Mitochondrial Dysfunction in the Aging Retina


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
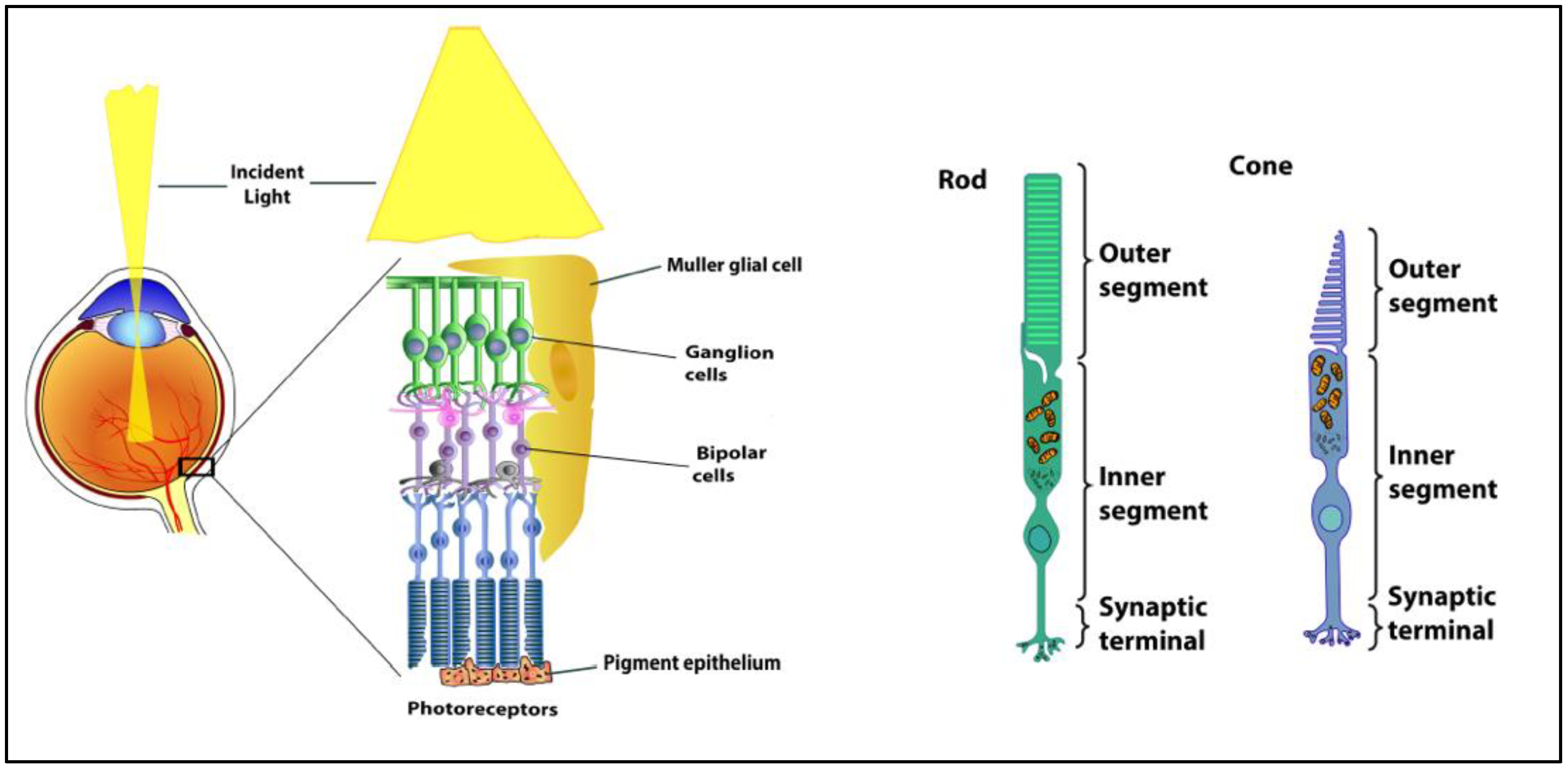
1. Retinal Anatomy and Physiology
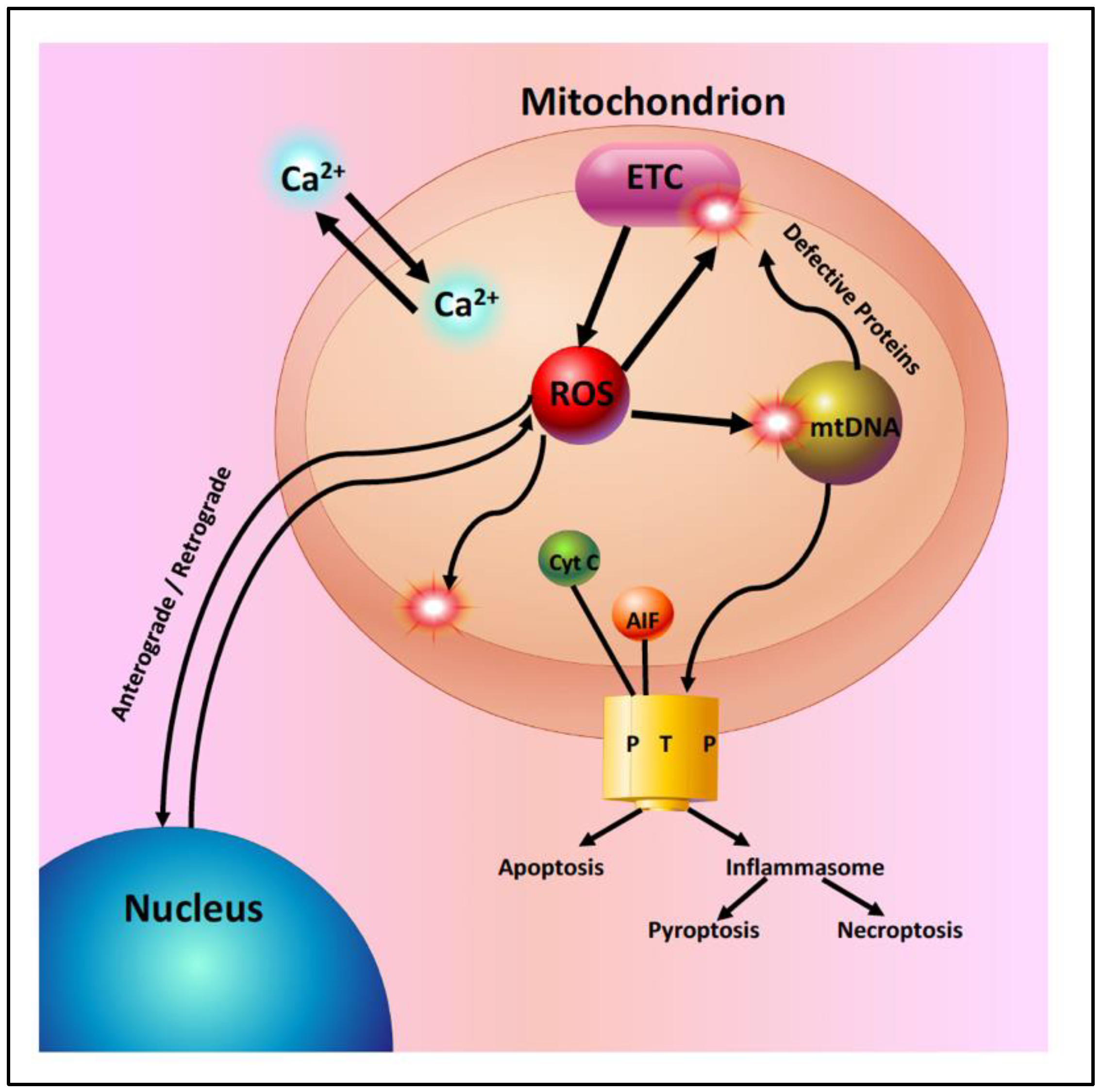
2. Mitochondrial Dysfunction with Aging and Disease
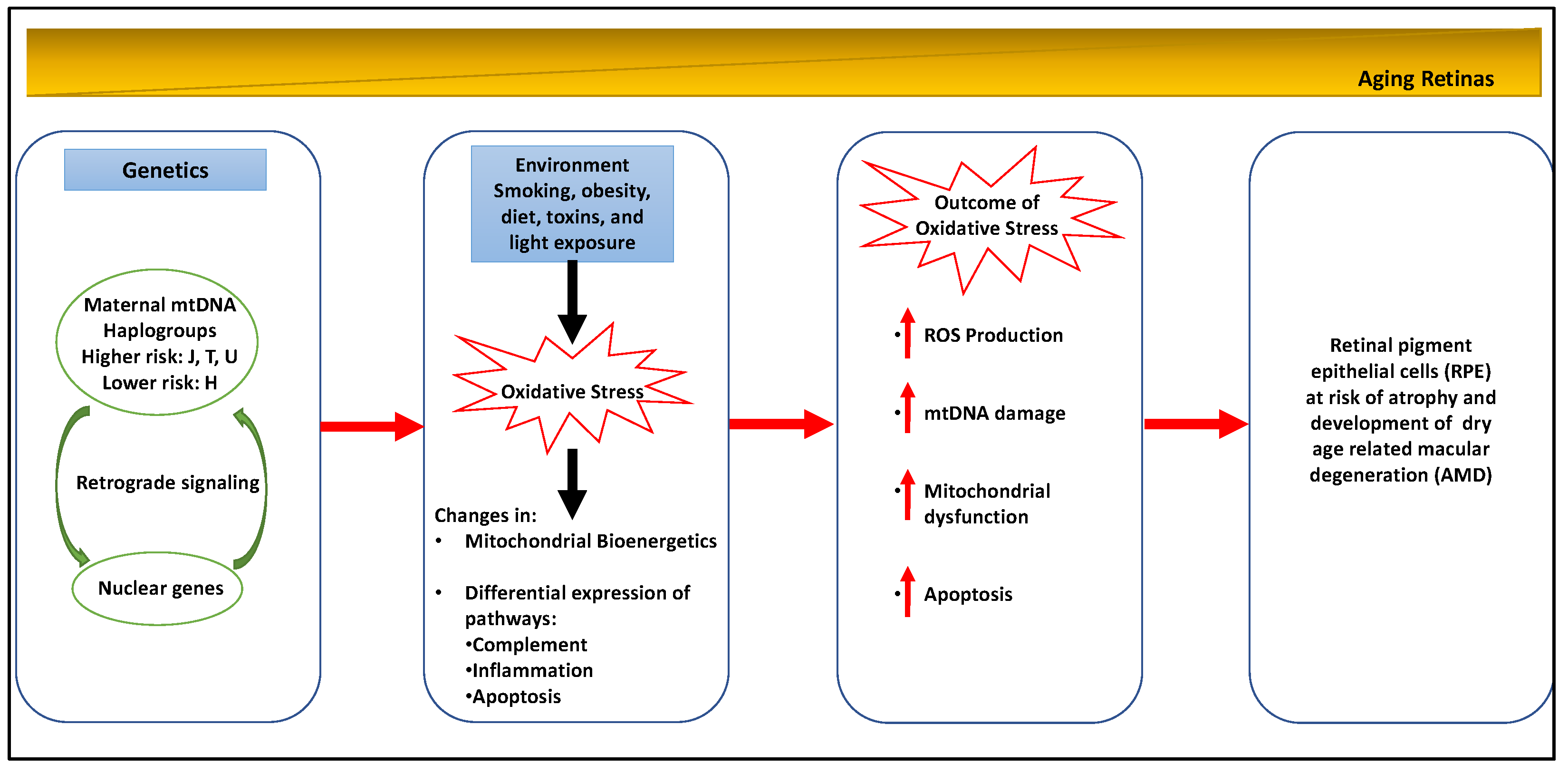
2.1. Age-Related Mitochondrial Changes
2.2. Age-Related Macular Degeneration
2.3. Diabetic Retinopathy
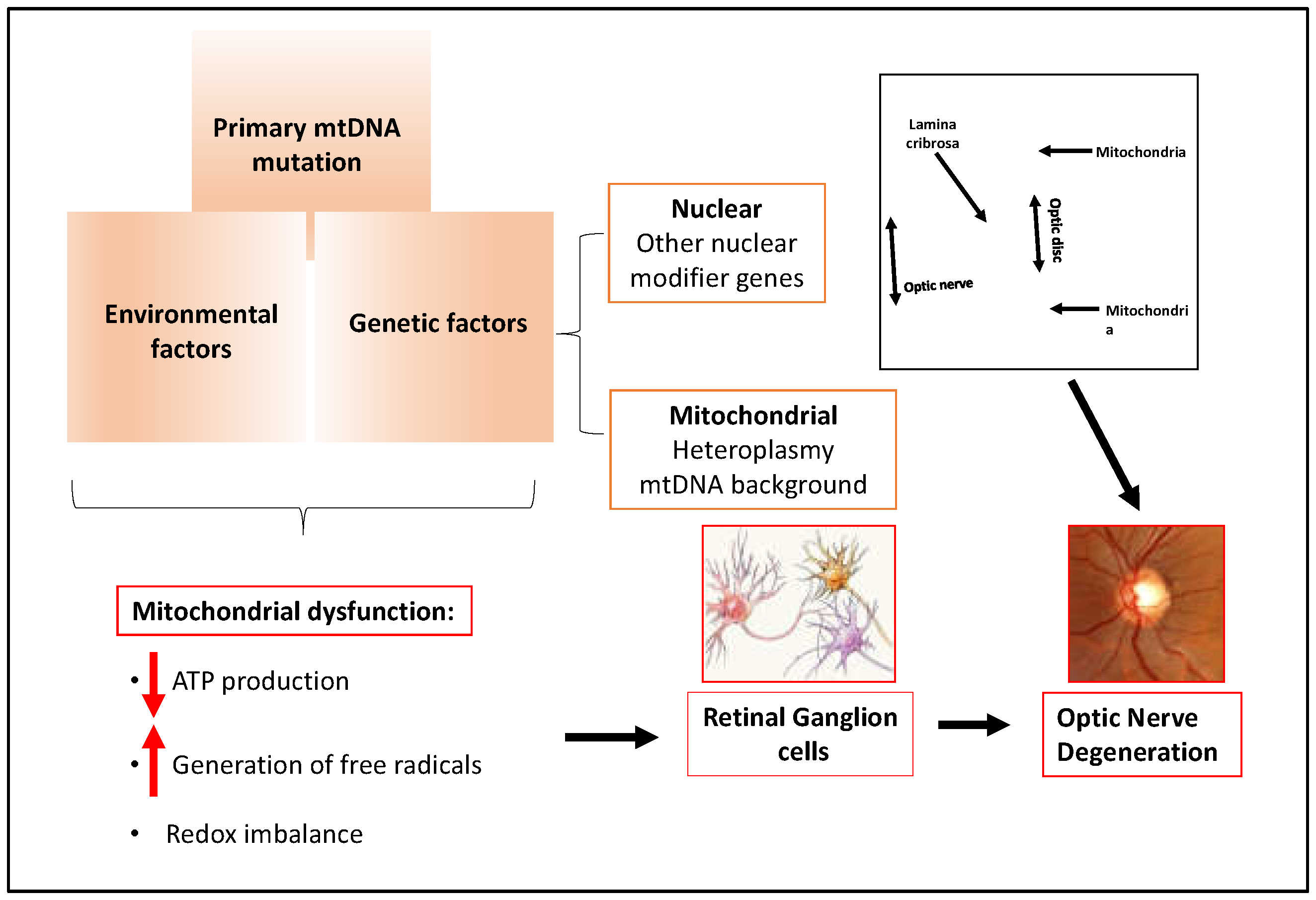
2.4. Glaucoma
3. Conclusions
Funding
Conflicts of Interest
References
- Sung, C.H.; Chuang, J.Z. The cell biology of vision. J. Cell Biol. 2010, 190, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S. Glycolytic and oxidative metabolism in relation to retinal function. J. Gen. Physiol. 1981, 77, 667–692. [Google Scholar] [CrossRef]
- Warbug, O. Uber die klassifizierung tierischer gewebe nach ihrem stoffwechsel. Biochem. Z. 1927, 184, 484–488. [Google Scholar]
- Ames, A., 3rd; Li, Y.Y.; Heher, E.C.; Kimble, C.R. Energy metabolism of rabbit retina as related to function: High cost of sodium transport. J. Neurosci. 1992, 12, 840–853. [Google Scholar] [CrossRef]
- Medrano, C.J.; Fox, D.A. Oxygen consumption in the rat outer and inner retina: Light- and pharmacologically-induced inhibition. Exp. Eye Res. 1995, 61, 273–284. [Google Scholar] [CrossRef]
- Hurley, J.B.; Lindsay, K.J.; Du, J. Glucose, lactate and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751. [Google Scholar] [CrossRef]
- Barot, M.; Gokulgandhi, M.R.; Mitra, A.K. Mitochondrial dysfunction in retinal diseases. Curr. Eye Res. 2011, 36, 1069–1077. [Google Scholar] [CrossRef]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Neufeld, A.H. Age-related increase in mitochondrial DNA damage and loss of DNA repair capacity in the neural retina. Neurobiol. Aging 2010, 31, 2002–2010. [Google Scholar] [CrossRef]
- Barron, M.J.; Johnson, M.A.; Andrews, R.M.; Clarke, M.P.; Griffiths, P.G.; Bristow, E.; He, L.-P.; Durham, S.; Turnbull, D.M. Mitochondrial abnormalities in aging macular photoreceptors. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3016–3022. [Google Scholar]
- Country, M.W. Retinal metabolism: A comparative look as energetics in the retina. Brain Res. 2017, 1672, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Lefevere, E.; Toft-Kehler, A.K.; Vohra, R.; Kolko, M.; Moons, L.; van Hovea, I. Mitochondrial dysfunction underlying outer retinal diseases. Mitochondrion 2017, 36, 66–76. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Lin, H.; Godley, B.F.; Boultonc, M.E. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res. 2008, 27, 596–607. [Google Scholar] [CrossRef]
- Kennedy, S.R.; Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013, 9, e1003794. [Google Scholar] [CrossRef]
- Ameur, A.; Stewart, J.B.; Freyer, C.; Hagström, E.; Ingman, M.; Larsson, N.-G.; Gyllensten, U. Ultra-deep sequencing of mouse mitochondrial DNA: Mutational patterns and their origins. PLoS Genet. 2011, 7, e1002028. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Gabrieli, C.B. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD pathobiology: A bioenergetic crisis in the RPE. Investig. Ophthalmol. Vis. Sci. 2018, 59, 41–55. [Google Scholar] [CrossRef]
- Turluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef]
- King, A.; Gottlieb, E.; Brooks, D.G.; Murphy, M.P.; Dunaief, J.L. Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochem. Photobiol. 2004, 79, 470475. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, H.; Yu, A.; Xie, J. Association between sunlight exposure and risk of age-related macular degeneration: A meta-analysis. BMC Ophthalmol. 2018, 18, 331–338. [Google Scholar] [CrossRef]
- Sivapathasuntharam, C.; Hayes, M.J.; Shinhmar, H.; Kam, J.H.; Sivaprasad, S.; Jeffery, G. Complement factor H regulates retinal development and its absence may establish a footprint for age related macular degeneration. Sci. Rep. 2019, 9, 1082–1092. [Google Scholar] [CrossRef]
- Calaza, K.C.; Kam, J.H.; Hogg, C.; Jeffery, G. Mitochondrial decline precedes phenotype development in the complement factor H mouse model of retinal degeneration but can be corrected by near infrared light. Neurobiol. Aging 2015, 36, 2869–2876. [Google Scholar] [CrossRef]
- Lee, W.-H.; Higuchi, H.; Ikeda, S.; Macke, E.L.; Takimoto, T.; Pattnaik, B.R.; Liu, C.; Chu, L.-F.; Siepka, S.M.; Krentz, K.J.; et al. Mouse Tmem135 mutation reveals a mechanism involving mitochondrial dynamics that leads to age-dependent retinal pathologies. eLife 2016, 5, e19264. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Mechanisms of disease diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications; a unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mirsha, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef]
- Roy, S.; Kern, T.S.; Song, B.; Stuebe, C. Mechanistic insights into pathological changes in the diabetic retina: Implications for targeting diabetic retinopathy. Am. J. Pathol. 2017, 187, 9–19. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Goncalves, R.L.S.; Hey-Mogensen, M.; Yadava, N.; Bunik, V.I.; Brand, M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 2014, 289, 8312–8325. [Google Scholar] [CrossRef]
- Kowluru, R.A. Diabetic retinopathy, metabolic memory and epigenetic modifications. Vis. Res. 2017, 139, 30–38. [Google Scholar] [CrossRef]
- Kowluru, R.A. Mitochondria damage in the pathogenesis of diabetic retinopathy and in the metabolic memory associated with its continued progression. Curr. Med. Chem. 2013, 20, 3226–3233. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Abbas, S.N. Diabetes-induced mitochondrial dysfunction in the retina. Investig. Ophthalmol. Vis. Sci. 2003, 12, 5327–5334. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Liu, Q.; Kim, K.-Y.; Crowston, J.G.; Lindsey, J.D.; Agarwal, N.; Ellisman, M.H.; Perkins, G.A.; Weinreb, R.N. Elevated hydrostatic pressure triggers mitochondrial fission and decreases cellular ATP in differentiated RGC-5 cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2145–2151. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2533–2541. [Google Scholar] [CrossRef]
- Moreno, M.C.; Campanelli, J.; Sande, P.; Sáenz, D.A.; Sarmiento, M.I.K.; Rosenstein, R.E. Retinal oxidative stress induced by high intraocular pressure. Free Radic. Biol. Med. 2004, 37, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.Y.; Van Bergen, N.J.; Trounce, I.A.; Crowston, J.G. Mitochondrial dysfunction and glaucoma. J. Glaucoma 2009, 18, 93–100. [Google Scholar] [CrossRef]
- Harun-or-Rashid, M.; Pappenhagen, N.; Palmer, P.G.; Smith, M.A.; Gevorgyan, V.; Wilson, G.N.; Crish, S.D.; Inman, D.M. Structural and functional rescue of chronic metabolically stressed optic nerves through respiration. J. Neurosci. 2018, 38, 5122–5139. [Google Scholar] [CrossRef] [PubMed]
- Libby, R.T.; Anderson, M.G.; Pang, I.H.; Robinson, Z.H.; Savinova, O.V.; Cosma, I.M.; Snow, A.; Wilson, L.A.; Smith, R.S.; Clark, A.F.; et al. Inherited glaucoma in DBA/2J mice: Pertinent disease features for studying the neurodegeneration. Vis. Neurosci. 2005, 22, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Baltan, S.; Inman, D.M.; Danilov, C.A.; Morrison, R.S.; Calkins, D.J.; Horner, P.J. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J. Neurosci. 2010, 30, 5644–5652. [Google Scholar] [CrossRef]
- Kleesattel, D.; Crish, S.D.; Inman, D.M. Decreased energy capacity and increased autophagic activity in optic nerve axons with defective anterograde transport. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8215–8227. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.-K.; Kim, K.-Y.; Lindsey, J.D.; Angert, M.; Duong-Polk, K.X.; Scott, R.T.; Kim, J.J.; Kukhmazov, I.; Ellisman, M.H.; Perkins, G.A.; et al. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4903–4911. [Google Scholar] [CrossRef]
- Coughlin, L.; Morrison, R.S.; Horner, P.J.; Inman, D.M. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1437–1446. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W.M. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 760, 756–760. [Google Scholar] [CrossRef]
- Ju, W.K.; Kim, K.Y.; Noh, Y.H.; Hoshijima, M.; Lukas, T.J.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A. Increased mitochondrial fission and volume density by blocking glutamate excitotoxicity protect glaucomatous optic nerve head astrocytes. Glia 2015, 63, 736–753. [Google Scholar] [CrossRef]
- Lee, S. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef]




© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eells, J.T. Mitochondrial Dysfunction in the Aging Retina. Biology 2019, 8, 31. https://doi.org/10.3390/biology8020031
Eells JT. Mitochondrial Dysfunction in the Aging Retina. Biology. 2019; 8(2):31. https://doi.org/10.3390/biology8020031
Chicago/Turabian StyleEells, Janis T. 2019. "Mitochondrial Dysfunction in the Aging Retina" Biology 8, no. 2: 31. https://doi.org/10.3390/biology8020031
APA StyleEells, J. T. (2019). Mitochondrial Dysfunction in the Aging Retina. Biology, 8(2), 31. https://doi.org/10.3390/biology8020031