Mitochondrial Dysfunction and Multiple Sclerosis


Abstract
1. Introduction
2. Mitochondria and Their Role in Neurodegeneration in Multiple Sclerosis
2.1. Mitochondria
2.2. Inflammation and Glia in Multiple Sclerosis
2.3. Neurodegeneration in Multiple Sclerosis and Evidence for Mitochondrial Involvement
2.3.1. Human Studies of Mitochondria Function in Multiple Sclerosis
2.3.2. Neurodegeneration in Multiple Sclerosis Animal Models
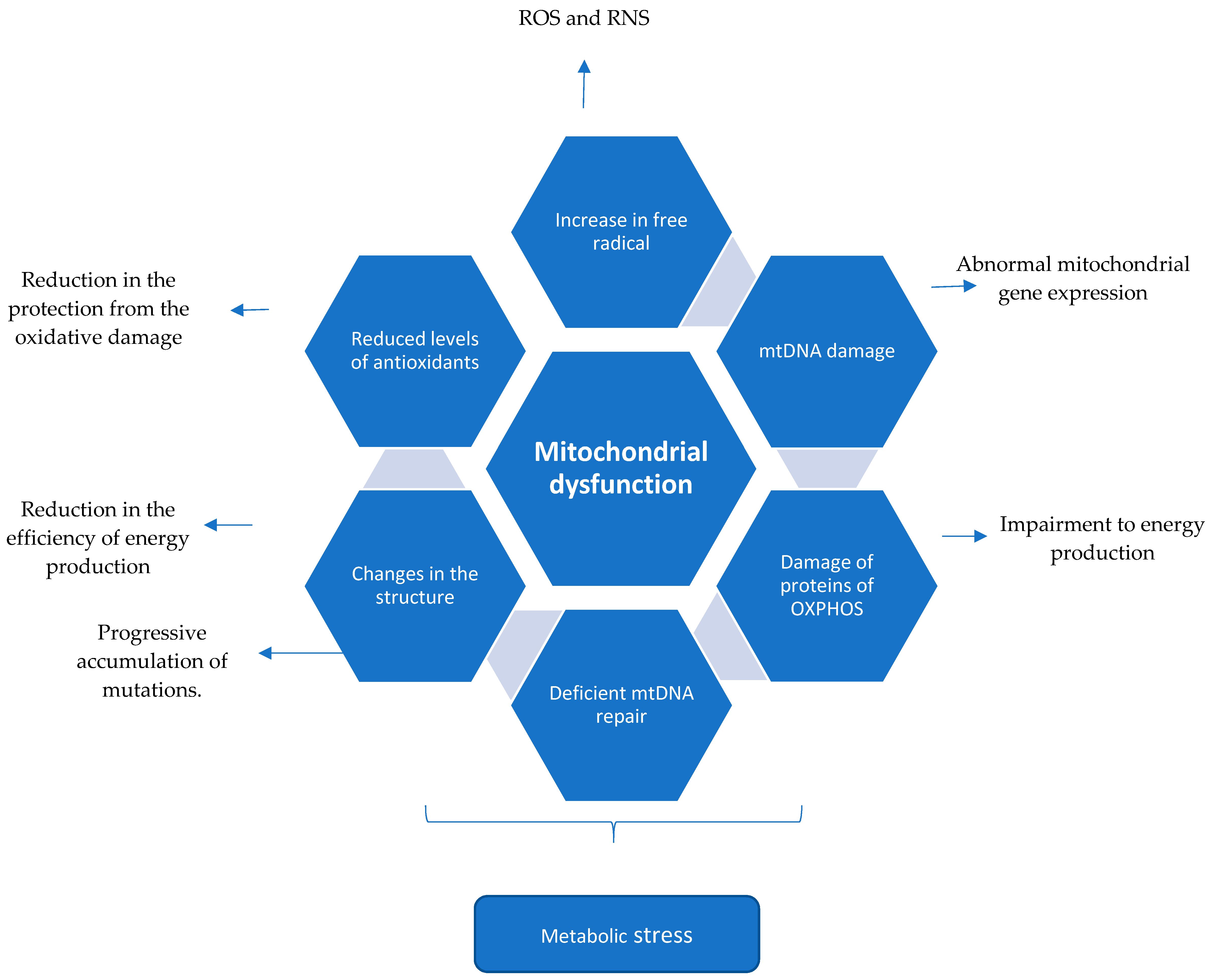
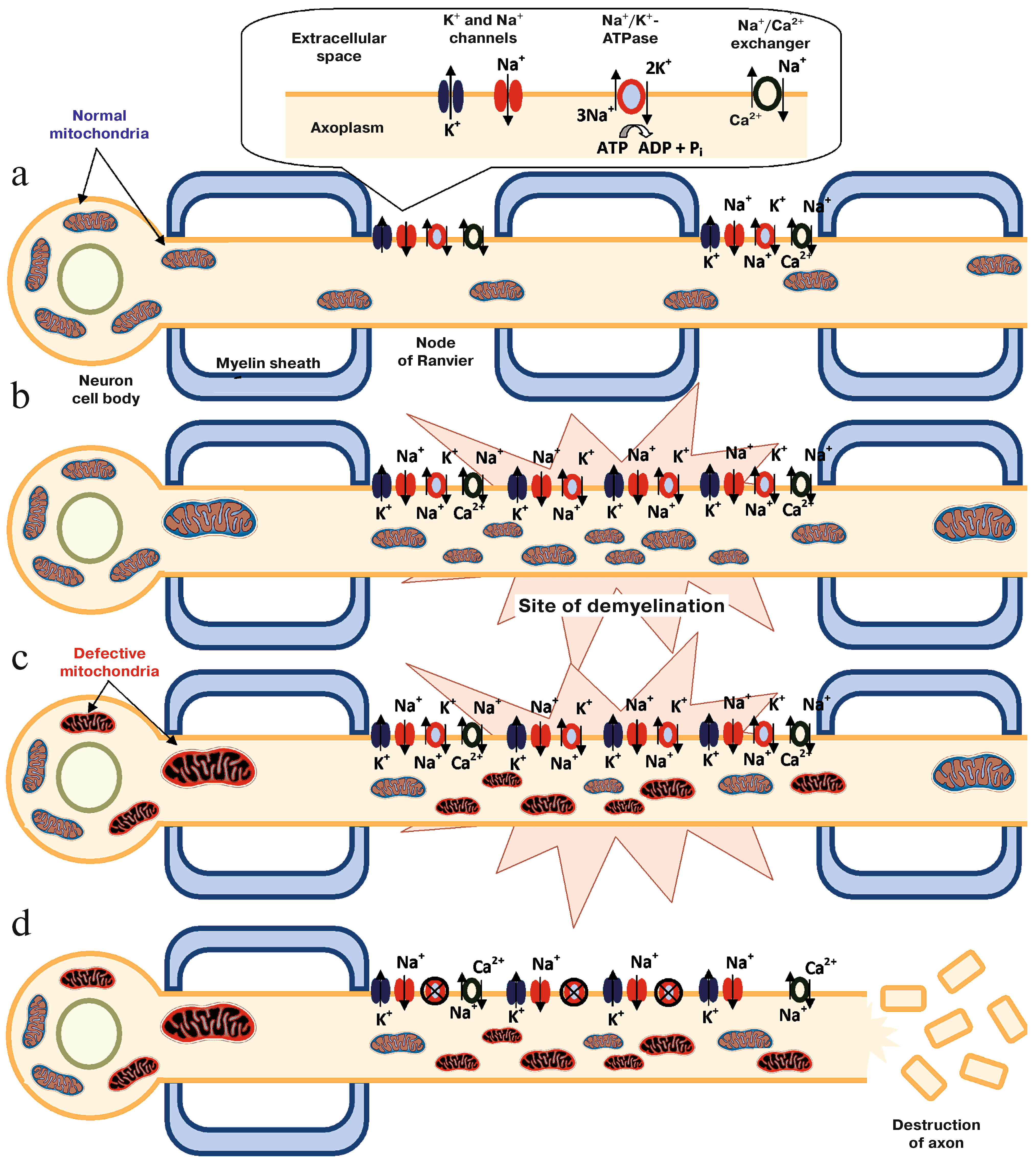
2.4. Summary (Mechanism of Mitochondrial Dysfunction Perpetuating the CNS Injury in Multiple Sclerosis)
3. Mitochondrial Mutations in Multiple Sclerosis and Overlapping Diseases
3.1. Mitochondrial Mutations and Multiple Sclerosis Risk
3.2. Leber’s Hereditary Optic Neuropathy
3.3. Dominant Optic Atrophy and OPA1 Mutations
3.4. POLG1 Mutations
4. Potential Therapies and Targets
4.1. Mitochondrial Metabolism and Chronic Neuroinflammation
4.2. Gene Therapy
5. Conclusions
Funding
Conflicts of Interest
References
- Rafael, H. Omental transplantation for neurodegenerative diseases. Am. J. Neurodegener. Dis. 2014, 3, 50–63. [Google Scholar] [PubMed]
- Burnside, S.W.; Hardingham, G.E. Transcriptional regulators of redox balance and other homeostatic processes with the potential to alter neurodegenerative disease trajectory. Biochem. Soc. Trans. 2017, 45, 1295–1303. [Google Scholar] [CrossRef]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Kozin, M.S.; Kulakova, O.G.; Favorova, O.O. Involvement of Mitochondria in Neurodegeneration in Multiple Sclerosis. Biochemistry (Mosc) 2018, 83, 813–830. [Google Scholar] [CrossRef]
- Ahlgren, C.; Oden, A.; Lycke, J. High nationwide incidence of multiple sclerosis in Sweden. PLoS ONE 2014, 9, e108599. [Google Scholar] [CrossRef]
- Koch, M.; Kingwell, E.; Rieckmann, P.; Tremlett, H. The natural history of primary progressive multiple sclerosis. Neurology 2009, 73, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Steinman, L. Multiple sclerosis: A two-stage disease. Nat. Immunol. 2001, 2, 762–764. [Google Scholar] [CrossRef]
- Ellwardt, E.; Zipp, F. Molecular mechanisms linking neuroinflammation and neurodegeneration in MS. Exp. Neurol. 2014, 262 Pt A, 8–17. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef]
- Haas, R.H.; Zolkipli, Z. Mitochondrial disorders affecting the nervous system. Semin. Neurol. 2014, 34, 321–340. [Google Scholar] [PubMed]
- Pitceathly, R.D.; McFarland, R. Mitochondrial myopathies in adults and children: Management and therapy development. Curr. Opin. Neurol. 2014, 27, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.M.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of mitochondrial disorders--past, present and future. Biochim. Biophys. Acta 2004, 1659, 115–120. [Google Scholar] [CrossRef]
- Mazunin, I.O.; Volod’ko, N.V.; Starikovskaia, E.B.; Sukernik, R.I. Mitochondrial genome and human mitochondrial diseases. Mol. Biol. (Mosk) 2010, 44, 755–772. [Google Scholar] [CrossRef]
- Holt, I.J.; Reyes, A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, 12. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef]
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient invasions: From endosymbionts to organelles. Science 2004, 304, 253–257. [Google Scholar] [CrossRef]
- Emes, R.D. The Logic of Evolution: Review of the Logic of Chance by Eugene V. Koonin. Front. Genet. 2012, 3, 135. [Google Scholar] [CrossRef][Green Version]
- Hunt, R.J.; Bateman, J.M. Mitochondrial retrograde signaling in the nervous system. FEBS Lett. 2017, 592, 663–678. [Google Scholar] [CrossRef]
- Mitra, K. Mitochondrial fission-fusion as an emerging key regulator of cell proliferation and differentiation. Bioessays 2013, 35, 955–964. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef]
- Suhaili, S.H.; Karimian, H.; Stellato, M.; Lee, T.H.; Aguilar, M.I. Mitochondrial outer membrane permeabilization: A focus on the role of mitochondrial membrane structural organization. Biophys. Rev. 2017, 9, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Walle, L.V.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ. 2008, 15, 453–460. [Google Scholar] [CrossRef]
- Ding, Z.J.; Chen, X.; Tang, X.X.; Wang, X.; Song, Y.L.; Chen, X.D.; Wang, J.; Wang, R.F.; Mi, W.J.; Chen, F.Q.; et al. Apoptosis-inducing factor and calpain upregulation in glutamate-induced injury of rat spiral ganglion neurons. Mol. Med. Rep. 2015, 12, 1685–1692. [Google Scholar] [CrossRef][Green Version]
- Jang, D.S.; Penthala, N.R.; Apostolov, E.O.; Wang, X.; Crooks, P.A.; Basnakian, A.G. Novel cytoprotective inhibitors for apoptotic endonuclease G. DNA Cell Biol. 2015, 34, 92–100. [Google Scholar] [CrossRef]
- Von Budingen, H.C.; Bar-Or, A.; Zamvil, S.S. B cells in multiple sclerosis: Connecting the dots. Curr. Opin. Immunol. 2011, 23, 713–720. [Google Scholar] [CrossRef]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Thomas, T.L.; Betmouni, S.; Scolding, N.; Love, S. Elevated activity and microglial expression of myeloperoxidase in demyelinated cerebral cortex in multiple sclerosis. Brain Pathol. 2008, 18, 86–95. [Google Scholar] [CrossRef]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef]
- Lassmann, H. Models of multiple sclerosis: New insights into pathophysiology and repair. Curr. Opin. Neurol. 2008, 21, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Bjartmar, C.; Wujek, J.R.; Trapp, B.D. Axonal loss in the pathology of MS: Consequences for understanding the progressive phase of the disease. J. Neurol. Sci. 2003, 206, 165–171. [Google Scholar] [CrossRef]
- Bruck, W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J. Neurol. 2005, 252 (Suppl. 5), v3–v9. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.; Lucchinetti, C.F. Pathology of demyelinating diseases. Annu. Rev. Pathol. 2012, 7, 185–217. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.T.; Wimmer, I.; Höftberger, R.; Gerlach, S.; Haider, L.; Zrzavy, T.; Hametner, S.; Mahad, D.; Binder, C.J.; Krumbholz, M.; et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain 2013, 136, 1799–1815. [Google Scholar] [CrossRef]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef]
- Scalfari, A.; Neuhaus, A.; Daumer, M.; Deluca, G.C.; Muraro, P.A.; Ebers, G.C. Early relapses, onset of progression, and late outcome in multiple sclerosis. JAMA Neurol. 2013, 70, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Barkhof, F.; Calabresi, P.A.; Miller, D.H.; Reingold, S.C. Imaging outcomes for neuroprotection and repair in multiple sclerosis trials. Nat. Rev. Neurol. 2009, 5, 256–266. [Google Scholar] [CrossRef]
- Chard, D.T.; Griffin, C.M.; Parker, G.J.; Kapoor, R.; Thompson, A.J.; Miller, D.H. Brain atrophy in clinically early relapsing-remitting multiple sclerosis. Brain 2002, 125, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Barkhof, F.; Frank, J.A.; Parker, G.J.; Thompson, A.J. Measurement of atrophy in multiple sclerosis: Pathological basis, methodological aspects and clinical relevance. Brain 2002, 125, 1676–1695. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, G.C.; Ebers, G.C.; Esiri, M.M. Axonal loss in multiple sclerosis: A pathological survey of the corticospinal and sensory tracts. Brain 2004, 127, 1009–1018. [Google Scholar] [CrossRef]
- Bitsch, A.; Schuchardt, J.; Bunkowski, S.; Kuhlmann, T.; Bruck, W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain 2000, 123 Pt 6, 1174–1183. [Google Scholar] [CrossRef]
- DeLuca, G.C.; Williams, K.; Evangelou, N.; Ebers, G.C.; Esiri, M.M. The contribution of demyelination to axonal loss in multiple sclerosis. Brain 2006, 129 Pt 6, 1507–1516. [Google Scholar] [CrossRef]
- Peterson, J.W.; Bo, L.; Mork, S.; Chang, A.; Trapp, B.D. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 2001, 50, 389–400. [Google Scholar] [CrossRef]
- Heidker, R.M.; Emerson, M.R.; LeVine, S.M. Metabolic pathways as possible therapeutic targets for progressive multiple sclerosis. Neural Regen. Res. 2017, 12, 1262–1267. [Google Scholar]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Patergnani, S.; Fossati, V.; Bonora, M.; Giorgi, C.; Marchi, S.; Missiroli, S.; Rusielewicz, T.; Wieckowski, M.R.; Pinton, P. Mitochondria in Multiple Sclerosis: Molecular Mechanisms of Pathogenesis. Int. Rev. Cell Mol. Biol. 2017, 328, 49–103. [Google Scholar] [PubMed]
- Schoenfeld, R.; Wong, A.; Silva, J.; Li, M.; Itoh, A.; Horiuchi, M.; Itoh, T.; Pleasure, D.; Cortopassi, G. Oligodendroglial differentiation induces mitochondrial genes and inhibition of mitochondrial function represses oligodendroglial differentiation. Mitochondrion 2010, 10, 143–150. [Google Scholar] [CrossRef]
- French, H.M.; Reid, M.; Mamontov, P.; Simmons, R.A.; Grinspan, J.B. Oxidative stress disrupts oligodendrocyte maturation. J. Neurosci. Res. 2009, 87, 3076–3087. [Google Scholar] [CrossRef]
- Madsen, P.M.; Pinto, M.; Patel, S.; McCarthy, S.; Gao, H.; Taherian, M.; Karmally, S.; Pereira, C.V.; Dvoriantchikova, G.; Ivanov, D.; et al. Mitochondrial DNA Double-Strand Breaks in Oligodendrocytes Cause Demyelination, Axonal Injury, and CNS Inflammatio. J. Neurosci., vol. 2017, 37, 10185–10199. [Google Scholar] [CrossRef]
- Li, S.; Clements, R.; Sulak, M.; Gregory, R.; Freeman, E.; McDonough, J. Decreased NAA in gray matter is correlated with decreased availability of acetate in white matter in postmortem multiple sclerosis cortex. Neurochem. Res. 2013, 38, 2385–2396. [Google Scholar] [CrossRef]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Nijland, P.G.; Drexhage, J.A.; Gerritsen, W.; Geerts, D.; van Het Hof, B.; Reijerkerk, A.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Reduced expression of PGC-1alpha partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta Neuropathol. 2013, 125, 231–243. [Google Scholar] [CrossRef]
- Pandit, A.; Vadnal, J.; Houston, S.; Freeman, E.; McDonough, J. Impaired regulation of electron transport chain subunit genes by nuclear respiratory factor 2 in multiple sclerosis. J. Neurol. Sci. 2009, 279, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Bø, L.; Rodenburg, R.J.; Belien, J.A.; Musters, R.; Hazes, T.; Wintjes, L.T.; Smeitink, J.A.; Geurts, J.J.; De Vries, H.E.; et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J. Pathol. 2009, 219, 193–204. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lee, P.; Adany, P.; Hughes, A.J.; Belliston, S.; Denney, D.R.; Lynch, S.G. In vivo evidence of oxidative stress in brains of patients with progressive multiple sclerosis. Mult. Scler. 2018, 24, 1029–1038. [Google Scholar] [CrossRef]
- Broadwater, L.; Pandit, A.; Clements, R.; Azzam, S.; Vadnal, J.; Sulak, M.; Yong, V.W.; Freeman, E.J.; Gregory, R.B.; McDonough, J. Analysis of the mitochondrial proteome in multiple sclerosis cortex. Biochim. Biophys. Acta 2011, 1812, 630–641. [Google Scholar] [CrossRef]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Zambonin, J.L.; Zhao, C.; Ohno, N.; Campbell, G.R.; Engeham, S.; Ziabreva, I.; Schwarz, N.; Lee, S.E.; Frischer, J.M.; Turnbull, D.M.; et al. Increased mitochondrial content in remyelinated axons: Implications for multiple sclerosis. Brain 2011, 134, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef]
- Hampton, D.W.; Serio, A.; Pryce, G.; Al-Izki, S.; Franklin, R.J.; Giovannoni, G.; Baker, D.; Chandran, S. Neurodegeneration progresses despite complete elimination of clinical relapses in a mouse model of multiple sclerosis. Acta Neuropathol. Commun. 2013, 1, 84. [Google Scholar] [CrossRef] [PubMed]
- Recks, M.S.; Stormanns, E.R.; Bader, J.; Arnhold, S.; Addicks, K.; Kuerten, S. Early axonal damage and progressive myelin pathology define the kinetics of CNS histopathology in a mouse model of multiple sclerosis. Clin. Immunol. 2013, 149, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Tao, T.; Yan, W.; Chen, C.S.; Qin, X. Curcumin inhibits mitochondrial injury and apoptosis from the early stage in EAE mice. Oxid. Med. Cell. Longev. 2014, 2014, 728751. [Google Scholar] [CrossRef]
- Sadeghian, M.; Mastrolia, V.; Rezaei Haddad, A.; Mosley, A.; Mullali, G.; Schiza, D.; Sajic, M.; Hargreaves, I.; Heales, S.; Duchen, M.R.; et al. Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci. Rep. 2016, 6, 33249. [Google Scholar] [CrossRef]
- Talla, V.; Yu, H.; Chou, T.H.; Porciatti, V.; Chiodo, V.; Boye, S.L.; Hauswirth, W.W.; Lewin, A.S.; Guy, J. NADH-dehydrogenase type-2 suppresses irreversible visual loss and neurodegeneration in the EAE animal model of MS. Mol. Ther. 2013, 21, 1876–1888. [Google Scholar] [CrossRef]
- Talla, V.; Koilkonda, R.; Porciatti, V.; Chiodo, V.; Boye, S.L.; Hauswirth, W.W.; Guy, J. Complex I subunit gene therapy with NDUFA6 ameliorates neurodegeneration in EAE. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1129–1140. [Google Scholar] [CrossRef]
- Fetisova, E.; Chernyak, B.; Korshunova, G.; Muntyan, M.; Skulachev, V. Mitochondria-targeted Antioxidants as a Prospective Therapeutic Strategy for Multiple Sclerosis. Curr. Med. Chem. 2017, 24, 2086–2114. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Manczak, M.; Shirendeb, U.P.; Reddy, P.H. MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim. Biophys. Acta. 2013, 1832, 2322–2331. [Google Scholar] [CrossRef]
- Andrews, H.; White, K.; Thomson, C.; Edgar, J.; Bates, D.; Griffiths, I.; Turnbull, D.; Nichols, P. Increased axonal mitochondrial activity as an adaptation to myelin deficiency in the Shiverer mouse. J. Neurosci. Res. 2006, 83, 1533–1539. [Google Scholar] [CrossRef]
- Procaccini, C.; de Rosa, V.; Pucino, V.; Formisano, L.; Matarese, G. Animal models of Multiple Sclerosis. Eur. J. Pharmacol. 2015, 759, 182–191. [Google Scholar] [CrossRef]
- Warford, J.; Robertson, G.S. New methods for multiple sclerosis drug discovery. Expert Opin. Drug Discov. 2011, 6, 689–699. [Google Scholar] [CrossRef]
- Furlan, R.; Poliani, P.L.; Marconi, P.C.; Bergami, A.; Ruffini, F.; Adorini, L.; Glorioso, J.C.; Comi, G.; Martino, G. Central nervous system gene therapy with interleukin-4 inhibits progression of ongoing relapsing-remitting autoimmune encephalomyelitis in Biozzi AB/H mice. Gene Ther. 2001, 8, 13–19. [Google Scholar] [CrossRef][Green Version]
- Al-Izki, S.; Pryce, G.; O’Neill, J.K.; Butter, C.; Giovannoni, G.; Amor, S.; Baker, D. Practical guide to the induction of relapsing progressive experimental autoimmune encephalomyelitis in the Biozzi ABH mouse. Mult. Scler. Relat. Disord. 2012, 1, 29–38. [Google Scholar] [CrossRef]
- Praet, J.; Guglielmetti, C.; Berneman, Z.; van der Linden, A.; Ponsaerts, P. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014, 47, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Heppner, F.L.; Becher, B. Modeling multiple sclerosis in laboratory animals. Semin. Immunopathol. 2009, 31, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Hemm, R.D.; Carlton, W.W.; Welser, J.R. Ultrastructural changes of cuprizone encephalopathy in mice. Toxicol. Appl. Pharmacol. 1971, 18, 869–882. [Google Scholar] [CrossRef]
- Bénardais, K.; Kotsiari, A.; Skuljec, J.; Koutsoudaki, P.N.; Gudi, V.; Singh, V.; Vulinović, F.; Skripuletz, T.; Stangel, M. Cuprizone [bis(cyclohexylidenehydrazide)] is selectively toxic for mature oligodendrocytes. Neurotox. Res. 2013, 24, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow Metab. 2012, 32, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, T.; Metz, I.; Dallenga, T.; König, F.B.; Müller, S.; Stadelmann, C.; Brück, W. Wallerian degeneration: A major component of early axonal pathology in multiple sclerosis. Brain Pathol. 2010, 20, 976–985. [Google Scholar] [CrossRef]
- Campbell, G.; Mahad, D.J. Mitochondrial dysfunction and axon degeneration in progressive multiple sclerosis. FEBS Lett. 2018, 592, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Kiryu-Seo, S.; Ohno, N.; Kidd, G.J.; Komuro, H.; Trapp, B.D. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J. Neurosci. 2010, 30, 6658–6666. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, S.; Stys, P.K. Metabolic injury to axons and myelin. Exp. Neurol. 2013, 246, 26–34. [Google Scholar] [CrossRef]
- Witte, M.E.; Mahad, D.J.; Lassmann, H.; van Horssen, J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 2014, 20, 179–187. [Google Scholar] [CrossRef]
- Willer, C.J.; Dyment, D.A.; Risch, N.J.; Sadovnick, A.D.; Ebers, G.C. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 12877–12882. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, J.A.; Oksenberg, J.R. The immunogenetics of multiple sclerosis: A comprehensive review. J. Autoimmun. 2015, 64, 13–25. [Google Scholar] [CrossRef]
- Yu, X.; Koczan, D.; Sulonen, A.M.; Akkad, D.A.; Kroner, A.; Comabella, M.; Costa, G.; Corongiu, D.; Goertsches, R.; Camina-Tato, M.; et al. mtDNA nt13708A variant increases the risk of multiple sclerosis. PLoS ONE 2008, 3, e1530. [Google Scholar] [CrossRef]
- Andalib, S.; Emamhadi, M.; Yousefzadeh-Chabok, S.; Salari, A.; Sigaroudi, A.E.; Vafaee, M.S. MtDNA T4216C variation in multiple sclerosis: A systematic review and meta-analysis. Acta Neurol. Belg. 2016, 116, 439–443. [Google Scholar] [CrossRef]
- Tranah, G.J.; Santaniello, A.; Caillier, S.J.; D’Alfonso, S.; Martinelli Boneschi, F.; Hauser, S.L.; Oksenberg, J.R. Mitochondrial DNA sequence variation in multiple sclerosis. Neurology 2015, 85, 325–330. [Google Scholar] [CrossRef]
- Falabella, M.; Forte, E.; Magnifico, M.C.; Santini, P.; Arese, M.; Giuffrè, A.; Radić, K.; Chessa, L.; Coarelli, G.; Buscarinu, M.C.; et al. Evidence for Detrimental Cross Interactions between Reactive Oxygen and Nitrogen Species in Leber’s Hereditary Optic Neuropathy Cells. Oxid. Med. Cell. Longev. 2016, 2016, 3187560. [Google Scholar] [CrossRef]
- Howell, N. Leber hereditary optic neuropathy: Mitochondrial mutations and degeneration of the optic nerve. Vision Res. 1997, 37, 3495–3507. [Google Scholar] [CrossRef]
- Harding, A.E.; Sweeney, M.G.; Miller, D.H.; Mumford, C.J.; Kellar-Wood, H.; Menard, D.; McDonald, W.I.; Compston, D.A. Occurrence of a multiple sclerosis-like illness in women who have a Leber’s hereditary optic neuropathy mitochondrial DNA mutation. Brain 1992, 115 Pt 4, 979–989. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef]
- Palace, J. Multiple sclerosis associated with Leber’s Hereditary Optic Neuropathy. J. Neurol. Sci. 2009, 286, 24–27. [Google Scholar] [CrossRef]
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Karbowski, M.; Youle, R.J.; Schimpf, S.; Wissinger, B.; Pinti, M.; Cossarizza, A.; Vidoni, S.; et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2008, 131, 352–367. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Chassagne, M.; de Sèze, J.; Mohr, M.; Clerc-Renaud, P.; Tranchant, C.; Mousson de Camaret, B. POLG1 variations presenting as multiple sclerosis. Arch. Neurol. 2010, 67, 1140–1143. [Google Scholar] [CrossRef]
- Spain, R.; Powers, K.; Murchison, C.; Heriza, E.; Winges, K.; Yadav, V.; Cameron, M.; Kim, E.; Horak, F.; Simon, J.; et al. Lipoic acid in secondary progressive MS: A randomized controlled pilot trial. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e374. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Pluchino, S. Targeting Mitochondrial Metabolism in Neuroinflammation: Towards a Therapy for Progressive Multiple Sclerosis. Trends Mol. Med. 2018, 24, 838–855. [Google Scholar] [CrossRef] [PubMed]
- Tourbah, A.; Lebrun-Frenay, C.; Edan, G.; Clanet, M.; Papeix, C.; Vukusic, S.; De Sèze, J.; Debouverie, M.; Gout, O.; Clavelou, P.; et al. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: A randomised, double-blind, placebo-controlled study. Mult. Scler. 2016, 22, 1719–1731. [Google Scholar] [CrossRef]
- Jang, Y.-H.; Lim, K.-I. Recent Advances in Mitochondria-Targeted Gene Delivery. Molecules 2018, 23, 2316. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Zarrouk-Mahjoub, S. Re: Guy et al.: Gene therapy for Leber hereditary optic neuropathy: Low-and medium-dose visual results (Ophthalmology. 2017; 124:1621–1634). Ophthalmology 2018, 125, e14–e15. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| MS Phenotype | Tissue | Cell Type | Mitochondria Pathology | Reference |
|---|---|---|---|---|
| 1 PP 9 SP 8 C | Motor cortex | Neurons | —decreased expression of mitochondrial nuclear gene DNA —functionally reduced complex I and III activities | Dutta, R. et al. 2006 [52] |
| 1 PP 9 SP 6 C | Chronic inactive lesions | Demyelinated axons | —increased total mitochondrial content and complex IV activity | Mahad, D.J. et al. 2009 [65] |
| 8 SP 5 C | Grey matter in Cortex | NCD | —epigenetic changes affected by ROS, through the reduced capacity of NRF-2 (a transcription factor for ETC proteins) | Pandit, A. et al. 2009 [61] |
| 5 PP 14 SP 7 ND 7 C | Active and chronic lesions | NCD | —increase in the levels of a heat shock protein (mtHSP70), a marker of mitochondrial stress —an increase in the number of mitochondria and in the translation of mitochondrial proteins | Witte, M.E. et al. 2009 [62] |
| 13 SP 10 C | NCD | Neurons | —accumulation of large mtDNA deletions, with some showing specific deletion in the subunits of complex IV | Campbell, G.R. et al. 2011 [59] |
| 2 PP 7 SP 1 RC | NCD | Acute and chronic demyelinated axons | —increased mitochondrial content and complex IV activity compared with remyelinating and myelinated axons | Zambonin, J.L. et al. 2011 [66] |
| 8 SP 8 C | NCD | NCD | —different patterns of mass spectrometry in human cytochrome c oxidase subunit 5b (COX5b), the brain-specific creatine kinase isoform, and the β-chain of hemoglobin | Broadwater, L. et al. 2011 [64] |
| 7 PP 7 SP 1 ND 9 C | NCD | Pyramidal neurons | —decrease in PGC-1α levels, OXPHOS subunits, antioxidants and uncoupling proteins 4 and 5 | Witte, M.E. et al. 2013 [60] |
| 20 PP 20 SP vs 21 RR | NCD | NCD | —decreased levels of glutathione (GSH), a potent antioxidant, signaling that oxidative stress more strongly affects the neurodegeneration phase than the neuroinflammation one | Choi, Y. et al. 2018 [63] |
| MS Animal Model | Type of MS Modeled | Indication for Research | Mitochondrial Findings |
|---|---|---|---|
| EAE-SJL/J mice -C57BL/6J mice -Biozzi chronic EAE | -RR -PP and SP -RR -> SP | Understanding of the neuroinflammatory process after immunologic activation of the mice (SJL/J with PLP or MBP and C57BL/6J with MOG) [77,78]. Accumulative damage of neuroinflammation with secondary progression of the disease [68,79,80]. | C57BL/6′s mitochondria morphology changes (swelling) [69], early mitochondrial dysfunction in EAE disease [70] and impairment of mitochondrial and axonal depolarization [71]. C57Bl/6 model did not reproduce the cortex respiratory protein’s alterations seen in MS patients [64]. |
| TCR transgenic mice | -RR [78] | Understanding spontaneous neuroinflammatory process after immunologic activation [77]. | - |
| TMEV | Demyelination and axonal damage | Infection mediated by Picornavirus inducing an encephalomyelitis (whole neuroaxis) [77]. | - |
| Toxin-induced demyelination (Cuprizone, Lysolecithin, Ethidium bromide) | Demyelination and remyelination | Reproducible onset of demyelination and start of remyelination after interruption of toxic exposure. If chronic exposure of cuprizone also possible to see impairment of remyelination [81]. | Cuprizone is a copper chelator an essential component of COX [82]. Mice’s brain treated with cuprizone presented “giant” mitochondria in oligodendroglial cells [83]. Oligodendrocytes treated with cuprizone presented with decreased mitochondrial potential (in vitro) [84]. |
| Disease | Gene Mutation | MS Overlap | Overlap in Potential Mechanism |
|---|---|---|---|
| MS | mtDNA nt13708A mtDNA T4216C nt 11778 (G→A) | NA | NA |
| LHON | nt 3460 nt 11778 (G→A) nt 14484 | 5% LHON have evidence of demyelinating lesion | Degeneration of optic nerve |
| DOA | over 90 gene mutations | OPA1 protein: known link to DOA, implicated in 3 patients with MS-like disease | OPA1 mutation and truncated protein |
| POLG1 | Not specified | Linked to cases of demyelination | Not specified |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barcelos, I.P.d.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. https://doi.org/10.3390/biology8020037
Barcelos IPd, Troxell RM, Graves JS. Mitochondrial Dysfunction and Multiple Sclerosis. Biology. 2019; 8(2):37. https://doi.org/10.3390/biology8020037
Chicago/Turabian StyleBarcelos, Isabella Peixoto de, Regina M. Troxell, and Jennifer S. Graves. 2019. "Mitochondrial Dysfunction and Multiple Sclerosis" Biology 8, no. 2: 37. https://doi.org/10.3390/biology8020037
APA StyleBarcelos, I. P. d., Troxell, R. M., & Graves, J. S. (2019). Mitochondrial Dysfunction and Multiple Sclerosis. Biology, 8(2), 37. https://doi.org/10.3390/biology8020037