Mitochondrial Transport and Turnover in the Pathogenesis of Amyotrophic Lateral Sclerosis


{kind=link}
Abstract
1. Introduction
2. Mitochondrial Axonal Transport in ALS
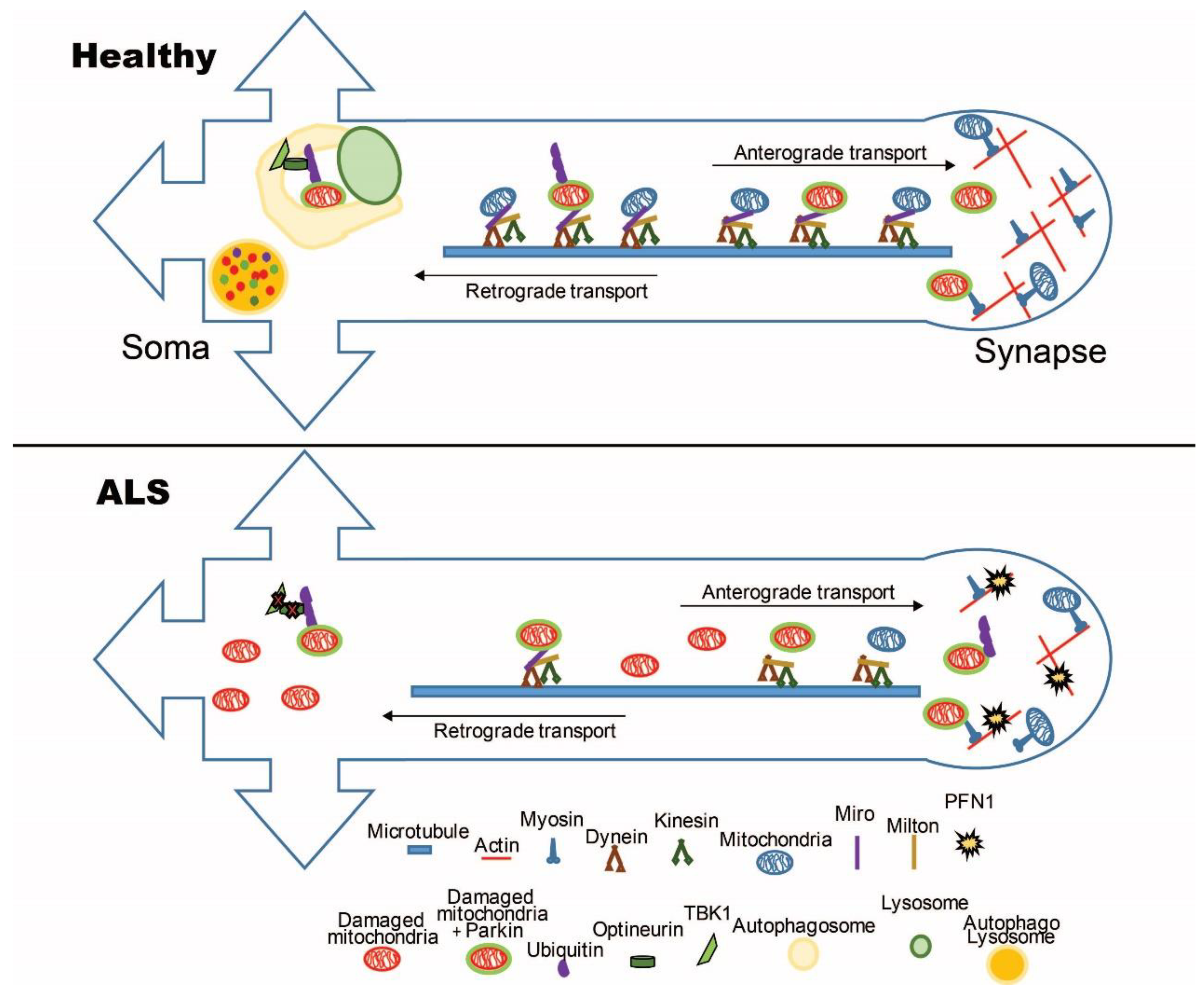
2.1. The Mitochondrial Transport Machinery
2.2. Alterations of the Mitochondrial Transport Machinery in ALS
3. Mitochondrial Turnover in ALS
3.1. Mitochondrial Quality Control Mechanisms
3.2. Mitochondrial Quality Control in ALS
4. Conclusions
Funding
Conflicts of Interest
References
- Hardiman, O.; van den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Emery, A.E.; Holloway, S. Familial motor neuron diseases. Adv. Neurol. 1982, 36, 139–147. [Google Scholar] [PubMed]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Andrus, P.K.; Fleck, T.J.; Gurney, M.E.; Hall, E.D. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 2041–2048. [Google Scholar] [CrossRef]
- Wood, J.D.; Beaujeux, T.P.; Shaw, P.J. Protein aggregation in motor neurone disorders. Neuropathol. Appl. Neurobiol. 2003, 29, 529–545. [Google Scholar] [CrossRef]
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.L. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 2008, 3, e2849. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Ince, P.G.; Falkous, G.; Mantle, D. Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann. Neurol. 1995, 38, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain J. Neurol. 2014, 137, 2329–2345. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007, 10, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Higginbottom, A.; Heath, P.R.; Barber, S.; Greenald, D.; Kirby, J.; Shaw, P.J. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain J. Neurol. 2011, 134, 2627–2641. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef]
- Sargsyan, S.A.; Monk, P.N.; Shaw, P.J. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia 2005, 51, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Bel Hadj, S.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Tian, J.H.; Pan, P.Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.H. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Holzbaur, E.L. Autophagosome assembly and cargo capture in the distal axon. Autophagy 2012, 8, 858–860. [Google Scholar] [CrossRef]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef]
- Fransson, A.; Ruusala, A.; Aspenstrom, P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J. Biol. Chem. 2003, 278, 6495–6502. [Google Scholar] [CrossRef]
- Fransson, S.; Ruusala, A.; Aspenstrom, P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 2006, 344, 500–510. [Google Scholar] [CrossRef]
- Mironov, S.L. ADP regulates movements of mitochondria in neurons. Biophys. J. 2007, 92, 2944–2952. [Google Scholar] [CrossRef]
- Wang, X.; Schwarz, T.L. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef]
- Macaskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef]
- Eisen, A.; Weber, M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001, 24, 564–573. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Martineau, E.; Di Polo, A.; Vande Velde, C.; Robitaille, R. Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. eLife 2018, 7. [Google Scholar] [CrossRef]
- De Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008, 31, 151–173. [Google Scholar] [CrossRef]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H., Jr.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Magrane, J.; Sahawneh, M.A.; Przedborski, S.; Estevez, A.G.; Manfredi, G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 229–242. [Google Scholar] [CrossRef]
- Marinkovic, P.; Reuter, M.S.; Brill, M.S.; Godinho, L.; Kerschensteiner, M.; Misgeld, T. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 4296–4301. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Shahidullah, M.; Le Marchand, S.J.; Fei, H.; Zhang, J.; Pandey, U.B.; Dalva, M.B.; Pasinelli, P.; Levitan, I.B. Defects in synapse structure and function precede motor neuron degeneration in Drosophila models of FUS-related ALS. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 19590–19598. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Krakora, D.; Macrander, C.; Suzuki, M. Neuromuscular junction protection for the potential treatment of amyotrophic lateral sclerosis. Neurol. Res. Int. 2012, 2012, 379657. [Google Scholar] [CrossRef]
- Bilsland, L.G.; Sahai, E.; Kelly, G.; Golding, M.; Greensmith, L.; Schiavo, G. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 20523–20528. [Google Scholar] [CrossRef]
- Ligon, L.A.; LaMonte, B.H.; Wallace, K.E.; Weber, N.; Kalb, R.G.; Holzbaur, E.L. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport 2005, 16, 533–536. [Google Scholar] [CrossRef]
- Zhang, F.; Strom, A.L.; Fukada, K.; Lee, S.; Hayward, L.J.; Zhu, H. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J. Biol. Chem. 2007, 282, 16691–16699. [Google Scholar] [CrossRef] [PubMed]
- Igoudjil, A.; Magrane, J.; Fischer, L.R.; Kim, H.J.; Hervias, I.; Dumont, M.; Cortez, C.; Glass, J.D.; Starkov, A.A.; Manfredi, G. In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 15826–15837. [Google Scholar] [CrossRef]
- Pickles, S.; Destroismaisons, L.; Peyrard, S.L.; Cadot, S.; Rouleau, G.A.; Brown, R.H., Jr.; Julien, J.P.; Arbour, N.; Vande Velde, C. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum. Mol. Genet. 2013, 22, 3947–3959. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Semmler, S.; Broom, H.R.; Destroismaisons, L.; Legroux, L.; Arbour, N.; Meiering, E.; Cashman, N.R.; Vande Velde, C. ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol. Commun. 2016, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Vande Velde, C.; Israelson, A.; Xie, J.; Bailey, A.O.; Dong, M.Q.; Chun, S.J.; Roy, T.; Winer, L.; Yates, J.R.; et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc. Natl. Acad. Sci. USA 2010, 107, 21146–21151. [Google Scholar] [CrossRef]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef]
- Magrane, J.; Hervias, I.; Henning, M.S.; Damiano, M.; Kawamata, H.; Manfredi, G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum. Mol. Genet. 2009, 18, 4552–4564. [Google Scholar] [CrossRef]
- Cozzolino, M.; Pesaresi, M.G.; Amori, I.; Crosio, C.; Ferri, A.; Nencini, M.; Carri, M.T. Oligomerization of mutant SOD1 in mitochondria of motoneuronal cells drives mitochondrial damage and cell toxicity. Antioxid. Redox Signal. 2009, 11, 1547–1558. [Google Scholar] [CrossRef]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Genet. 2008, 17, 3303–3317. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.L.; Hollenbeck, P.J. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 1995, 131, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2018, 24, 54–72. [Google Scholar] [CrossRef]
- Wu, C.H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, L.; Nystrom, L.E.; Sundkvist, I.; Markey, F.; Lindberg, U. Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J. Mol. Biol. 1977, 115, 465–483. [Google Scholar] [CrossRef]
- Birbach, A. Profilin, a multi-modal regulator of neuronal plasticity. Bioessays News Rev. Mol. Cell. Dev. Biol. 2008, 30, 994–1002. [Google Scholar] [CrossRef]
- Kang, F.; Purich, D.L.; Southwick, F.S. Profilin promotes barbed-end actin filament assembly without lowering the critical concentration. J. Biol. Chem. 1999, 274, 36963–36972. [Google Scholar] [CrossRef]
- Yarmola, E.G.; Bubb, M.R. How depolymerization can promote polymerization: The case of actin and profilin. Bioessays News Rev. Mol. Cell. Dev. Biol. 2009, 31, 1150–1160. [Google Scholar] [CrossRef]
- Smith, B.N.; Vance, C.; Scotter, E.L.; Troakes, C.; Wong, C.H.; Topp, S.; Maekawa, S.; King, A.; Mitchell, J.C.; Lund, K.; et al. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol. Aging 2015, 36, 1602.e17-27. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nonaka, T.; Suzuki, G.; Kametani, F.; Hasegawa, M. Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum. Mol. Genet. 2016, 25, 1420–1433. [Google Scholar] [CrossRef]
- Boopathy, S.; Silvas, T.V.; Tischbein, M.; Jansen, S.; Shandilya, S.M.; Zitzewitz, J.A.; Landers, J.E.; Goode, B.L.; Schiffer, C.A.; Bosco, D.A. Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc. Natl. Acad. Sci. USA 2015, 112, 7984–7989. [Google Scholar] [CrossRef]
- Yang, C.; Danielson, E.W.; Qiao, T.; Metterville, J.; Brown, R.H., Jr.; Landers, J.E.; Xu, Z. Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc. Natl. Acad. Sci. USA 2016, 113, E6209–E6218. [Google Scholar] [CrossRef]
- Fil, D.; DeLoach, A.; Yadav, S.; Alkam, D.; MacNicol, M.; Singh, A.; Compadre, C.M.; Goellner, J.J.; O’Brien, C.A.; Fahmi, T.; et al. Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 2017, 26, 686–701. [Google Scholar] [CrossRef]
- Shneyer, B.I.; Usaj, M.; Henn, A. Myo19 is an outer mitochondrial membrane motor and effector of starvation-induced filopodia. J. Cell Sci. 2016, 129, 543–556. [Google Scholar] [CrossRef]
- Lopez-Domenech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. Embo J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Oeding, S.J.; Majstrowicz, K.; Hu, X.P.; Schwarz, V.; Freitag, A.; Honnert, U.; Nikolaus, P.; Bahler, M. Identification of Miro1 and Miro2 as mitochondrial receptors for myosin XIX. J. Cell Sci. 2018, 131, jcs219469. [Google Scholar] [CrossRef]
- Gerdes, F.; Tatsuta, T.; Langer, T. Mitochondrial AAA proteases—Towards a molecular understanding of membrane-bound proteolytic machines. Biochim. Biophys. Acta 2012, 1823, 49–55. [Google Scholar] [CrossRef]
- Matsushima, Y.; Kaguni, L.S. Matrix proteases in mitochondrial DNA function. Biochim. Biophys. Acta 2012, 1819, 1080–1087. [Google Scholar] [CrossRef]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef]
- Radke, S.; Chander, H.; Schafer, P.; Meiss, G.; Kruger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Youle, R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 2011, 23, 476–482. [Google Scholar] [CrossRef]
- Heo, J.M.; Rutter, J. Ubiquitin-dependent mitochondrial protein degradation. Int. J. Biochem. Cell Biol. 2011, 43, 1422–1426. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; de Vries, R.L.; Tocilescu, M.; Przedborski, S. PINK1/Parkin direct mitochondria to autophagy. Autophagy 2010, 6, 315–316. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Li, W.; Bengtson, M.H.; Ulbrich, A.; Matsuda, A.; Reddy, V.A.; Orth, A.; Chanda, S.K.; Batalov, S.; Joazeiro, C.A. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS ONE 2008, 3, e1487. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, R.; Ishido, S.; Kyo, S.; Fukuda, T.; Goto, E.; Matsuki, Y.; Ohmura-Hoshino, M.; Sada, K.; Hotta, H.; Yamamura, H.; et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. Embo J. 2006, 25, 3618–3626. [Google Scholar] [CrossRef]
- Karbowski, M.; Neutzner, A.; Youle, R.J. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 2007, 178, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Yonashiro, R.; Sugiura, A.; Miyachi, M.; Fukuda, T.; Matsushita, N.; Inatome, R.; Ogata, Y.; Suzuki, T.; Dohmae, N.; Yanagi, S. Mitochondrial ubiquitin ligase MITOL ubiquitinates mutant SOD1 and attenuates mutant SOD1-induced reactive oxygen species generation. Mol. Biol. Cell 2009, 20, 4524–4530. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Aerts, L.; Craessaerts, K.; De Strooper, B.; Morais, V.A. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J. Biol. Chem. 2015, 290, 2798–2811. [Google Scholar] [CrossRef]
- Soubannier, V.; McLelland, G.L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 2010, 5, e10054. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Muller, K.; Marroquin, N.; Nordin, F.; Hubers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Rubino, E.; Rainero, I.; Chio, A.; Rogaeva, E.; Galimberti, D.; Fenoglio, P.; Grinberg, Y.; Isaia, G.; Calvo, A.; Gentile, S.; et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012, 79, 1556–1562. [Google Scholar] [CrossRef]
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J.; et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12, 1094–1104. [Google Scholar] [CrossRef]
- Ye, Y.; Shibata, Y.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Rapoport, T.A. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14132–14138. [Google Scholar] [CrossRef]
- Kim, N.C.; Tresse, E.; Kolaitis, R.M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 2013, 78, 65–80. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef]
- Weihofen, A.; Thomas, K.J.; Ostaszewski, B.L.; Cookson, M.R.; Selkoe, D.J. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 2009, 48, 2045–2052. [Google Scholar] [CrossRef]
- Shlevkov, E.; Kramer, T.; Schapansky, J.; LaVoie, M.J.; Schwarz, T.L. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. USA 2016, 113, E6097–E6106. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, W.; Siedlak, S.L.; Liu, Y.; Liu, J.; Jiang, K.; Perry, G.; Zhu, X.; Wang, X. Miro1 deficiency in amyotrophic lateral sclerosis. Front. Aging Neurosci. 2015, 7, 100. [Google Scholar] [CrossRef]
- Palomo, G.M.; Granatiero, V.; Kawamata, H.; Konrad, C.; Kim, M.; Arreguin, A.J.; Zhao, D.; Milner, T.A.; Manfredi, G. Parkin is a disease modifier in the mutant SOD1 mouse model of ALS. Embo Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Moller, A.; Bauer, C.S.; Cohen, R.N.; Webster, C.P.; De Vos, K.J. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 2017, 26, 4668–4679. [Google Scholar] [CrossRef]
- Kruppa, A.J.; Kishi-Itakura, C.; Masters, T.A.; Rorbach, J.E.; Grice, G.L.; Kendrick-Jones, J.; Nathan, J.A.; Minczuk, M.; Buss, F. Myosin VI-Dependent Actin Cages Encapsulate Parkin-Positive Damaged Mitochondria. Dev. Cell 2018, 44, 484–499 e486. [Google Scholar] [CrossRef]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef]
- Pizzasegola, C.; Caron, I.; Daleno, C.; Ronchi, A.; Minoia, C.; Carri, M.T.; Bendotti, C. Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2009, 10, 221–228. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011, 7, 412–425. [Google Scholar] [CrossRef]
- Zhang, K.; Shi, P.; An, T.; Wang, Q.; Wang, J.; Li, Z.; Duan, W.; Li, C.; Guo, Y. Food restriction-induced autophagy modulates degradation of mutant SOD1 in an amyotrophic lateral sclerosis mouse model. Brain Res. 2013, 1519, 112–119. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Song, L.; Tang, Y.; Shen, Y.; Jia, L.; Le, W. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014, 10, 588–602. [Google Scholar] [CrossRef]
- Kim, J.; Kim, T.Y.; Cho, K.S.; Kim, H.N.; Koh, J.Y. Autophagy activation and neuroprotection by progesterone in the G93A-SOD1 transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 59, 80–85. [Google Scholar] [CrossRef]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Le, W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 2008, 4, 290–293. [Google Scholar] [CrossRef]
- Morimoto, N.; Nagai, M.; Ohta, Y.; Miyazaki, K.; Kurata, T.; Morimoto, M.; Murakami, T.; Takehisa, Y.; Ikeda, Y.; Kamiya, T.; et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007, 1167, 112–117. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, B.; Lin, M.Y.; Wang, S.; Foust, K.D.; Sheng, Z.H. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron 2015, 87, 355–370. [Google Scholar] [CrossRef]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granatiero, V.; Manfredi, G. Mitochondrial Transport and Turnover in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biology 2019, 8, 36. https://doi.org/10.3390/biology8020036
Granatiero V, Manfredi G. Mitochondrial Transport and Turnover in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biology. 2019; 8(2):36. https://doi.org/10.3390/biology8020036
Chicago/Turabian StyleGranatiero, Veronica, and Giovanni Manfredi. 2019. "Mitochondrial Transport and Turnover in the Pathogenesis of Amyotrophic Lateral Sclerosis" Biology 8, no. 2: 36. https://doi.org/10.3390/biology8020036
APA StyleGranatiero, V., & Manfredi, G. (2019). Mitochondrial Transport and Turnover in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biology, 8(2), 36. https://doi.org/10.3390/biology8020036