, P.; Reungjui, S.; Zhang, L.; Roncal, C.; Sautin, Y.; Johnson, R.; Nakagawa, T. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am. J. Physiol. Renal. 2007, 293, F1256–F1261. [Google Scholar] [CrossRef]
- Bartley, C.; Brun, T.; Oberhauser, L.; Grimaldi, M.; Molica, F.; Kwak, B.R.; Bosco, D.; Chanson, M.; Maechler, P. Chronic fructose renders pancreatic β-cells hyper-responsive to glucose-stimulated insulin secretion through extracellular ATP signaling. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E25–E41. [Google Scholar] [CrossRef]
- Froesch, E.R. Fructose metabolism in adipose tissue. Acta Med. Scand. Suppl. 1972, 542, 37–46. [Google Scholar] [CrossRef]
- Zierath, J.R.; Nolte, L.A.; Wahlström, E.; Galuska, D.; Shepherd, P.R.; Kahn, B.B.; Wallberg-Henriksson, H. Carrier-mediated fructose uptake significantly contributes to carbohydrate metabolism in human skeletal muscle. Biochem. J. 1995, 311, 517–521. [Google Scholar] [CrossRef]
- Roncal-Jimenez, C.A.; Ishimoto, T.; Lanaspa, M.A.; Milagres, T.; Hernando, A.A.; Jensen, T.; Miyazaki, M.; Doke, T.; Hayasaki, T.; Nakagawa, T.; et al. Aging-associated renal disease in mice is fructokinase dependent. Am. J. Physiol. Renal. Physiol. 2016, 311, F722–F730. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Romero, R. Mechanistically different effects of fat and sugar on insulin resistance, hypertension, and gut microbiota in rats. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E552–E563. [Google Scholar] [CrossRef] [PubMed]
- Astbury, S.; Song, A.; Zhou, M.; Nielsen, B.; Hoedl, A.; Willing, B.P.; Symonds, M.E.; Bell, R.C. High fructose intake during pregnancy in rats influences the maternal microbiome and gut development in the offspring. Front. Genet. 2018, 9, 203. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875. [Google Scholar] [CrossRef]
- Beyer, P.L.; Caviar, E.M.; McCallum, R.W. Fructose intake at current levels in the United States may cause gastrointestinal distress in normal adults. J. Am. Diet. Assoc. 2005, 105, 1559–1566. [Google Scholar] [CrossRef]
- Rumessen, J.J.; Gudmand-Høyer, E. Absorption capacity of fructose in healthy adults. Comparison with sucrose and its constituent monosaccharides. Gut 1986, 27, 1161–1168. [Google Scholar] [CrossRef]
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef]
- Ravich, W.J.; Bayless, T.M.; Thomas, M. Fructose: Incomplete intestinal absorption in humans. Gastroenterology 1983, 84, 26–29. [Google Scholar] [CrossRef]
- Ureta, T.; Medina, C.; Preller, A. The evolution of hexokinases. Arch. Biol. Med. Exp. 1987, 20, 343–357. [Google Scholar]
- Rakoff-Nahoum, S.; Foster, K.; Comstock, L. The evolution of cooperation within the gut microbiota. Nature 2016, 533, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Ahn, I.S.; Lang, J.M.; Olson, C.A.; Diamante, G.; Zhang, G.; Ying, Z.; Byun, H.R.; Cely, I.; Ding, J.; Cohn, P. Host genetic background and gut microbiota contribute to differential metabolic responses to fructose consumption in mice. J. Nutr. 2020, 150, 2716–2728. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Alderete, T.L.; Kim, J.S.; Millstein, J.; Gilliland, F.D.; Goran, M.I. High intake of dietary fructose in overweight/obese teenagers associated with depletion of Eubacterium and Streptococcus in the gut microbiome. Gut Microbes 2019, 10, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef]
- Whitehead, R.H.; Young, G.P.; Bhathal, P.S. Effects of short chain fatty acids on a new human colon carcinoma cell line (LIM1215). Gut 1986, 27, 1457–1463. [Google Scholar] [CrossRef]
- D’Argenio, G.; Mazzacca, G. Short-chain fatty acid in the human colon. Relation to inflammatory bowel diseases and colon cancer. Adv. Exp. Med. Biol. 1999, 472, 149–158. [Google Scholar] [CrossRef]
- Hong, Y.; Nishimura, Y.; Hishikawa, D.; Tsuzuki, H.; Miyahara, H.; Gotoh, C.; Choi, K.; Feng, D.; Chen, C.; Lee, H.; et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 2005, 146, 5092–5099. [Google Scholar] [CrossRef]
- Fellows, R.; Denizot, J.; Stellato, C.; Cuomo, A.; Jain, P.; Stoyanova, E.; Balázsi, S.; Hajnády, Z.; Liebert, A.; Kazakevych, J. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat. Commun. 2018, 9, 105. [Google Scholar] [CrossRef]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef]
- Wang, J.; Ma, J.; Nie, H.; Zhang, X.J.; Zhang, P.; She, Z.G.; Li, H.; Ji, Y.X.; Cai, J. Hepatic regulator of G protein Signaling 5 ameliorates NAFLD by suppressing TAK1-JNK/p38 Signaling. Hepatology 2020. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, Y.J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Müller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M.; et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef] [PubMed]
- di Luccia, B.; Crescenzo, R.; Mazzoli, A.; Cigliano, L.; Venditti, P.; Walser, J.C.; Widmer, A.; Baccigalupi, L.; Ricca, E.; Iossa, S. Rescue of fructose-induced metabolic syndrome by antibiotics or faecal transplantation in a rat model of obesity. PLoS ONE 2015, 10, e0134893. [Google Scholar] [CrossRef] [PubMed]
- Brandt, A.; Jin, C.J.; Nolte, K.; Sellmann, C.; Engstler, A.J.; Bergheim, I. Short-term intake of a fructose-, fat- and cholesterol-rich diet causes hepatic steatosis in mice: Effect of antibiotic treatment. Nutrients 2017, 9, 1013. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H. Increased intestinal permeability and decreased barrier function: Does it really influence the risk of inflammation? Inflamm. Intest. Dis. 2016, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Mirmonsef, P.; Zariffard, M.R.; Gilbert, D.; Makinde, H.; Landay, A.L.; Spear, G.T. Short-chain fatty acids induce pro-inflammatory cytokine production alone and in combination with toll-like receptor ligands. Am. J. Reprod. Immunol. 2012, 67, 391–400. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Ferreira, D.F.; Fiamoncini, J.; Prist, I.H.; Ariga, S.K.; de Souza, H.P.; de Lima, T.M. Novel role of TLR4 in NAFLD development: Modulation of metabolic enzymes expression. BBA Mol. Cell Biol. Lipids 2015, 1851, 1353–1359. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Liu, J.; Zhuang, Z.J.; Bian, D.X.; Ma, X.J.; Xun, Y.H.; Yang, W.J.; Luo, Y.; Liu, Y.L.; Jia, L.; Wang, Y.; et al. Toll-like receptor-4 signalling in the progression of non-alcoholic fatty liver disease induced by high-fat and high-fructose diet in mice. Clin. Exp. Pharmacol.Physiol. 2014, 41, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Asipu, A.; Hayward, B.E.; O’Reilly, J.; Bonthron, D.T. Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenesis of essential fructosuria. Diabetes 2003, 52, 2426–2432. [Google Scholar] [CrossRef] [PubMed]
- Maryanoff, B.E.; O’Neill, J.C.; McComsey, D.F.; Yabut, S.C.; Luci, D.K.; Jordan, A.D.; Masucci, J.A.; Jones, W.J.; Abad, M.C.; Gibbs, A.C.; et al. Inhibitors of ketohexokinase: Discovery of pyrimidinopyrimidines with specific substitution that complements the ATP-Binding Site. ACS Med. Chem. Lett. 2011, 2, 538–543. [Google Scholar] [CrossRef]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, F.; Kuo, G.-H.; Xiang, A.; Gibbs, A.C.; Abad, M.C.; Sun, W.; Kuo, L.C.; Sui, Z. Optimization of a pyrazole hit from FBDD into a novel series of indazoles as ketohexokinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 4762–4767. [Google Scholar] [CrossRef]
- Huard, K.; Ahn, K.; Amor, P.; Beebe, D.A.; Borzilleri, K.A.; Chrunyk, B.A.; Coffey, S.B.; Cong, Y.; Conn, E.L.; Culp, J.S.; et al. Discovery of fragment-derived small molecules for in vivo inhibition of Ketohexokinase (KHK). J. Med. Chem. 2017, 60, 7835–7849. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Aarsland, A.; Chinkes, D.; Wolfe, R.R. Hepatic and whole-body fat synthesis in humans during carbohydrate overfeeding. Am. J. Clin. Nutr. 1997, 65, 1774–1782. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: A bedside to bench investigation [published correction appears in Cell Metab. 2017 Sep 5;26(3):576]. Cell Metab. 2017, 26, 394–406.e6. [Google Scholar] [CrossRef] [PubMed]
- Ritze, Y.; Bárdos, G.; D’Haese, J.G.; Ernst, B.; Thurnheer, M.; Schultes, B.; Bischoff, S.C. Effect of high sugar intake on glucose transporter and weight regulating hormones in mice and humans. PLoS ONE 2014, 9, e101702. [Google Scholar] [CrossRef]
- Janssens, J.P.; Shapira, N.; Debeuf, P.; Michiels, L.; Putman, R.; Bruckers, L.; Renard, D.; Molenberghs, G. Effects of soft drink and table beer consumption on insulin response in normal teenagers and carbohydrate drink in youngsters. Eur. J. Cancer Prev. 1999, 8, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Bergkvist, L.; Wolk, A. Consumption of sugar and sugar-sweetened foods and the risk of pancreatic cancer in a prospective study. Am. J. Clin. Nutr. 2006, 84, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Ferrocino, I.; Liberto, E.; Chiazza, F.; Cento, A.S.; Collotta, D.; Querio, G.; Nigro, D.; Bitonto, V.; Cutrin, J.C.; et al. Fructose liquid and solid formulations differently affect gut integrity, microbiota composition and related liver toxicity: A comparative in vivo study. J. Nutr. Chem. 2018, 55, 185–199. [Google Scholar] [CrossRef]
- Olsen, N.J.; Andersen, L.B.; Wedderkopp, N.; Kristensen, P.L.; Heitmann, B.L. Intake of liquid and solid sucrose in relation to changes in body fatness over 6 years among 8- to 10-year-old children: The European youth heart study. Obes. Facts 2012, 5, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Harbottle, B.; Hernandez, G.; Smith, V.; Coffin, M.; Noland, T.; Dillard, K.; Glanz, H.; Fanter, R.; Burrin, D.; et al. Comparison of high fructose corn syrup versus sucrose consumption on non-alcoholic fatty liver disease in juvenile iberian pigs. Curr. Dev. Nutr. 2020, 4, 691. [Google Scholar] [CrossRef]
- Yu, Z.; Lowndes, J.; Rippe, J. High-fructose corn syrup and sucrose have equivalent effects on energy-regulating hormones at normal human consumption levels. Nutr. Res. 2013, 33, 1043–1052. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skenderian, S.; Park, G.; Jang, C. Organismal Fructose Metabolism in Health and Non-Alcoholic Fatty Liver Disease. Biology 2020, 9, 405. https://doi.org/10.3390/biology9110405
Skenderian S, Park G, Jang C. Organismal Fructose Metabolism in Health and Non-Alcoholic Fatty Liver Disease. Biology. 2020; 9(11):405. https://doi.org/10.3390/biology9110405
Chicago/Turabian StyleSkenderian, Shea, Grace Park, and Cholsoon Jang. 2020. "Organismal Fructose Metabolism in Health and Non-Alcoholic Fatty Liver Disease" Biology 9, no. 11: 405. https://doi.org/10.3390/biology9110405
APA StyleSkenderian, S., Park, G., & Jang, C. (2020). Organismal Fructose Metabolism in Health and Non-Alcoholic Fatty Liver Disease. Biology, 9(11), 405. https://doi.org/10.3390/biology9110405