ApoA-I-Mediated Lipoprotein Remodeling Monitored with a Fluorescent Phospholipid



Abstract
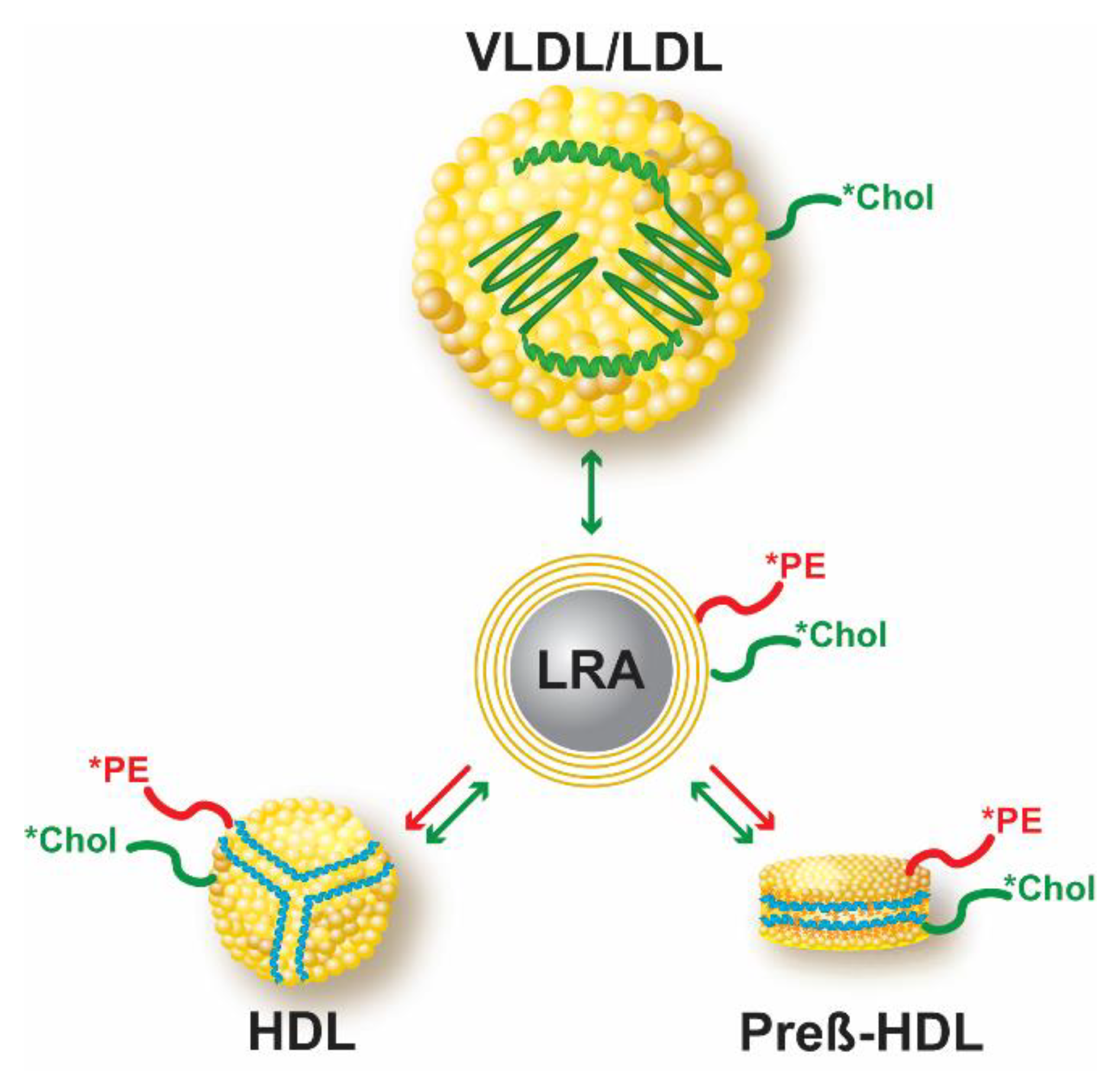
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.3. Fluorescent Lipid Exchange between Lipoproteins
2.4. Synthetic Lipoprotein X Preparation
2.5. Lipoprotein X Metabolism In Vitro
2.6. rHDL Preparation
2.7. Lipid-Coated LRA Preparation
2.8. In Vitro Studies of Fluorescent Lipid Transfer from Lipid-Coated LRA Particles to Lipoproteins
2.9. Fluorescent Lipid Agarose Gel Electrophoresis
2.10. FPLC Analysis
2.11. Total PE and Cholesterol Fluorescence Measurements
2.12. In-Gel Proteomic Analysis
3. Results
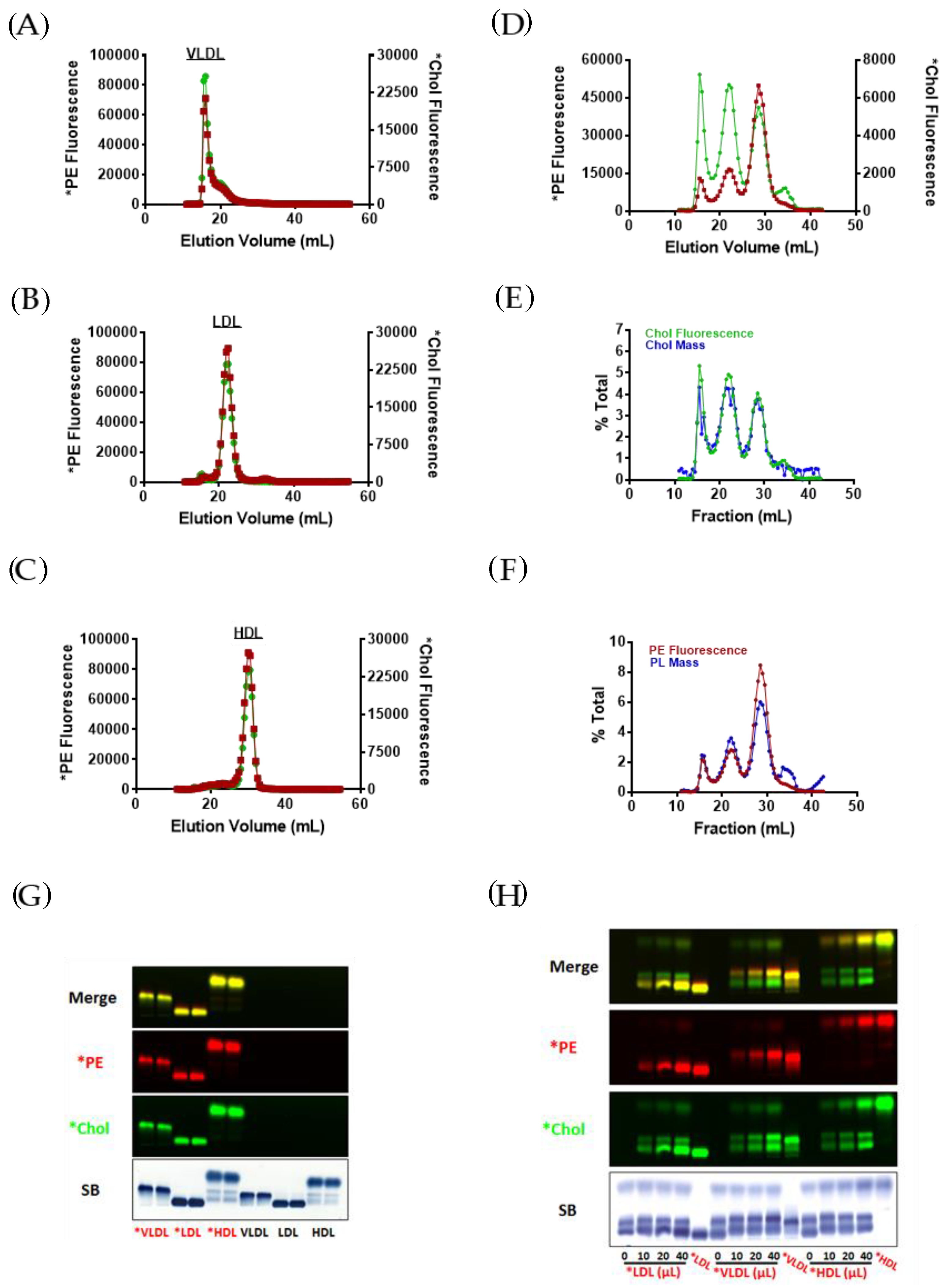
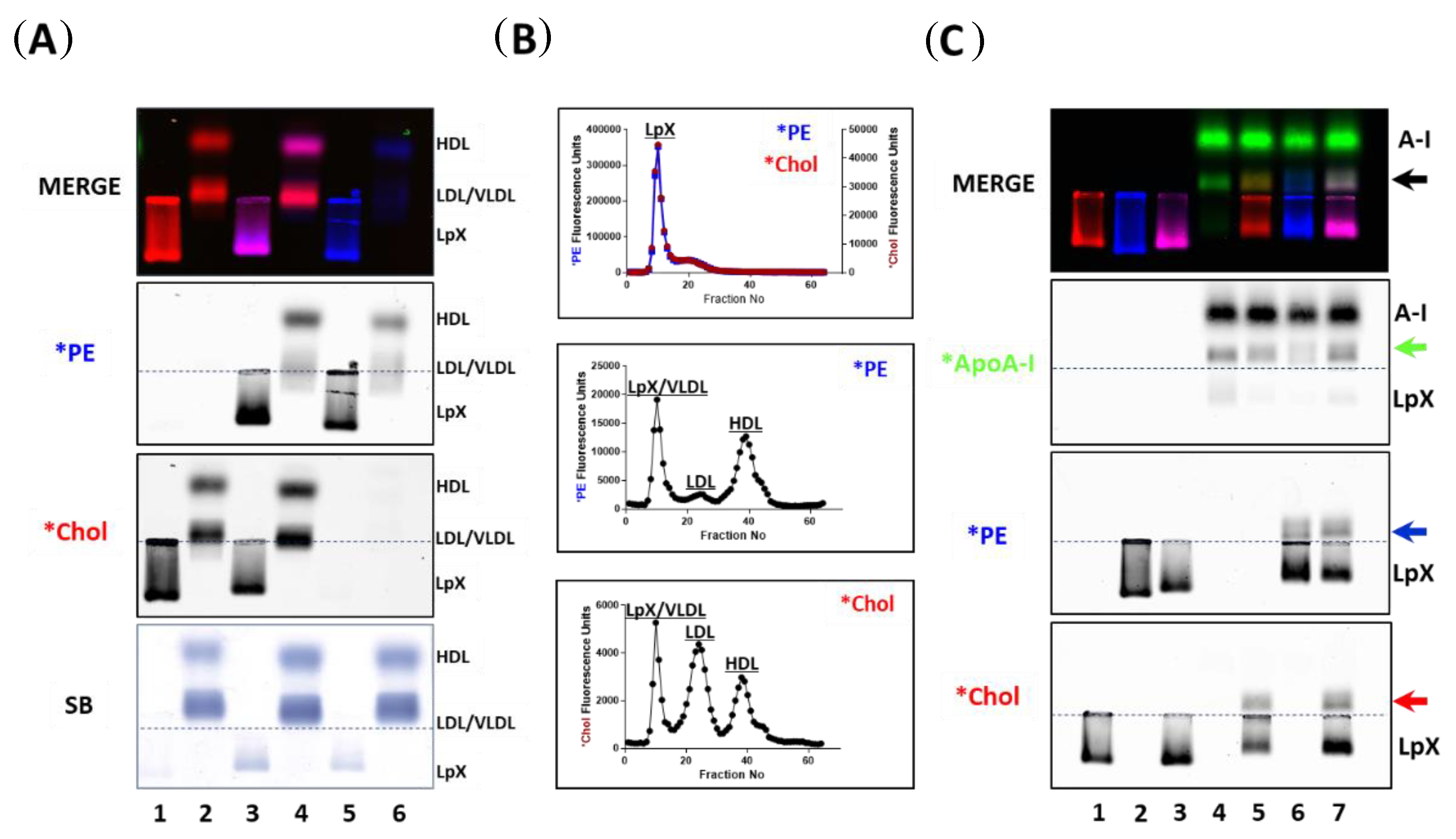
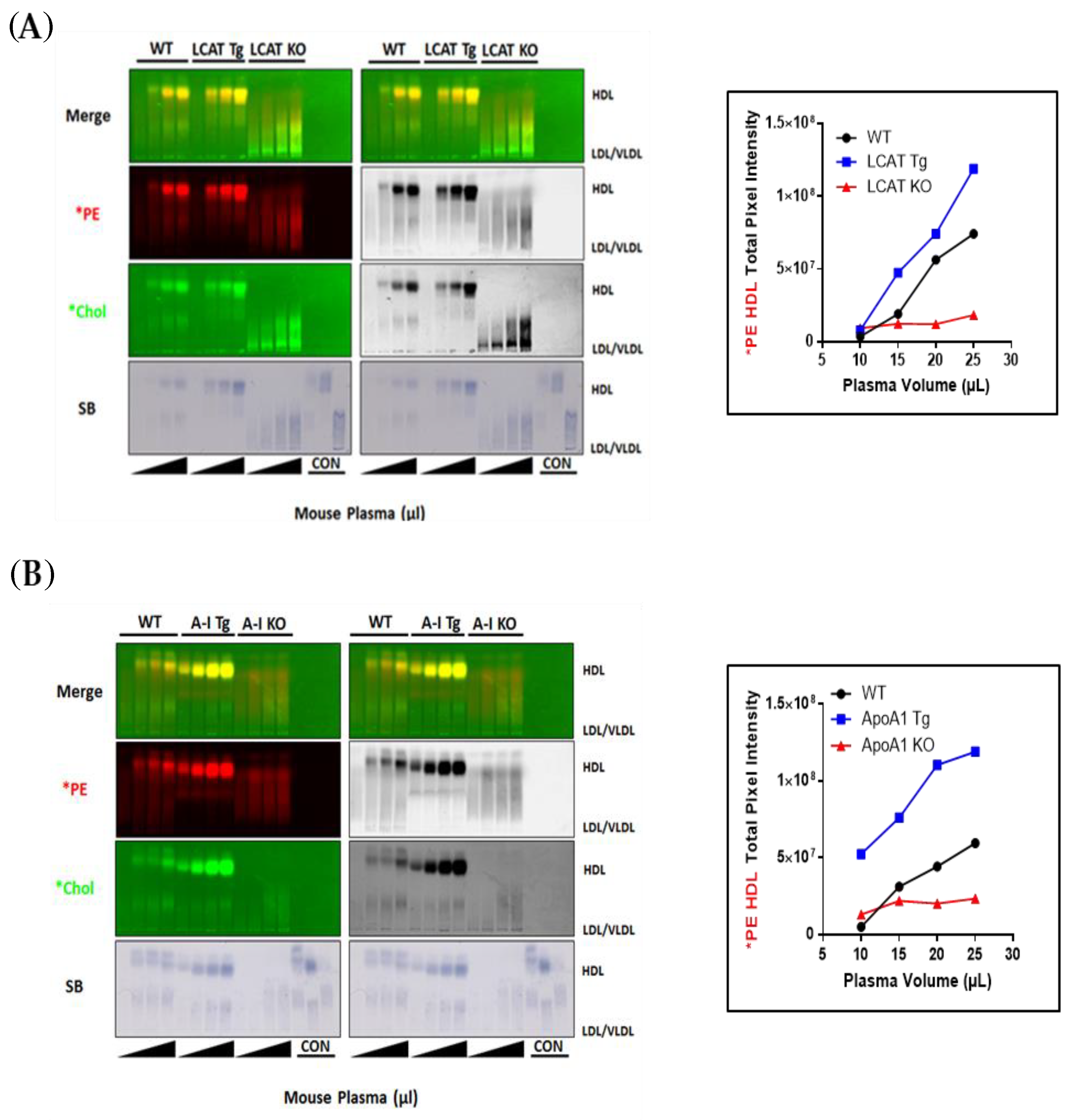
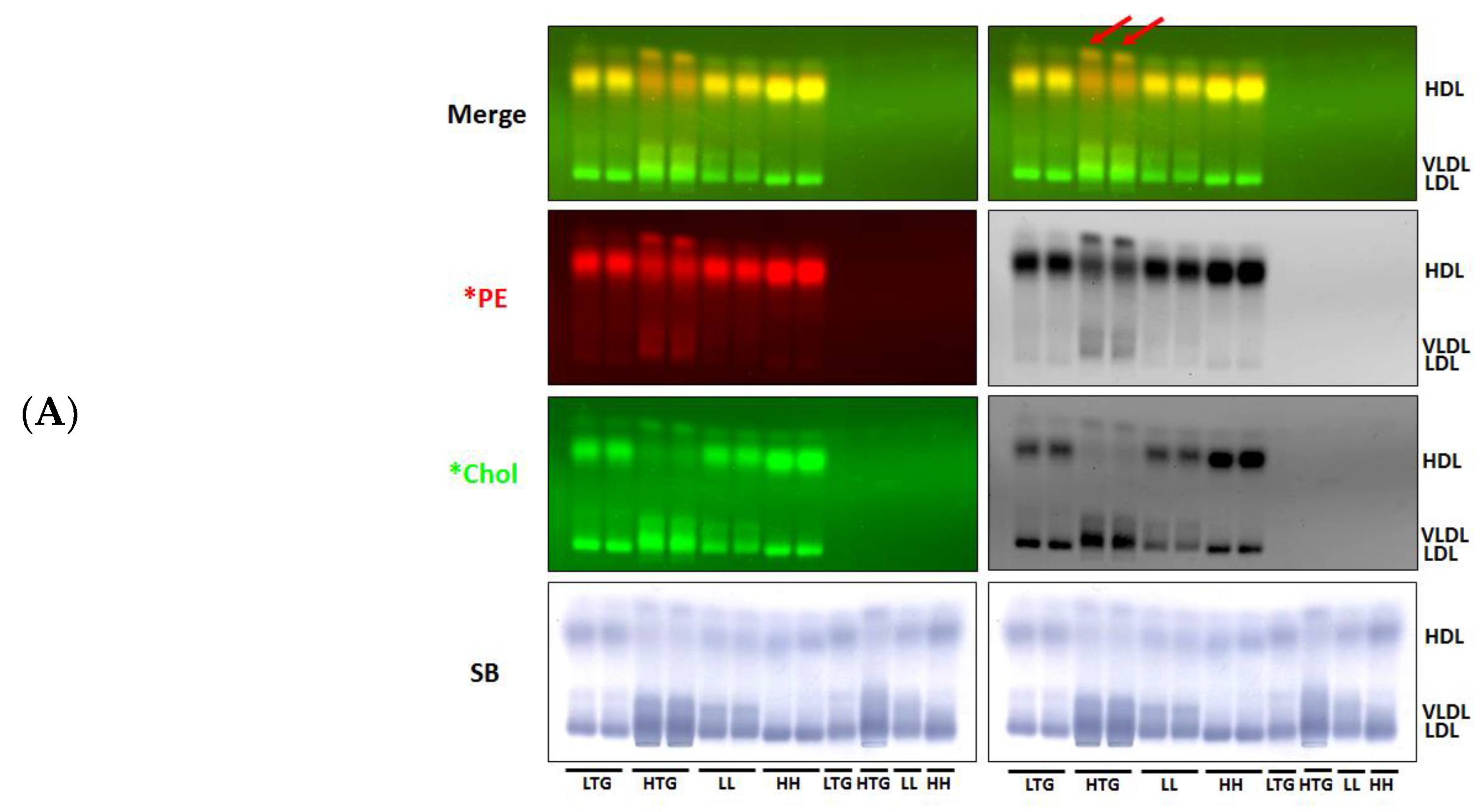
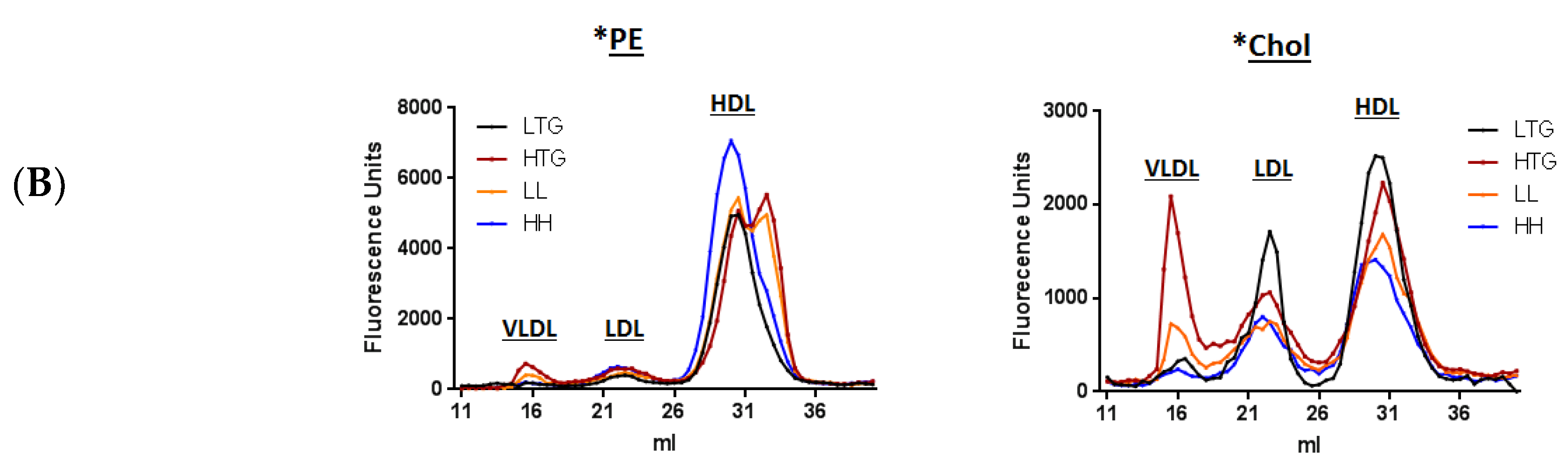
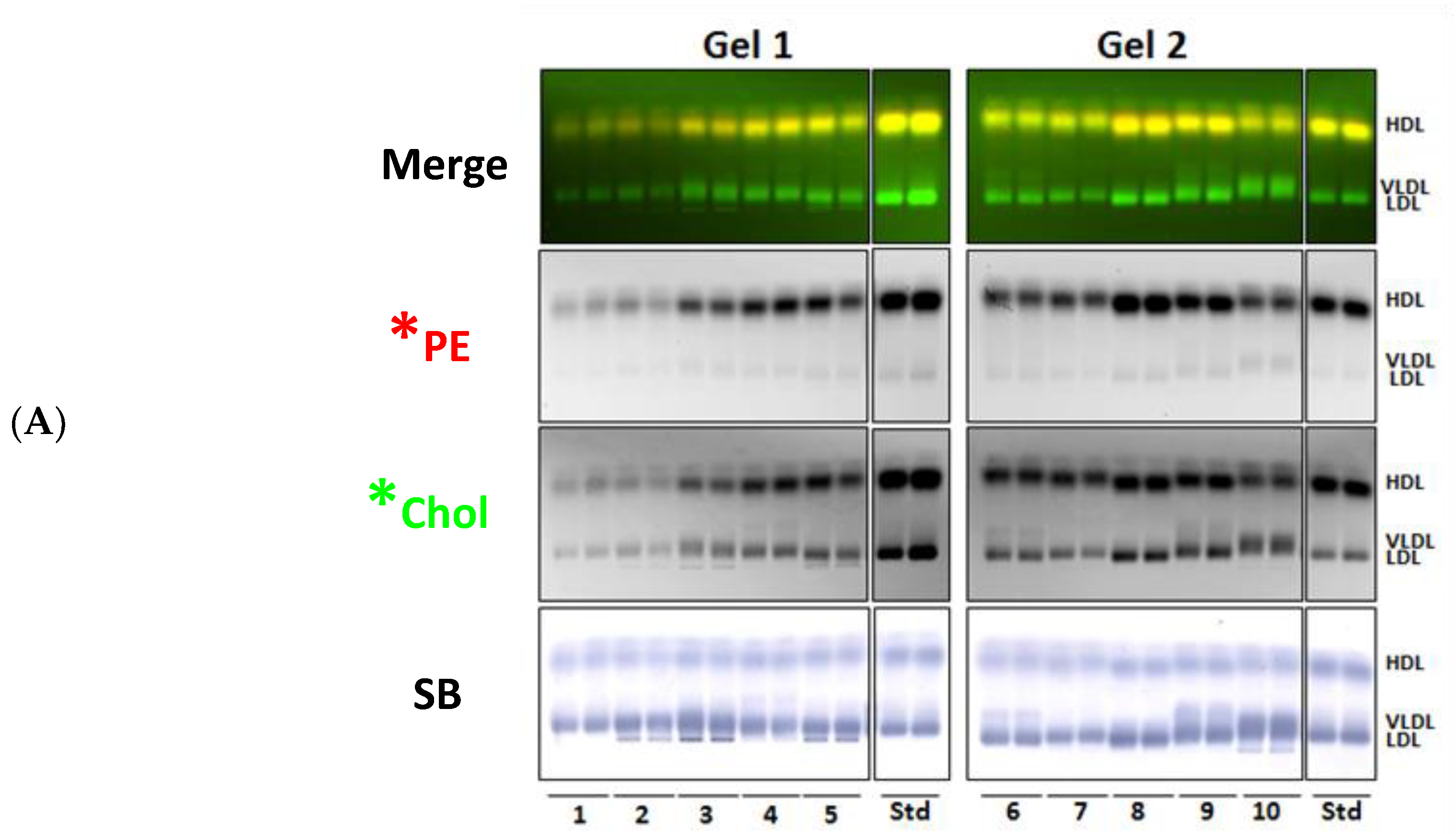
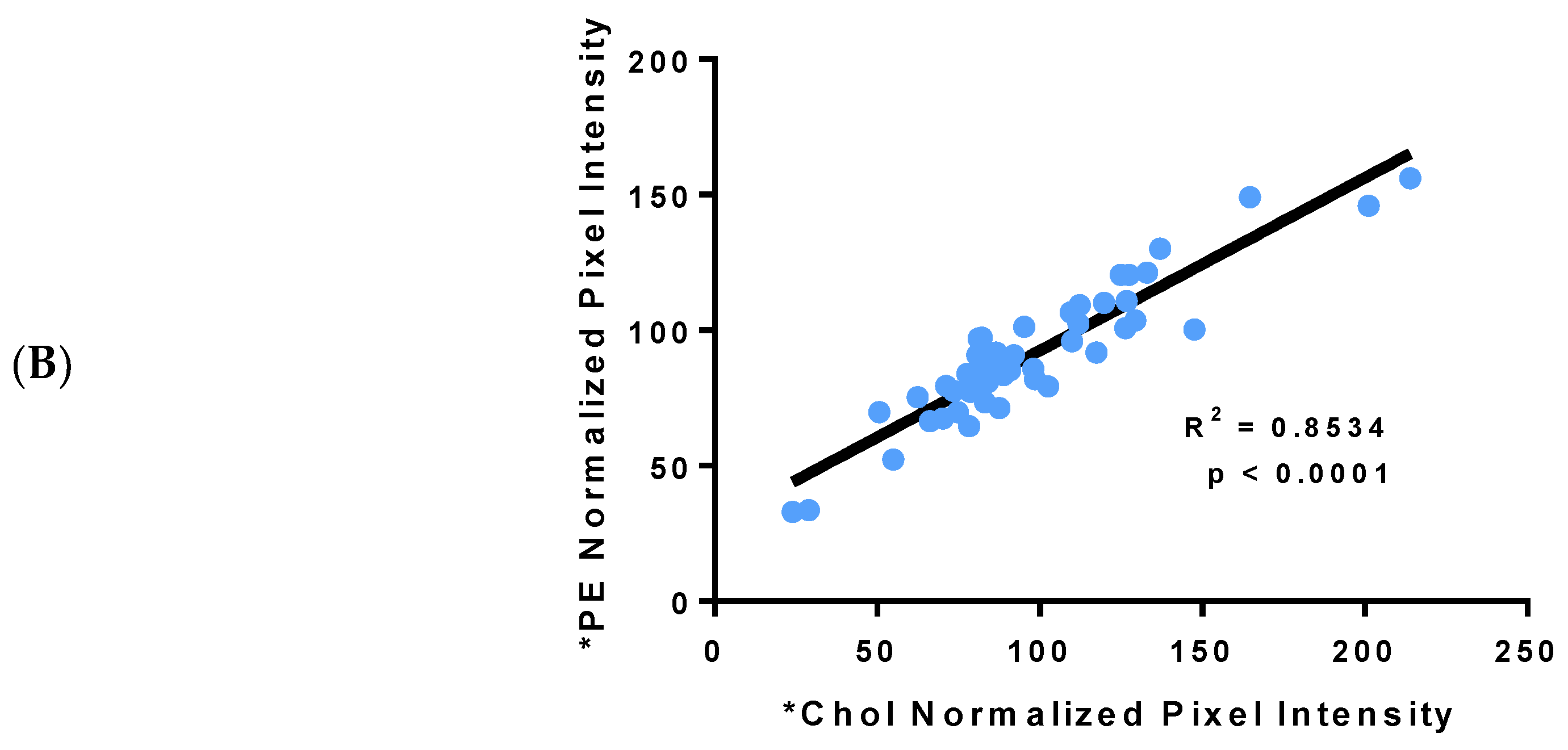
3.1. Incorporation of Fluorescent PE and Cholesterol into Lipoproteins
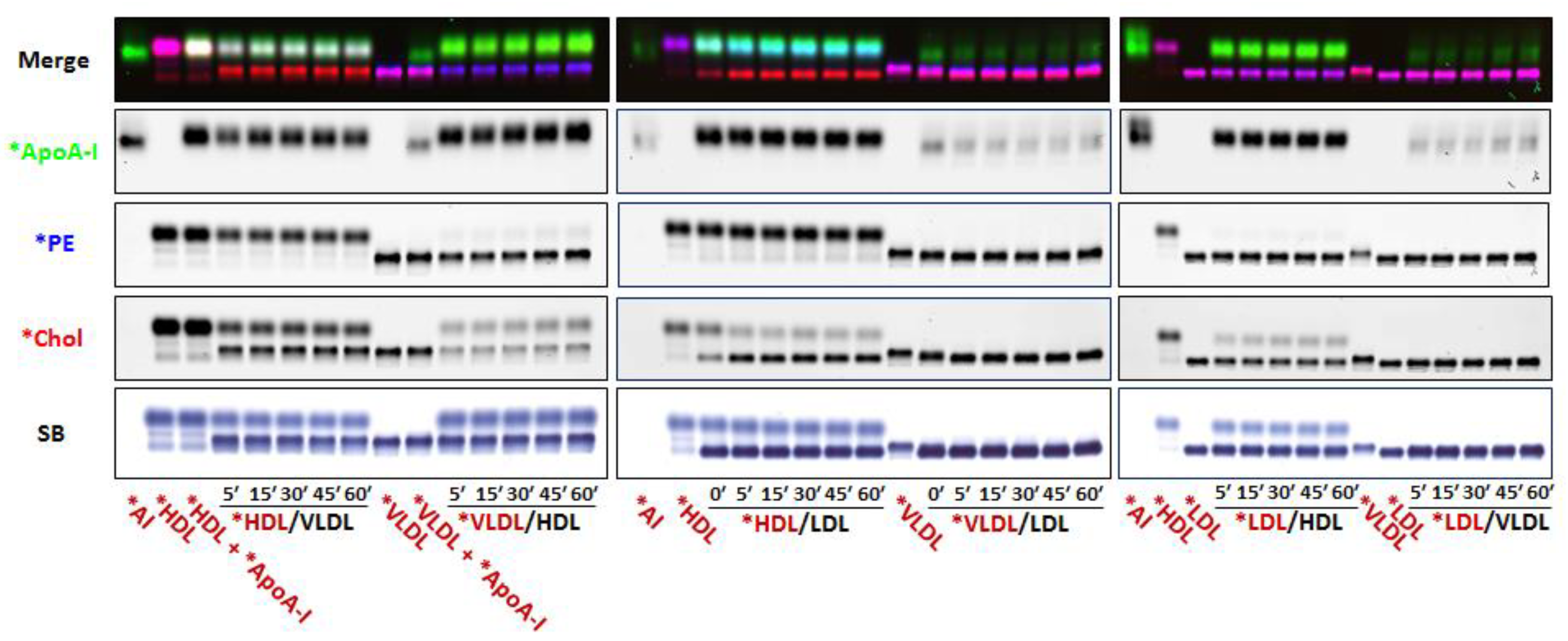
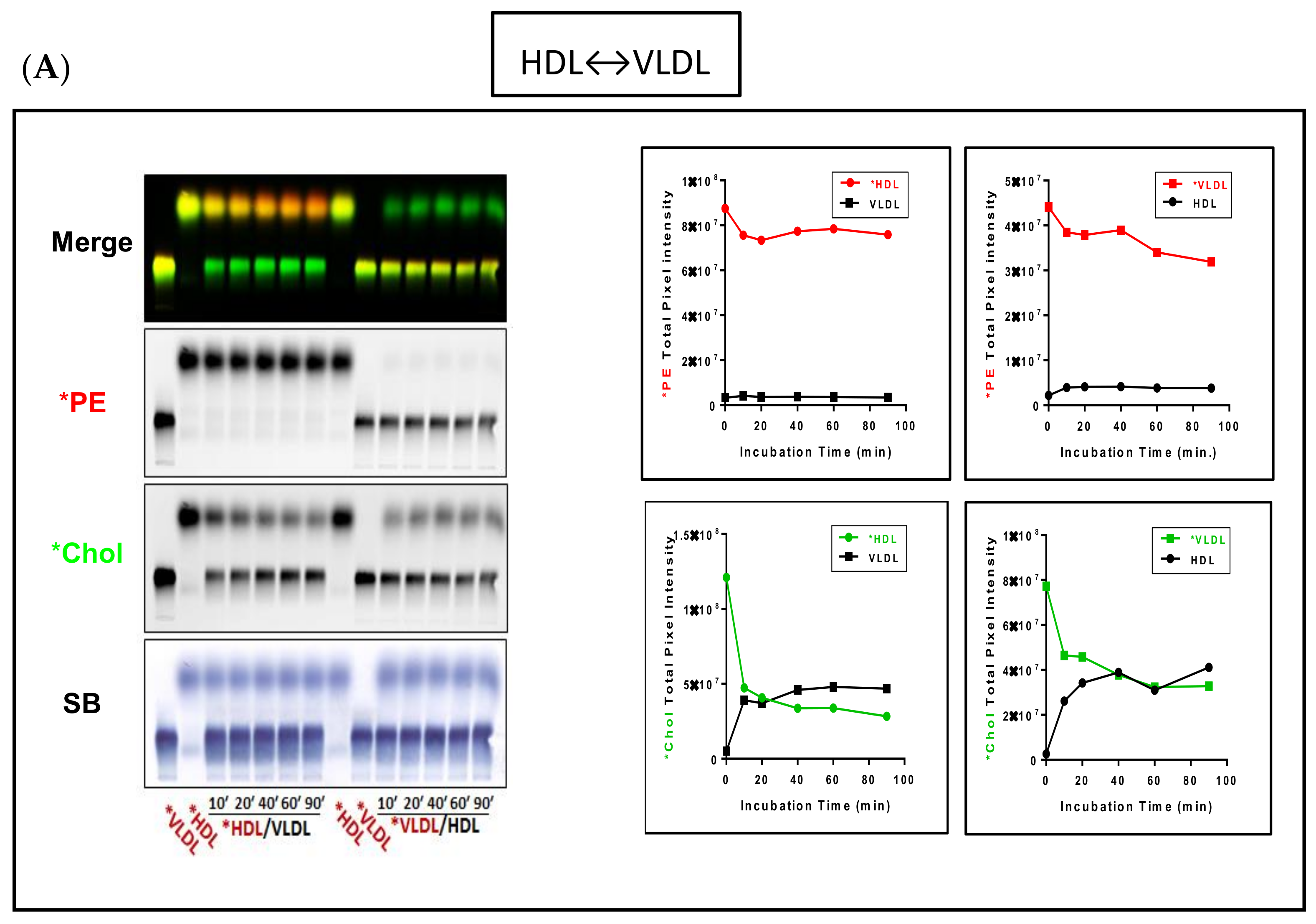
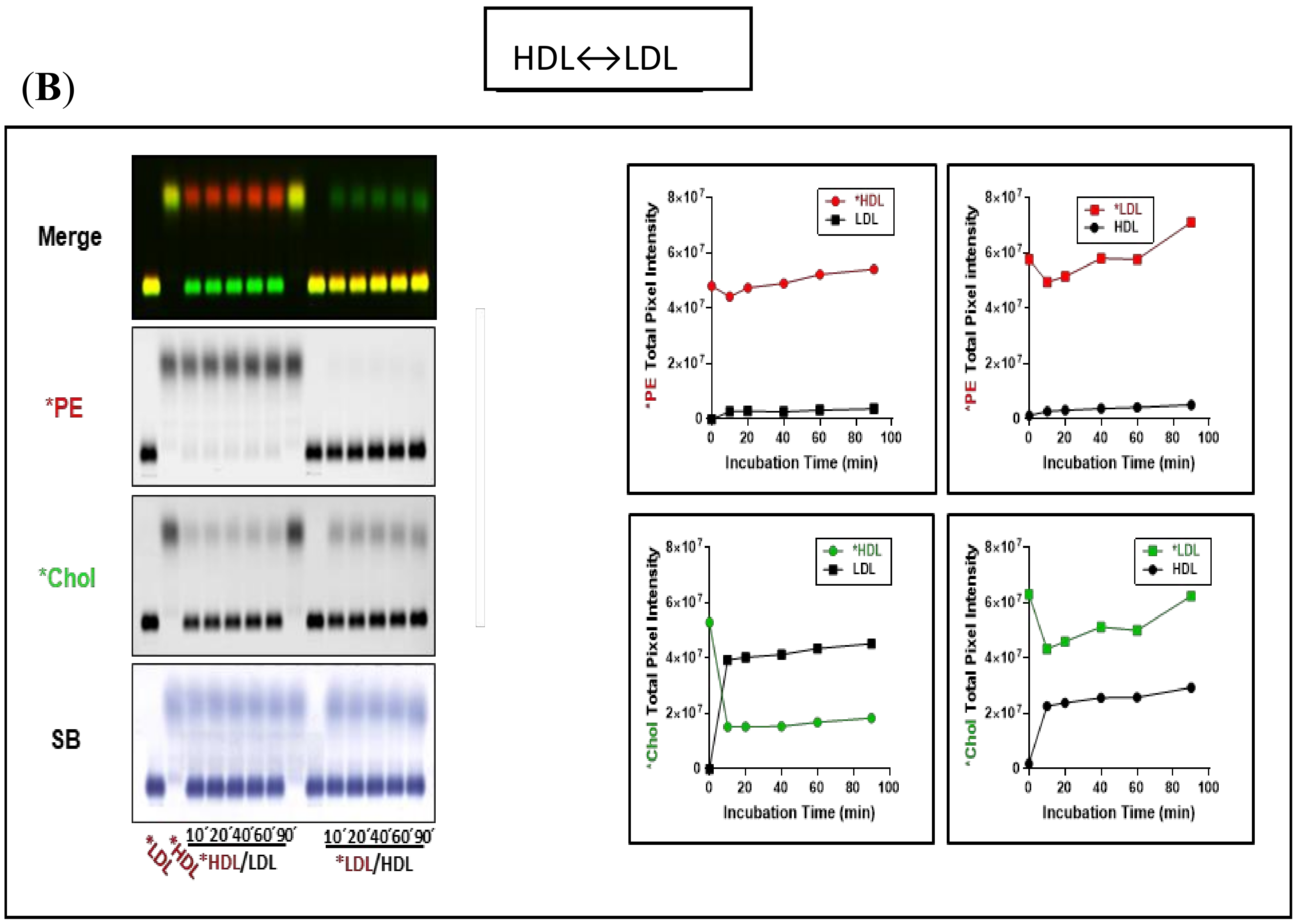
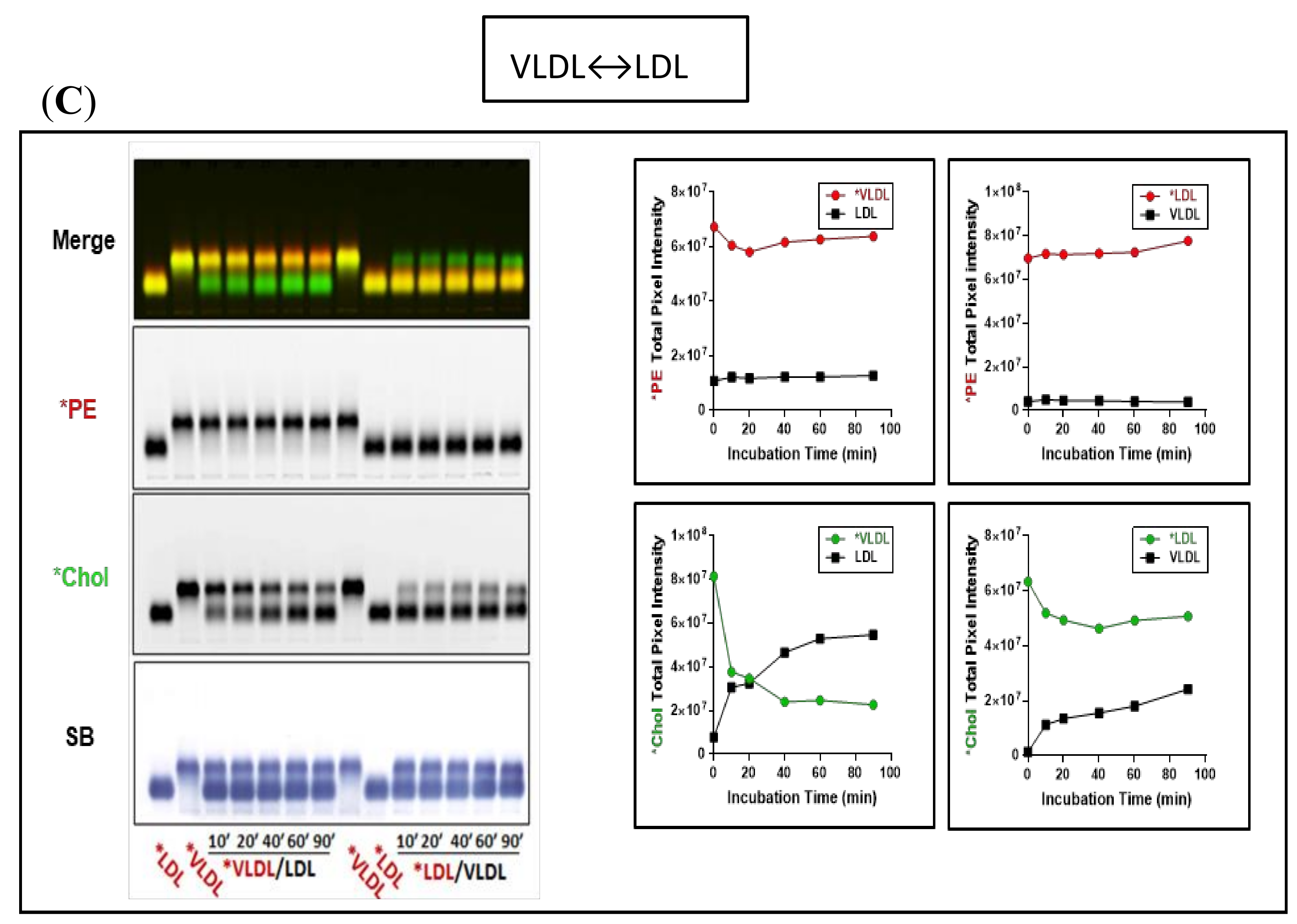
3.2. Fluorescent PE Can Be Used to Monitor Lipoprotein Remodeling
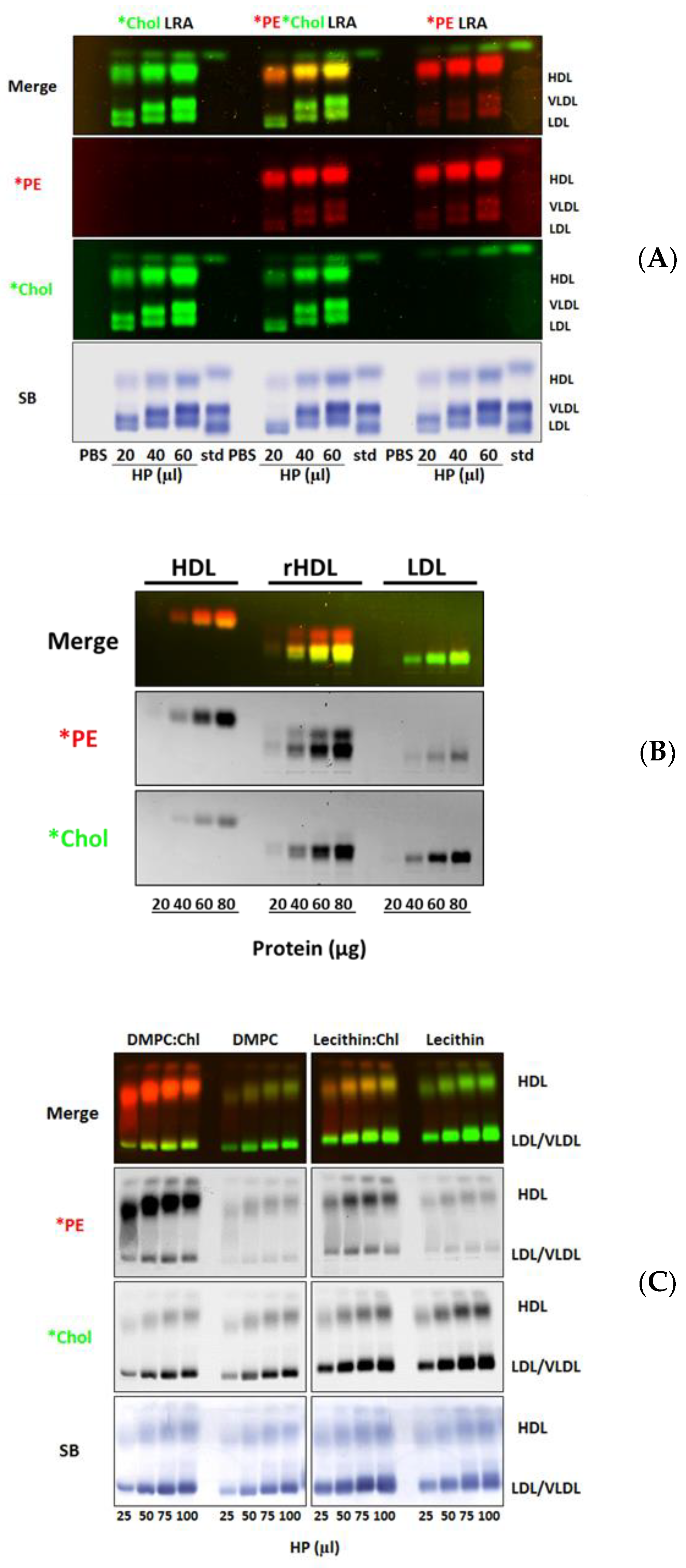
3.3. Fluorescent Lipid Transfer to Lipoproteins from Substrate-Bound Lipid Donor Membranes
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dashty, M.; Motazacker, M.M.; Levels, J.; de Vries, M.; Mahmoudi, M.; Peppelenbosch, M.P.; Rezaee, F. Proteome of human plasma very low-density lipoprotein and low-density lipoprotein exhibits a link with coagulation and lipid metabolism. Thromb. Haemost. 2014, 111, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Von Zychlinski, A.; Williams, M.; McCormick, S.; Kleffmann, T. Absolute quantification of apolipoproteins and associated proteins on human plasma lipoproteins. J. Proteom. 2014, 106, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.M.; Deng, J.; Lu, L.J.; Davidson, W.S. Proteomic characterization of human plasma high density lipoprotein fractionated by gel filtration chromatography. J. Proteome Res. 2010, 9, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.; Tan, L.; Long, J.L.; Davidson, W.S. Proteomic diversity of high density lipoproteins: Our emerging understanding of its importance in lipid transport and beyond. J. Lipid Res. 2013, 54, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [PubMed]
- Jove, M.; Naudi, A.; Portero-Otin, M.; Cabre, R.; Rovira-Llopis, S.; Banuls, C.; Rocha, M.; Hernandez-Mijares, A.; Victor, V.M.; Pamplona, R. Plasma lipidomics discloses metabolic syndrome with a specific HDL phenotype. FASEB J. 2014, 28, 5163–5171. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.; Ruel, I.; Hafiane, A.; Cochrane, H.; Iatan, I.; Jauhiainen, M.; Ehnholm, C.; Krimbou, L.; Genest, J. Analysis of lipid transfer activity between model nascent HDL particles and plasma lipoproteins: Implications for current concepts of nascent HDL maturation and genesis. J. Lipid Res. 2010, 51, 785–797. [Google Scholar] [CrossRef]
- Borja, M.S.; Ng, K.F.; Irwin, A.; Hong, J.; Wu, X.; Isquith, D.; Zhao, X.Q.; Prazen, B.; Gildengorin, V.; Oda, M.N.; et al. HDL-apolipoprotein A-I exchange is independently associated with cholesterol efflux capacity. J. Lipid Res. 2015, 56, 2002–2009. [Google Scholar] [CrossRef] [PubMed]
- Borja, M.S.; Zhao, L.; Hammerson, B.; Tang, C.; Yang, R.; Carson, N.; Fernando, G.; Liu, X.; Budamagunta, M.S.; Genest, J.; et al. HDL-apoA-I exchange: Rapid detection and association with atherosclerosis. PLoS ONE 2013, 8, e71541. [Google Scholar] [CrossRef]
- Zhang, P.; Gao, J.; Pu, C.; Feng, G.; Wang, L.; Huang, L.; Zhang, Y. ApoM/HDL-C and apoM/apoA-I ratios are indicators of diabetic nephropathy in healthy controls and type 2 diabetes mellitus. Clin. Chim. Acta 2017, 466, 31–37. [Google Scholar] [CrossRef]
- Meikle, P.J.; Wong, G.; Barlow, C.K.; Kingwell, B.A. Lipidomics: Potential role in risk prediction and therapeutic monitoring for diabetes and cardiovascular disease. Pharmacol. Ther. 2014, 143, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Parks, J.S. Dietary cholesterol effects on adipose tissue inflammation. Curr. Opin. Lipidol. 2016, 27, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Mika, A.; Sledzinski, T. Alterations of specific lipid groups in serum of obese humans: A review. Obes. Rev. 2017, 18, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Ossoli, A.; Neufeld, E.B.; Thacker, S.G.; Vaisman, B.; Pryor, M.; Freeman, L.A.; Brantner, C.A.; Baranova, I.; Francone, N.O.; Demosky, S.J., Jr.; et al. Lipoprotein X Causes Renal Disease in LCAT Deficiency. PLoS ONE 2016, 11, e0150083. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. Disorders of lipid metabolism in nephrotic syndrome: Mechanisms and consequences. Kidney Int. 2016, 90, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, S.; Kellner-Weibel, G.; de la Llera-Moya, M.; Phillips, M.C.; Asztalos, B.F.; Bittman, R.; Rothblat, G.H. A sensitive assay for ABCA1-mediated cholesterol efflux using BODIPY-cholesterol. J. Lipid Res. 2011, 52, 2332–2340. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Toh, R.; Murkami, K.; Kiriyama, M.; Yoshikawa, K.; Miwa, K.; Kubo, T.; Irino, Y.; Mori, K.; Tanaka, N.; et al. Cholesterol Uptake Capacity: A New Measure of HDL Functionality for Coronary Risk Assessment. J. Appl. Lab. Med. 2017, 2, 186–200. [Google Scholar] [CrossRef]
- Horiuchi, Y.; Lai, S.J.; Yamazaki, A.; Nakamura, A.; Ohkawa, R.; Yano, K.; Kameda, T.; Okubo, S.; Shimano, S.; Hagihara, M.; et al. Validation and application of a novel cholesterol efflux assay using immobilized liposomes as a substitute for cultured cells. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Sakurai, A.; Vaisman, B.L.; Nishida, T.; Neufeld, E.B.; Demosky, S.J., Jr.; Sampson, M.L.; Shamburek, R.D.; Freeman, L.A.; Remaley, A.T. Development of a novel fluorescent activity assay for lecithin:cholesterol acyltransferase. Ann. Clin. Biochem. 2018, 55, 414–421. [Google Scholar] [CrossRef]
- Lund-Katz, S.; Hammerschlag, B.; Phillips, M.C. Kinetics and mechanism of free cholesterol exchange between human serum high- and low-density lipoproteins. Biochemistry 1982, 21, 2964–2969. [Google Scholar] [CrossRef]
- Rousset, X.; Vaisman, B.; Auerbach, B.; Krause, B.R.; Homan, R.; Stonik, J.; Csako, G.; Shamburek, R.; Remaley, A.T. Effect of recombinant human lecithin cholesterol acyltransferase infusion on lipoprotein metabolism in mice. J. Pharm. Exp. Ther. 2010, 335, 140–148. [Google Scholar] [CrossRef]
- Sakai, N.; Vaisman, B.L.; Koch, C.A.; Hoyt, R.F., Jr.; Meyn, S.M.; Talley, G.D.; Paiz, J.A.; Brewer, H.B., Jr.; Santamarina-Fojo, S. Targeted disruption of the mouse lecithin:cholesterol acyltransferase (LCAT) gene. Generation of a new animal model for human LCAT deficiency. J. Biol. Chem. 1997, 272, 7506–7510. [Google Scholar] [CrossRef]
- Vaisman, B.L.; Klein, H.G.; Rouis, M.; Berard, A.M.; Kindt, M.R.; Talley, G.D.; Meyn, S.M.; Hoyt, R.F., Jr.; Marcovina, S.M.; Albers, J.J.; et al. Overexpression of human lecithin cholesterol acyltransferase leads to hyperalphalipoproteinemia in transgenic mice. J. Biol. Chem. 1995, 270, 12269–12275. [Google Scholar] [CrossRef]
- Brewer, H.B., Jr.; Ronan, R.; Meng, M.; Bishop, C. Isolation and characterization of apolipoproteins A-I, A-II, and A-IV. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1986; Volume 128, pp. 223–246. [Google Scholar]
- Lambert, G.; Sakai, N.; Vaisman, B.L.; Neufeld, E.B.; Marteyn, B.; Chan, C.C.; Paigen, B.; Lupia, E.; Thomas, A.; Striker, L.J.; et al. Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J. Biol. Chem. 2001, 276, 15090–15098. [Google Scholar] [CrossRef]
- Lerch, P.G.; Fortsch, V.; Hodler, G.; Bolli, R. Production and characterization of a reconstituted high density lipoprotein for therapeutic applications. Vox Sang. 1996, 71, 155–164. [Google Scholar] [CrossRef]
- Thacker, S.G.; Zarzour, A.; Chen, Y.; Alcicek, M.S.; Freeman, L.A.; Sviridov, D.O.; Demosky, S.J., Jr.; Remaley, A.T. High-density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology 2016, 149, 306–319. [Google Scholar] [CrossRef]
- Neufeld, E.B.; Stonik, J.A.; Demosky, S.J., Jr.; Knapper, C.L.; Combs, C.A.; Cooney, A.; Comly, M.; Dwyer, N.; Blanchette-Mackie, J.; Remaley, A.T.; et al. The ABCA1 transporter modulates late endocytic trafficking: Insights from the correction of the genetic defect in Tangier disease. J. Biol. Chem. 2004, 279, 15571–15578. [Google Scholar] [CrossRef]
- Frohlich, J. Role of lecithin:cholesterol acyltransferase and apolipoprotein A-I in cholesterol esterification in lipoprotein-X in vitro. J. Lipid Res. 1995, 36, 2344–2354. [Google Scholar]
- Rosenson, R.S.; Brewer, H.B., Jr.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef]
- Chen, Z.; O′Neill, E.A.; Meurer, R.D.; Gagen, K.; Luell, S.; Wang, S.P.; Ichetovkin, M.; Frantz-Wattley, B.; Eveland, S.; Strack, A.M.; et al. Reconstituted HDL elicits marked changes in plasma lipids following single-dose injection in C57Bl/6 mice. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 315–323. [Google Scholar] [CrossRef]
- Rubin, E.M.; Ishida, B.Y.; Clift, S.M.; Krauss, R.M. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc. Natl. Acad. Sci. USA 1991, 88, 434–438. [Google Scholar] [CrossRef]
- The Davidson/Shah Lab. Available online: http://homepages.uc.edu/~davidswm/HDLproteome.html (accessed on 14 August 2015).
- Asztalos, B.F.; Tani, M.; Schaefer, E.J. Metabolic and functional relevance of HDL subspecies. Curr. Opin. Lipidol. 2011, 22, 176–185. [Google Scholar] [CrossRef]
- Sankaranarayanan, S.; de la Llera-Moya, M.; Drazul-Schrader, D.; Phillips, M.C.; Kellner-Weibel, G.; Rothblat, G.H. Serum albumin acts as a shuttle to enhance cholesterol efflux from cells. J. Lipid Res. 2013, 54, 671–676. [Google Scholar] [CrossRef]
- Schwartz, C.C.; Zech, L.A.; VandenBroek, J.M.; Cooper, P.S. Cholesterol kinetics in subjects with bile fistula. Positive relationship between size of the bile acid precursor pool and bile acid synthetic rate. J. Clin. Investig. 1993, 91, 923–938. [Google Scholar] [CrossRef]
- Hoang, A.; Drew, B.G.; Low, H.; Remaley, A.T.; Nestel, P.; Kingwell, B.A.; Sviridov, D. Mechanism of cholesterol efflux in humans after infusion of reconstituted high-density lipoprotein. Eur. Heart J. 2012, 33, 657–665. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identified Proteins (30) | Accession Number | Molecular Weight | ALB (*TSC) | HDL (*TSC) |
| Serum albumin | ALBU | 69 kDa | 938 | 421 |
| Apolipoprotein A-I | APOA1 | 31 kDa | 58 | 4 |
| Vitamin D-binding protein | VTDB | 53 kDa | 0 | 50 |
| Prothrombin | THRB | 70 kDa | 0 | 49 |
| Inter-alpha-trypsin inhibitor heavy chain H2 | ITIH2 | 106 kDa | 0 | 39 |
| Cluster of Serum amyloid A-I protein | SAA1 | 14 kDa | 10 | 23 |
| Apolipoprotein A-II | APOA2 | 11 kDa | 14 | 19 |
| Afamin | AFAM | 69 kDa | 0 | 21 |
| Clusterin | CLUS | 52 kDa | 0 | 23 |
| Alpha-1-antitrypsin | A1AT | 47 kDa | 0 | 23 |
| Haptoglobin | HPT | 45 kDa | 0 | 30 |
| Apolipoprotein C-III | APOC3 | 11 kDa | 9 | 13 |
| Vitronectin | VTNC | 54 kDa | 0 | 19 |
| Antithrombin-III | ANT3 | 53 kDa | 0 | 14 |
| Apolipoprotein E | APOE | 36 kDa | 3 | 14 |
| Alpha-1β-glycoprotein | A1BG | 54 kDa | 0 | 13 |
| Protein AMBP | AMBP | 39 kDa | 5 | 10 |
| Serum paraxonase/arylesterase 1 | PON1 | 40 kDa | 10 | 0 |
| Serum amyloid A-4 protein | SAA4 | 15 kDa | 0 | 9 |
| Ceruloplasmin | CERU | 122 kDa | 0 | 8 |
| Kininogen-1 | KNG1 | 72 kDa | 0 | 8 |
| Retinol-binding protein 4 | RET4 | 23 kDa | 0 | 9 |
| Inter-alpha-trypsin inhibitor heavy chain H1 | ITIH2 | 101 kDa | 0 | 9 |
| Alpha-2-HS-glycoprotein | FETUA | 39 kDa | 0 | 5 |
| Apolipoprotein A-IV | APOA4 | 45 kDa | 0 | 6 |
| Apolipoprotein C-II | APOC2 | 11 kDa | 0 | 2 |
| Apolipoprotein D | APOD | 21 kDa | 0 | 4 |
| Transthyretin | TTHY | 16 kDa | 0 | 3 |
| Coagulation factor X | FA10 | 55 kDa | 0 | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neufeld, E.B.; Sato, M.; Gordon, S.M.; Durbhakula, V.; Francone, N.; Aponte, A.; Yilmaz, G.; Sviridov, D.; Sampson, M.; Tang, J.; et al. ApoA-I-Mediated Lipoprotein Remodeling Monitored with a Fluorescent Phospholipid. Biology 2019, 8, 53. https://doi.org/10.3390/biology8030053
Neufeld EB, Sato M, Gordon SM, Durbhakula V, Francone N, Aponte A, Yilmaz G, Sviridov D, Sampson M, Tang J, et al. ApoA-I-Mediated Lipoprotein Remodeling Monitored with a Fluorescent Phospholipid. Biology. 2019; 8(3):53. https://doi.org/10.3390/biology8030053
Chicago/Turabian StyleNeufeld, Edward B., Masaki Sato, Scott M. Gordon, Vinay Durbhakula, Nicolas Francone, Angel Aponte, Gizem Yilmaz, Denis Sviridov, Maureen Sampson, Jingrong Tang, and et al. 2019. "ApoA-I-Mediated Lipoprotein Remodeling Monitored with a Fluorescent Phospholipid" Biology 8, no. 3: 53. https://doi.org/10.3390/biology8030053
APA StyleNeufeld, E. B., Sato, M., Gordon, S. M., Durbhakula, V., Francone, N., Aponte, A., Yilmaz, G., Sviridov, D., Sampson, M., Tang, J., Pryor, M., & Remaley, A. T. (2019). ApoA-I-Mediated Lipoprotein Remodeling Monitored with a Fluorescent Phospholipid. Biology, 8(3), 53. https://doi.org/10.3390/biology8030053