Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions

Abstract
1. Introduction
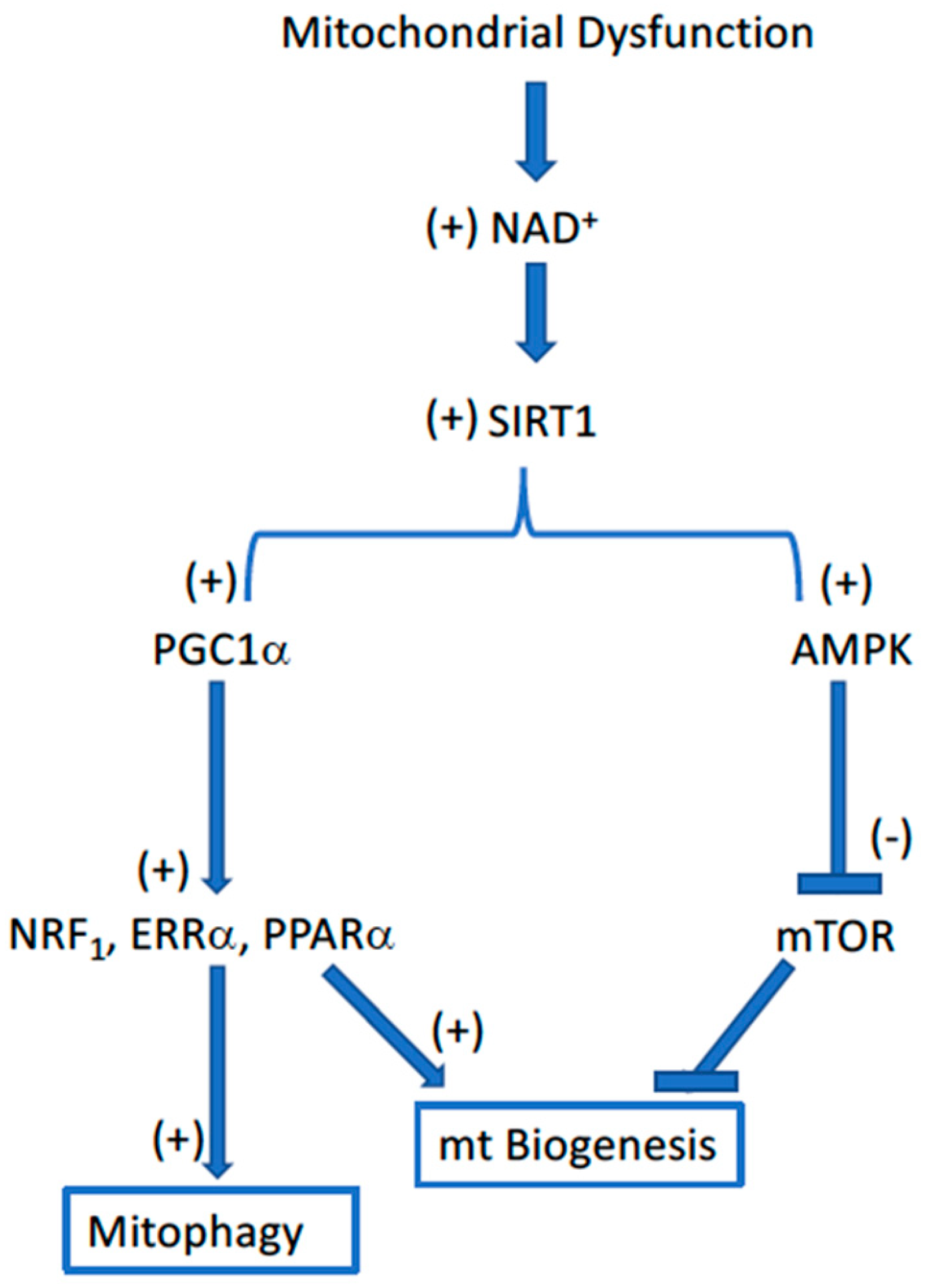
2. The Mitochondrial Theory of Aging
3. Drug-Induced Mitochondrial Toxicity
4. Polypharmacy in the Geriatric Population
5. Conclusions
Funding
Conflicts of Interest
References
- Dimauro, S.; Davidzon, G. Mitochondrial DNA and disease. Ann. Med. 2005, 37, 222–232. [Google Scholar] [CrossRef]
- Blake, R.; Trounce, I.A. Mitochondrial dysfunction and complications associated with diabetes. Biochim. Biophys. Acta 2014, 1840, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in non-alcoholic fatty liver disease progression. J. Lipid Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Lesnefsky, E.J.; Chen, Q.; Tandler, B. Mitochondrial Dysfunction in Cardiovascular Aging. Adv. Exp. Med. Biol. 2017, 982, 451–464. [Google Scholar] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Anupama, N.; Sindhu, G.; Raghu, K.G. Significance of mitochondria on cardiometabolic syndromes. Fundam. Clin. Pharmacol. 2018, 32, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef]
- Falah, M.; Houshmand, M.; Najafi, M.; Balali, M.; Mahmoudian, S.; Asghari, A.; Emamdjomeh, H.; Farhadi, M. The potential role for use of mitochondrial DNA copy number as predictive biomarker in presbycusis. Ther. Clin. Risk Manag. 2016, 12, 1573–1578. [Google Scholar] [CrossRef]
- Leruez, S.; Marill, A.; Bresson, T.; de Saint Martin, G.; Buisset, A.; Muller, J.; Tessier, L.; Gadras, C.; Verny, C.; Gohier, P.; et al. A Metabolomics Profiling of Glaucoma Points to Mitochondrial Dysfunction, Senescence, and Polyamines Deficiency. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4355–4361. [Google Scholar] [CrossRef]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive Oxygen Species, Aging and Articular Cartilage Homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Ng Kee Kwong, F.; Nicholson, A.G.; Harrison, C.L.; Hansbro, P.M.; Adcock, I.M.; Chung, K.F. Is mitochondrial dysfunction a driving mechanism linking COPD to nonsmall cell lung carcinoma? Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef]
- Zhu, Y.; Dean, A.E.; Horikoshi, N.; Heer, C.; Spitz, D.R.; Gius, D. Emerging evidence for targeting mitochondrial metabolic dysfunction in cancer therapy. J. Clin. Investig. 2018, 128, 3682–3691. [Google Scholar] [CrossRef]
- Always, S.E.; Mohamed, J.S.; Myers, M.J. Mitochondria Initiate and Regulate Sarcopenia. Exerc. Sport Sci. Rev. 2017, 45, 58–69. [Google Scholar] [CrossRef]
- Hara, H.; Kuwano, K.; Araya, J. Mitochondrial Quality Control in COPD and IPF. Cells 2018, 7, 86. [Google Scholar] [CrossRef]
- Raymond, P. The Rate of Living; Johns Hopkins University: New York, NY, USA, 1928. [Google Scholar]
- Lanza, I.R.; Nair, K.S. Mitochondrial function as a determinant of life span. Pflugers Arch. 2010, 459, 277–289. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Conley, K.E.; Jubrias, S.A.; Esselman, P.C. Oxidative capacity and ageing in human muscle. J. Physiol. 2000, 526, 203–210. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Harman, D. The biological clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Harman, D. Free radical theory of aging: Consequences of mitochondrial aging. Age 1983, 6, 86–94. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef]
- Tauchi, H.; Sato, T. Age changes in size and number of mitochondria of human hepatic cells. J. Gerontol. 1968, 23, 454–461. [Google Scholar] [CrossRef]
- Lopez-Lluch, G. Mitochondrial activity and dynamics changes regarding metabolism in ageing and obesity. Mech. Ageing Dev. 2017, 162, 108–121. [Google Scholar] [CrossRef]
- Welle, S.; Bhatt, K.; Shah, B.; Needler, N.; Delehanty, J.M.; Thornton, C.A. Reduced amount of mitochondrial DNA in aged human muscle. J. Appl. Physiol. (1985) 2003, 94, 1479–1484. [Google Scholar] [CrossRef][Green Version]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Lanza, I.R.; Short, D.K.; Short, K.R.; Raghavakaimal, S.; Basu, R.; Joyner, M.J.; McConnell, J.P.; Nair, K.S. Endurance exercise as a countermeasure for aging. Diabetes 2008, 57, 2933–2942. [Google Scholar] [CrossRef]
- Bailey, P.J.; Webster, G.C. Lowered rates of protein synthesis by mitochondria isolated from organisms of increasing age. Mech. Ageing Dev. 1984, 24, 233–241. [Google Scholar] [CrossRef]
- Marcus, D.L.; Ibrahim, N.G.; Freedman, M.L. Age-related decline in the biosynthesis of mitochondrial inner membrane proteins. Exp. Gerontol. 1982, 17, 333–341. [Google Scholar] [CrossRef]
- Rooyackers, O.E.; Adey, D.B.; Ades, P.A.; Nair, K.S. Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc. Natl. Acad. Sci. USA 1996, 93, 15364–15369. [Google Scholar] [CrossRef]
- Rooyackers, O.E.; Kersten, A.H.; Wagenmakers, A.J. Mitochondrial protein content and in vivo synthesis rates in skeletal muscle from critically ill rats. Clin. Sci. (Lond.) 1996, 91, 475–481. [Google Scholar] [CrossRef]
- Mammucari, C.; Rizzuto, R. Signaling pathways in mitochondrial dysfunction and aging. Mech. Ageing Dev. 2010, 131, 536–543. [Google Scholar] [CrossRef]
- Vendelbo, M.H.; Nair, K.S. Mitochondrial longevity pathways. Biochim. Biophys. Acta 2011, 1813, 634–644. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Karin, M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013, 18, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Introduction: Sirtuins in aging and diseases. Methods Mol. Biol. 2013, 1077, 3–10. [Google Scholar] [PubMed]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.A.; Reichert, A.S. Impaired quality control of mitochondria: Aging from a new perspective. Exp. Gerontol. 2010, 45, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Will, Y.; Dykens, J. Mitochondrial toxicity assessment in industry--a decade of technology development and insight. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1061–1067. [Google Scholar] [CrossRef]
- Swiss, R.; Will, Y. Assessment of mitochondrial toxicity in HepG2 cells cultured in high-glucose- or galactose-containing media. Curr. Protoc. Toxicol. 2011, 49, 2.20.1–2.20.14. [Google Scholar] [CrossRef]
- Kamalian, L.; Chadwick, A.E.; Bayliss, M.; French, N.S.; Monshouwer, M.; Snoeys, J.; Park, B.K. The utility of HepG2 cells to identify direct mitochondrial dysfunction in the absence of cell death. Toxicol. In Vitro 2015, 29, 732–740. [Google Scholar] [CrossRef]
- Hynes, J.; Swiss, R.L.; Will, Y. High-Throughput Analysis of Mitochondrial Oxygen Consumption. Methods Mol. Biol. 2018, 1782, 71–87. [Google Scholar]
- Nadanaciva, S.; Rana, P.; Beeson, G.C.; Chen, D.; Ferrick, D.A.; Beeson, C.C.; Will, Y. Assessment of drug-induced mitochondrial dysfunction via altered cellular respiration and acidification measured in a 96-well platform. J. Bioenerg. Biomembr. 2012, 44, 421–437. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Dykens, J.A.; Bernal, A.; Capaldi, R.A.; Will, Y. Mitochondrial impairment by PPAR agonists and statins identified via immunocaptured OXPHOS complex activities and respiration. Toxicol. Appl. Pharmacol. 2007, 223, 277–287. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.D.; Marroquin, L.D.; Nadanaciva, S.; Xu, J.J.; Dunn, M.C.; Smith, A.R.; Will, Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol. Sci. 2008, 103, 335–345. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Aleo, M.D.; Strock, C.J.; Stedman, D.B.; Wang, H.; Will, Y. Toxicity assessments of nonsteroidal anti-inflammatory drugs in isolated mitochondria, rat hepatocytes, and zebrafish show good concordance across chemical classes. Toxicol. Appl. Pharmacol. 2013, 272, 272–280. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Dillman, K.; Gebhard, D.F.; Shrikhande, A.; Will, Y. High-content screening for compounds that affect mtDNA-encoded protein levels in eukaryotic cells. J. Biomol. Screen. 2010, 15, 937–948. [Google Scholar] [CrossRef]
- Solem, L.E.; Henry, T.R.; Wallace, K.B. Disruption of mitochondrial calcium homeostasis following chronic doxorubicin administration. Toxicol. Appl. Pharmacol. 1994, 129, 214–222. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Serrano, J.; Kuehl, D.W.; Wallace, K.B. Preferential oxidation of cardiac mitochondrial DNA following acute intoxication with doxorubicin. Biochim. Biophys. Acta 1997, 1321, 101–106. [Google Scholar] [CrossRef]
- Wallace, K.B. Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol. Toxicol. 2003, 93, 105–115. [Google Scholar] [CrossRef]
- Sardao, V.A.; Oliveira, P.J.; Holy, J.; Oliveira, C.R.; Wallace, K.B. Doxorubicin-induced mitochondrial dysfunction is secondary to nuclear p53 activation in H9c2 cardiomyoblasts. Cancer Chemother. Pharmacol. 2009, 64, 811–827. [Google Scholar] [CrossRef]
- Qato, D.M.; Wilder, J.; Schumm, L.P.; Gillet, V.; Alexander, G.C. Changes in Prescription and Over-the-Counter Medication and Dietary Supplement Use among Older Adults in the United States, 2005 vs 2011. JAMA Intern. Med. 2016, 176, 473–482. [Google Scholar] [CrossRef]
- Saraf, A.A.; Petersen, A.W.; Simmons, S.F.; Schnelle, J.F.; Bell, S.P.; Kripalani, S.; Myers, A.P.; Mixon, A.S.; Long, E.A.; Jacobsen, J.M.; et al. Medications associated with geriatric syndromes and their prevalence in older hospitalized adults discharged to skilled nursing facilities. J. Hosp. Med. 2016, 11, 694–700. [Google Scholar] [CrossRef]
- Merel, S.E.; Paauw, D.S. Common Drug Side Effects and Drug-Drug Interactions in Elderly Adults in Primary Care. J. Am. Geriatr. Soc. 2017, 65, 1578–1585. [Google Scholar] [CrossRef]
- Chatterjee, S.S.; Stefanovich, V. Influence of anti-inflammatory agents on rat liver mitochondrial ATPase. Arzneimittel-Forschung 1976, 26, 499–502. [Google Scholar]
- Nulton-Persson, A.C.; Szweda, L.I.; Sadek, H.A. Inhibition of cardiac mitochondrial respiration by salicylic acid and acetylsalicylate. J. Cardiovasc. Pharmacol. 2004, 44, 591–595. [Google Scholar] [CrossRef]
- Raza, H.; John, A. Implications of altered glutathione metabolism in aspirin-induced oxidative stress and mitochondrial dysfunction in HepG2 cells. PLoS ONE 2012, 7, e36325. [Google Scholar] [CrossRef]
- Deschamps, D.; Fisch, C.; Fromenty, B.; Berson, A.; Degott, C.; Pessayre, D. Inhibition by salicylic acid of the activation and thus oxidation of long chain fatty acids. Possible role in the development of Reye’s syndrome. J. Pharmacol. Exp. Ther. 1991, 259, 894–904. [Google Scholar]
- Oh, K.W.; Qian, T.; Brenner, D.A.; Lemasters, J.J. Salicylate enhances necrosis and apoptosis mediated by the mitochondrial permeability transition. Toxicol. Sci. 2003, 73, 44–52. [Google Scholar] [CrossRef]
- Zimmerman, H.J. Effects of aspirin and acetaminophen on the liver. Arch. Intern. Med. 1981, 141, 333–342. [Google Scholar] [CrossRef]
- Bonifacio, A.; Mullen, P.J.; Mityko, I.S.; Navegantes, L.C.; Bouitbir, J.; Krahenbuhl, S. Simvastatin induces mitochondrial dysfunction and increased atrogin-1 expression in H9c2 cardiomyocytes and mice in vivo. Arch. Toxicol. 2016, 90, 203–215. [Google Scholar] [CrossRef]
- Fisar, Z.; Hroudova, J.; Singh, N.; Koprivova, A.; Maceckova, D. Effect of Simvastatin, Coenzyme Q10, Resveratrol, Acetylcysteine and Acetylcarnitine on Mitochondrial Respiration. Folia Biol. 2016, 62, 53–66. [Google Scholar]
- van Diemen, M.P.J.; Berends, C.L.; Akram, N.; Wezel, J.; Teeuwisse, W.M.; Mik, B.G.; Kan, H.E.; Webb, A.; Beenakker, J.W.M.; Groeneveld, G.J. Validation of a pharmacological model for mitochondrial dysfunction in healthy subjects using simvastatin: A randomized placebo-controlled proof-of-pharmacology study. Eur. J. Pharmacol. 2017, 815, 290–297. [Google Scholar] [CrossRef]
- Busanello, E.N.B.; Figueira, T.R.; Marques, A.C.; Navarro, C.D.C.; Oliveira, H.C.F.; Vercesi, A.E. Facilitation of Ca(2+) -induced opening of the mitochondrial permeability transition pore either by nicotinamide nucleotide transhydrogenase deficiency or statins treatment. Cell Biol. Int. 2018, 42, 742–746. [Google Scholar] [CrossRef]
- Sirvent, P.; Bordenave, S.; Vermaelen, M.; Roels, B.; Vassort, G.; Mercier, J.; Raynaud, E.; Lacampagne, A. Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem. Biophys. Res. Commun. 2005, 338, 1426–1434. [Google Scholar] [CrossRef]
- Sirvent, P.; Mercier, J.; Vassort, G.; Lacampagne, A. Simvastatin triggers mitochondria-induced Ca2+ signaling alteration in skeletal muscle. Biochem. Biophys. Res. Commun. 2005, 329, 1067–1075. [Google Scholar] [CrossRef]
- Wagner, B.K.; Kitami, T.; Gilbert, T.J.; Peck, D.; Ramanathan, A.; Schreiber, S.L.; Golub, T.R.; Mootha, V.K. Large-scale chemical dissection of mitochondrial function. Nat. Biotechnol. 2008, 26, 343–351. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Indiveri, C. Mitochondrial carnitine/acylcarnitine translocase: Insights in structure/function relationships. Basis for drug therapy and side effects prediction. Mini Rev. Med. Chem. 2015, 15, 396–405. [Google Scholar] [CrossRef]
- Urbano, F.; Bugliani, M.; Filippello, A.; Scamporrino, A.; Di Mauro, S.; Di Pino, A.; Scicali, R.; Noto, D.; Rabuazzo, A.M.; Averna, M.; et al. Atorvastatin but Not Pravastatin Impairs Mitochondrial Function in Human Pancreatic Islets and Rat beta-Cells. Direct Effect of Oxidative Stress. Sci. Rep. 2017, 7, 11863. [Google Scholar] [CrossRef]
- Godoy, J.C.; Niesman, I.R.; Busija, A.R.; Kassan, A.; Schilling, J.M.; Schwarz, A.; Alvarez, E.A.; Dalton, N.D.; Drummond, J.C.; Roth, D.M.; et al. Atorvastatin, but not pravastatin, inhibits cardiac Akt/mTOR signaling and disturbs mitochondrial ultrastructure in cardiac myocytes. FASEB J. 2019, 33, 1209–1225. [Google Scholar] [CrossRef]
- Broniarek, I.; Jarmuszkiewicz, W. Atorvastatin affects negatively respiratory function of isolated endothelial mitochondria. Arch. Biochem. Biophys. 2018, 637, 64–72. [Google Scholar] [CrossRef]
- Hinson, J.A.; Reid, A.B.; McCullough, S.S.; James, L.P. Acetaminophen-induced hepatotoxicity: Role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metab. Rev. 2004, 36, 805–822. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Suda, C.; Horie, T. Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J. Hepatol. 2005, 42, 110–116. [Google Scholar] [CrossRef]
- Prill, S.; Bavli, D.; Levy, G.; Ezra, E.; Schmalzlin, E.; Jaeger, M.S.; Schwarz, M.; Duschl, C.; Cohen, M.; Nahmias, Y. Real-time monitoring of oxygen uptake in hepatic bioreactor shows CYP450-independent mitochondrial toxicity of acetaminophen and amiodarone. Arch. Toxicol. 2016, 90, 1181–1191. [Google Scholar] [CrossRef]
- Moles, A.; Torres, S.; Baulies, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial-Lysosomal Axis in Acetaminophen Hepatotoxicity. Front. Pharmacol. 2018, 9, 453. [Google Scholar] [CrossRef]
- Manuel, M.A.; Weiner, M.W. Effects of ethacrynic acid and furosemide on isolated rat kidney mitochondria: Inhibition of electron transport in the region of phosphorylation site II. J. Pharmacol. Exp. Ther. 1976, 198, 209. [Google Scholar]
- Orita, Y.; Fukuhara, Y.; Yanase, M.; Ando, A.; Okada, N.; Abe, H. Effect of furosemide on mitochondrial electron transport system and oxidative phosphorylation. Arzneimittel-Forschung 1983, 33, 1446–1450. [Google Scholar]
- Wong, S.G.W.; Card, J.W.; Racz, W.J. The role of mitochondrial injury in bromobenzene and furosemide induced hepatotoxicity. Toxicol. Lett. 2000, 116, 171–181. [Google Scholar] [CrossRef]
- Tai, Y.K.; Cheong, Y.M.; Almsherqi, Z.A.; Chia, S.H.; Deng, Y.; McLachlan, C.S. High dose clopidogrel decreases mice liver mitochondrial respiration function in vitro. Int. J. Cardiol. 2009, 133, 250–252. [Google Scholar] [CrossRef]
- Maseneni, S.; Donzelli, M.; Brecht, K.; Krahenbuhl, S. Toxicity of thienopyridines on human neutrophil granulocytes and lymphocytes. Toxicology 2013, 308, 11–19. [Google Scholar] [CrossRef]
- Zahno, A.; Bouitbir, J.; Maseneni, S.; Lindinger, P.W.; Brecht, K.; Krahenbuhl, S. Hepatocellular toxicity of clopidogrel: Mechanisms and risk factors. Free Radic. Biol. Med. 2013, 65, 208–216. [Google Scholar] [CrossRef]
- Felix, L.; Oliveira, M.M.; Videira, R.; Maciel, E.; Alves, N.D.; Nunes, F.M.; Alves, A.; Almeida, J.M.; Domingues, M.R.; Peixoto, F.P. Carvedilol exacerbate gentamicin-induced kidney mitochondrial alterations in adult rat. Exp. Toxicol. Pathol. 2017, 69, 83–92. [Google Scholar] [CrossRef]
- Busanello, E.N.B.; Marques, A.C.; Lander, N.; de Oliveira, D.N.; Catharino, R.R.; Oliveira, H.C.F.; Vercesi, A.E. Pravastatin Chronic Treatment Sensitizes Hypercholesterolemic Mice Muscle to Mitochondrial Permeability Transition: Protection by Creatine or Coenzyme Q10. Front. Pharmacol. 2017, 8, 185. [Google Scholar] [CrossRef]
- Westwood, F.R.; Scott, R.C.; Marsden, A.M.; Bigley, A.; Randall, K. Rosuvastatin: Characterization of induced myopathy in the rat. Toxicol. Pathol. 2008, 36, 345–352. [Google Scholar] [CrossRef]
- van Leeuwen, J.S.; Unlu, B.; Vermeulen, N.P.; Vos, J.C. Differential involvement of mitochondrial dysfunction, cytochrome P450 activity, and active transport in the toxicity of structurally related NSAIDs. Toxicol. In Vitro 2012, 26, 197–205. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Bravo, C.; Vasquez, C.; Ayala, G.; Silveira, L.H.; Martinez-Lavin, M. Inhibition and uncoupling of oxidative phosphorylation by nonsteroidal anti-inflammatory drugs: Study in mitochondria, submitochondrial particles, cells, and whole heart. Biochem. Pharmacol. 1999, 57, 743–752. [Google Scholar] [CrossRef]
- Porceddu, M.; Buron, N.; Roussel, C.; Labbe, G.; Fromenty, B.; Borgne-Sanchez, A. Prediction of liver injury induced by chemicals in human with a multiparametric assay on isolated mouse liver mitochondria. Toxicol. Sci. 2012, 129, 332–345. [Google Scholar] [CrossRef]
- Aleo, M.D.; Luo, Y.; Swiss, R.; Bonin, P.D.; Potter, D.M.; Will, Y. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 2014, 60, 1015–1022. [Google Scholar] [CrossRef]
- Mendes, P.; Robles, P.G.; Mathur, S. Statin-induced rhabdomyolysis: A comprehensive review of case reports. Physiother. Can. 2014, 66, 124–132. [Google Scholar] [CrossRef]
- Harper, C.R.; Jacobson, T.A. Evidence-based management of statin myopathy. Curr. Atheroscler. Rep. 2010, 12, 322–330. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Duncan, A.J.; Heales, S.J.; Land, J.M. The effect of HMG-CoA reductase inhibitors on coenzyme Q10: Possible biochemical/clinical implications. Drug Saf. 2005, 28, 659–676. [Google Scholar] [CrossRef]
- Paiva, H.; Thelen, K.M.; Van Coster, R.; Smet, J.; De Paepe, B.; Mattila, K.M.; Laakso, J.; Lehtimaki, T.; von Bergmann, K.; Lutjohann, D.; et al. High-dose statins and skeletal muscle metabolism in humans: A randomized, controlled trial. Clin. Pharmacol. Ther. 2005, 78, 60–68. [Google Scholar] [CrossRef]
- Avis, H.J.; Hargreaves, I.P.; Ruiter, J.P.; Land, J.M.; Wanders, R.J.; Wijburg, F.A. Rosuvastatin lowers coenzyme Q10 levels, but not mitochondrial adenosine triphosphate synthesis, in children with familial hypercholesterolemia. J. Pediatr. 2011, 158, 458–462. [Google Scholar] [CrossRef]
- Neergheen, V.; Dyson, A.; Wainwright, L.; Hargreaves, I.P. Statin and Fibrate-induced Dichotomy of Mitochondrial Function. In Mitochondrial Dysfunction Caused by Drugs and Environmental Toxicants; Will, Y., Dykens, J., Eds.; Wiley: New York, NY, USA, 2018; pp. 459–473. [Google Scholar]
- Hargreaves, I.P.; Al Shahrani, M.; Wainwright, L.; Heales, S.J. Drug-Induced Mitochondrial Toxicity. Drug Saf. 2016, 39, 661–674. [Google Scholar] [CrossRef]
- Johnson, T.E.; Zhang, X.; Shi, S.; Umbenhauer, D.R. Statins and PPARalpha agonists induce myotoxicity in differentiated rat skeletal muscle cultures but do not exhibit synergy with co-treatment. Toxicol. Appl. Pharmacol. 2005, 208, 210–221. [Google Scholar] [CrossRef]
- Salimi, A.; Eybagi, S.; Seydi, E.; Naserzadeh, P.; Kazerouni, N.P.; Pourahmad, J. Toxicity of macrolide antibiotics on isolated heart mitochondria: A justification for their cardiotoxic adverse effect. Xenobiotica 2016, 46, 82–93. [Google Scholar] [CrossRef]
- Mathis, A.; Wild, P.; Boettger, E.C.; Kapel, C.M.; Deplazes, P. Mitochondrial ribosome as the target for the macrolide antibiotic clarithromycin in the helminth Echinococcus multilocularis. Antimicrob. Agents Chemother. 2005, 49, 3251–3255. [Google Scholar] [CrossRef]
- Ding, L.; Zang, L.; Zhang, Y.; Zhang, Y.; Wang, X.; Ai, W.; Ding, N.; Wang, H. Joint toxicity of fluoroquinolone and tetracycline antibiotics to zebrafish (Danio rerio) based on biochemical biomarkers and histopathological observation. J. Toxicol. Sci. 2017, 42, 267–280. [Google Scholar] [CrossRef]
- Kaur, K.; Fayad, R.; Saxena, A.; Frizzell, N.; Chanda, A.; Das, S.; Chatterjee, S.; Hegde, S.; Baliga, M.S.; Ponemone, V.; et al. Fluoroquinolone-related neuropsychiatric and mitochondrial toxicity: A collaborative investigation by scientists and members of a social network. J. Community Support. Oncol. 2016, 14, 54–65. [Google Scholar] [CrossRef]
- Finsterer, J.; Zarrouk-Mahjoub, S. Mitochondrial toxicity of cardiac drugs and its relevance to mitochondrial disorders. Expert Opin. Drug Metab. Toxicol. 2015, 11, 15–24. [Google Scholar] [CrossRef]
- De Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef]
- Miotto, P.M.; LeBlanc, P.J.; Holloway, G.P. High-Fat Diet Causes Mitochondrial Dysfunction as a Result of Impaired ADP Sensitivity. Diabetes 2018, 67, 2199–2205. [Google Scholar] [CrossRef]
- Jorgensen, W.; Rud, K.A.; Mortensen, O.H.; Frandsen, L.; Grunnet, N.; Quistorff, B. Your mitochondria are what you eat: A high-fat or a high-sucrose diet eliminates metabolic flexibility in isolated mitochondria from rat skeletal muscle. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Gram, M.; Dahl, R.; Dela, F. Physical inactivity and muscle oxidative capacity in humans. Eur. J. Sport Sci. 2014, 14, 376–383. [Google Scholar] [CrossRef]
- Kim, Y.; Triolo, M.; Hood, D.A. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxid. Med. Cell. Longev. 2017, 2017, 3165396. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; Berger, A. Added sugars drive nutrient and energy deficit in obesity: A new paradigm. Open Heart 2016, 3, e000469. [Google Scholar] [CrossRef]
- Chen, W.J.; Du, J.K.; Hu, X.; Yu, Q.; Li, D.X.; Wang, C.N.; Zhu, X.Y.; Liu, Y.J. Protective effects of resveratrol on mitochondrial function in the hippocampus improves inflammation-induced depressive-like behavior. Physiol. Behav. 2017, 182, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Assuncao, M.; Andrade, J.P. Protective action of green tea catechins in neuronal mitochondria during aging. Front. Biosci. 2015, 20, 247–262. [Google Scholar]
- Lim, S.; Ahn, S.Y.; Song, I.C.; Chung, M.H.; Jang, H.C.; Park, K.S.; Lee, K.U.; Pak, Y.K.; Lee, H.K. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS ONE 2009, 4, e5186. [Google Scholar] [CrossRef]
- Rai, P.K.; Russell, O.M.; Lightowlers, R.N.; Turnbull, D.M. Potential compounds for the treatment of mitochondrial disease. Br. Med. Bull. 2015, 116, 5–18. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Class | Rank order of Toxicity Observed (High to Low) | Target Organ |
|---|---|---|
| Anti-diabetic (thiazolidinediones) | Trovan * (troglitazone), Avandia (rosiglitazone), Actos (pioglitazone) | Liver [53] |
| Cholesterol lowering (statins) | Baycol * (cerivastatin), Zocor (simvastatin), Lipitor (atorvastatin), Lescol (fluvastatin) | Muscle [53] |
| Anti-diabetic (biguanides) | Phenformin * (N-phenethylbiguanide), Buformin *(1-butylbiguanide), Glucophage (metformin) | Lactic acidosis [55] |
| Anti-depressant/anxiety (SARIs) | Zerzone * (nefazodone), Desyrel (trazodone), Buspar (buspirone) | Liver [54] |
| Anti-lipidemic (fibrates) | Lopid (gemfibrozil), Lipanor (ciprofibrate), Trilepix (fenofibrate) | Liver [53] |
| Pain medication (NSAIDs) | Avalanche * (celebrex), Mobic (meloxicam), Voltaran (dichlofenac), Felden (piroxicam), Aspirin (acetylsalicylic acid) | Liver, intestine [56] |
| Antibiotics (fluoroquinolones) | Trovan * (trovafloxicin), Levaquin (levafloxicin), Cetraxal (ciprofloxicin) | Liver [57] |
| Anti-cancer (topoisomerase inhibitors) | Adriamycin (doxorubicin) | Heart [58,59,60,61] |
| Medication | Brand Name/Drug Name | % Prescribed | References | Mitochondrial Toxicity Reported |
|---|---|---|---|---|
| Pain medication (OTC) | Aspirin (acetylsalisylic acid) | 40 | [65,66,67,68,69,70] | Inhibition of Respiration, Uncoupling of Oxidative Phosphorylation, Opening of MPT Pore, Inhibition of ATPase, Alteration of Glutathione Status |
| Cholesterol lowering | Zocor (simvastatin) | 22 | [53,71,72,73,74,75,76,77] | Inhibition of Respiration, Uncoupling of oxidative Phosphorylation, Inhibition of ETC Complexes I, IV, V, Decrease in Membrane Potential, Increase Ca++ Release, Decrease ATP Levels |
| Blood pressure medication | Zestril (lisinopril) | 20 | No reports | |
| Diuretic | Microzide (hydrochlorothiazidine) | 19 | No reports | |
| Thyroid medication | Synthroid (levothyroxin) | 15 | No reports | |
| Heart medication | Lopressor (metoprolol) | 15 | No reports | |
| Heartburn (OTC) | Prilosec (omeprazole) | 10 | [78] | Inhibition of Carnitine/Acylcarnitie Transporter |
| Cholesterol lowering | Lipitor (atorvastatin) | 9 | [79,80,81] | Inhibits Mitochondrial Respiration in Pancreatic, Cardiomyocytes and Endothelial Cells |
| Pain medication (OTC) | Tylenol (acetaminophen) | 9 | [82,83,84,85] | Opening of MPT Pore, Formation of Reactive Oxygen Species, Depletion of mtDNA |
| Heart medication | Tenormin (atenolol) | 8 | No reports | |
| Diuretic | Lasix (furosemide) | 7 | [86,87,88] | Inhibition of Respiration, Uncoupling of Oxidative Phosphorylation |
| Blood thinner | Plavix (clopidogrel) | 7 | [89,90,91] | Inhibition of Respiration, Depletion of Glutathione, Induction of Oxidative Stress, Reduction in Membrane Potential |
| Blood thinner | Coumadin (warfarin) | 6 | No reports | |
| Heart medication | Coreg (carvedilol) | 5 | [92] | Mixed Reports of Mitochondrial Toxicity vs. Preventive Mode |
| Cholesterol lowering | Pravachol (pravastatin) | 5 | [93] | Opening MPT Pore, Inhibition of Respiration at High Concentrations |
| Cholesterol lowering | Crestor (rosuvastatin) | 5 | [94] | Mixed Reports of Mitochondrial Toxicity vs. Preventive Mode |
| Pain medication (OTC) | Aleve (naproxen) | 5 | [95,96] | Inhibition of Respiration, Inhibition of Ca++ Flux |
| Cholesterol lowering | Zetia (ezetimibe) | 5 | No reports |
| Variable | Patient 1 | Patient 2 |
|---|---|---|
| Age | 75 | 75 |
| Body Mass Index | 18 | 28 |
| Breakfast | Yogurt with granola, fresh fruit, poached egg, green tea | Eggs, bacon and potatoes |
| Exercise | 1 h walk daily, swimming once a week | From the car to the house, around the grocery store |
| Lunch | Hummus and vegetables, banana, oat meal cookie | French fries and hot dogs, ice cream sundae |
| Medications | None | Zocor, Lopid, Prilosec, Voltaren (oral), Lasix, Nexteron |
| Dinner | Wild caught salmon and salad | Pasta with sausage and cheese |
| Drink Consumption | Red wine | Coke |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Will, Y.; Shields, J.E.; Wallace, K.B. Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology 2019, 8, 32. https://doi.org/10.3390/biology8020032
Will Y, Shields JE, Wallace KB. Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology. 2019; 8(2):32. https://doi.org/10.3390/biology8020032
Chicago/Turabian StyleWill, Yvonne, Jefry E. Shields, and Kendall B. Wallace. 2019. "Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions" Biology 8, no. 2: 32. https://doi.org/10.3390/biology8020032
APA StyleWill, Y., Shields, J. E., & Wallace, K. B. (2019). Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology, 8(2), 32. https://doi.org/10.3390/biology8020032