
A Cell Biological Perspective on Past, Present and Future Investigations of the Spindle Assembly Checkpoint


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Early Hints of a Pathway that Monitors Chromosome Alignment and Controls Anaphase Onset
3. Discovery of the Spindle Assembly Checkpoint
4. Elucidation of the Design and Operation of the SAC
5. Molecular Mechanisms Underlying SAC Activation and Inactivation
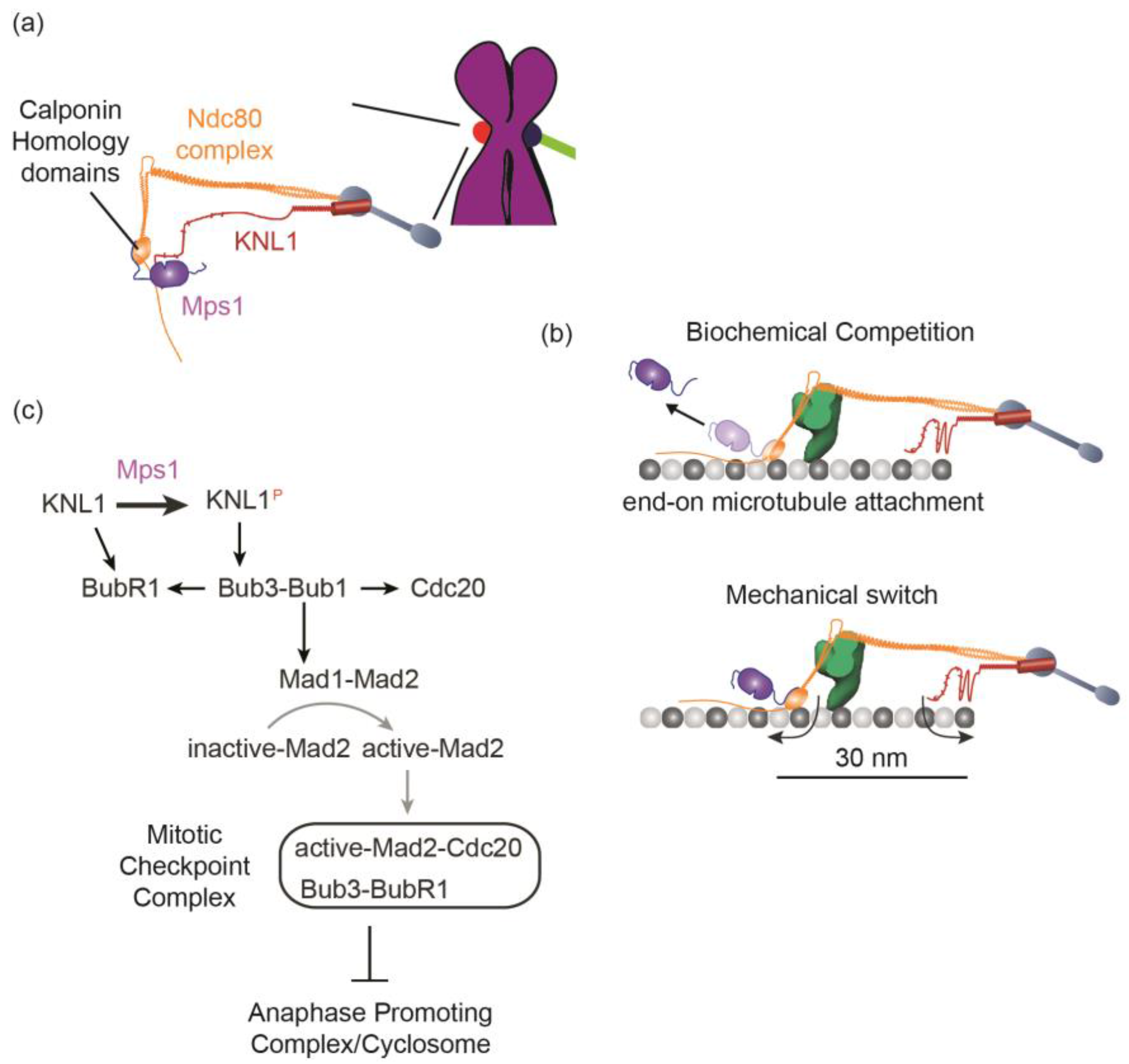
5.1. Detection of the Lack of “End-On” Microtubule Attachment to the Kinetochore
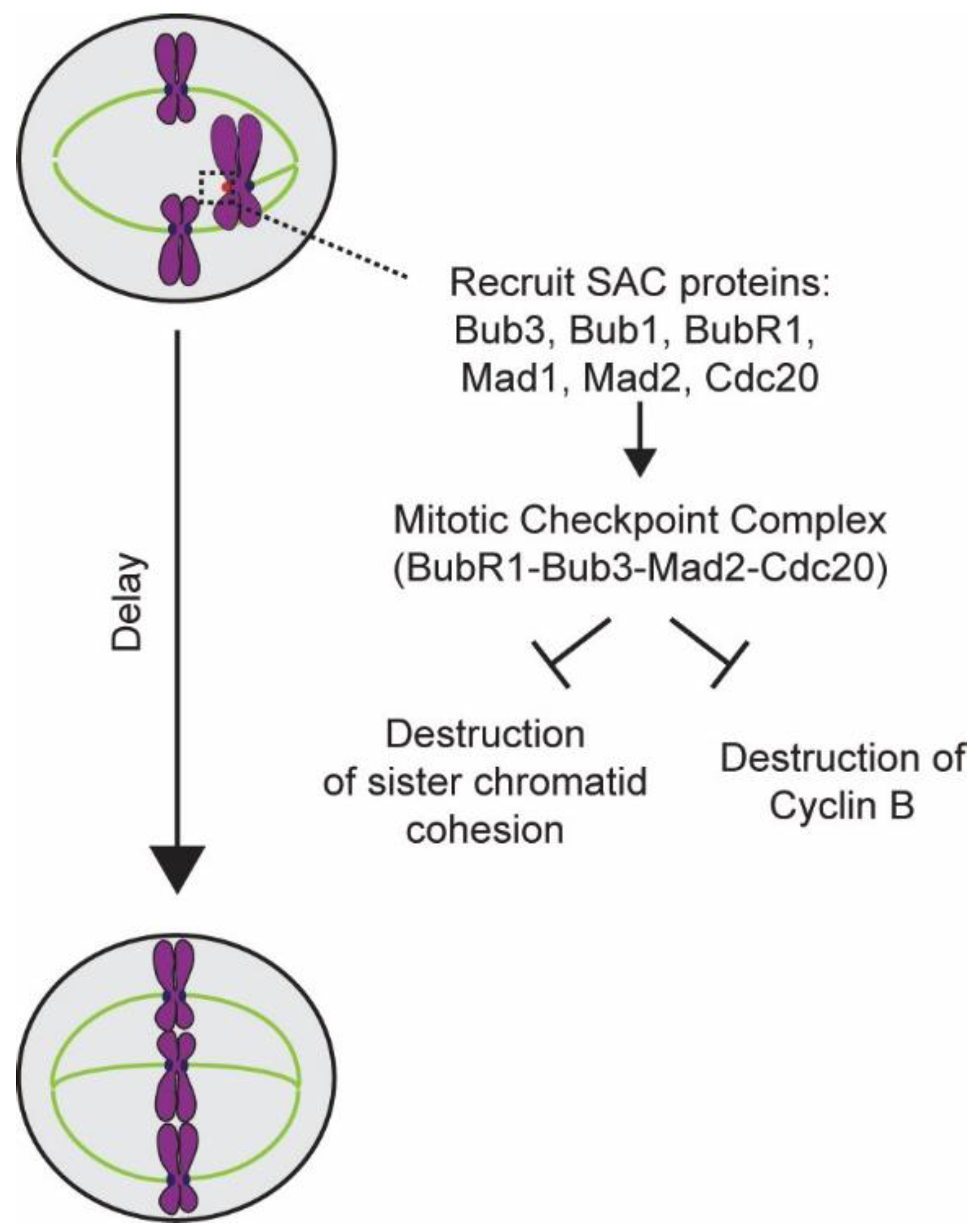
5.2. Generation of the Mitotic Checkpoint Complex
5.3. Inactivation of the SAC
6. Directions for Future Investigations of the SAC
6.1. What Does the Kinetochore Respond to—End-On Attachment to the Kinetochore, an Architectural Change within the Kinetochore Induced by Such Attachment, or Both?
6.2. How Does the Kinetochore Generate a Sufficiently Large Quantity of MCC at a High Rate?
6.3. Is the SAC a Switch or a Rheostat?
6.4. Can a Defective SAC Cause Aneuploidy?
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Musacchio, A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr. Biol. 2015, 25, R1002–R1018. [Google Scholar] [CrossRef] [PubMed]
- Wieser, S.; Pines, J. The biochemistry of mitosis. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Bajer, A.; Molè-Bajer, J. Cine-micrographic studies on mitosis in endosperm. Chromosoma 1955, 7, 558–607. [Google Scholar] [CrossRef]
- Callan, H.G.; Jacobs, P.A. The meiotic process in Mantis religiosa L. males. J. Genet. 1957, 55, 200–217. [Google Scholar] [CrossRef]
- Zirkle, R.E. Ultraviolet-microbeam irradiation of newt-cell cytoplasm: Spindle destruction, false anaphase, and delay of true anaphase. Radiat. Res. 1970, 41, 516–537. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, R.B. Chromosome micromanipulation. II. Induced reorientation and the experimental control of segregation in meiosis. Chromosoma 1967, 21, 17–50. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.R. Structural and mechanical control of mitotic progression. Cold Spring Harb. Symp. Quant. Biol. 1991, 56, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Neskovic, B.A. Developmental phases in intermitosis and the preparation for mitosis of mammalian cells in vitro. Int. Rev. Cytol. 1968, 24, 71–97. [Google Scholar] [PubMed]
- Mazia, D. Mitosis and the physiology of cell division. In The Cell; Brachet, J., Mirsky, A.E., Eds.; Academic Press: Cambridge, MA, USA, 1961; Volume III, pp. 77–413. [Google Scholar]
- Pringle, J.R. The use of conditional lethal cell cycle mutants for temporal and functional sequence mapping of cell cycle events. J. Cell. Physiol. 1978, 95, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Simchen, G. Cell cycle mutants. Annu. Rev. Genet. 1978, 12, 161–191. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [CrossRef]
- Weinert, T.A.; Hartwell, L.H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 1988, 241, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Zieve, G.W.; Turnbull, D.; Mullins, J.M.; McIntosh, J.R. Production of large numbers of mitotic mammalian cells by use of the reversible microtubule inhibitor nocodazole. Nocodazole accumulated mitotic cells. Exp. Cell Res. 1980, 126, 397–405. [Google Scholar] [CrossRef]
- Umesono, K.; Toda, T.; Hayashi, S.; Yanagida, M. Cell division cycle genes NDA2 and NDA3 of the fission yeast Schizosaccharomyces pombe control microtubular organization and sensitivity to anti-mitotic benzimidazole compounds. J. Mol. Biol. 1983, 168, 271–284. [Google Scholar] [CrossRef]
- Jacobs, C.W.; Adams, A.E.; Szaniszlo, P.J.; Pringle, J.R. Functions of microtubules in the Saccharomyces cerevisiae cell cycle. J. Cell Biol. 1988, 107, 1409–1426. [Google Scholar] [CrossRef] [PubMed]
- Bernat, R.L.; Borisy, G.G.; Rothfield, N.F.; Earnshaw, W.C. Injection of anticentromere antibodies in interphase disrupts events required for chromosome movement at mitosis. J. Cell Biol. 1990, 111, 1519–1533. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Murray, A.W. Feedback control of mitosis in budding yeast. Cell 1991, 66, 519–531. [Google Scholar] [CrossRef]
- Hoyt, M.A.; Totis, L.; Roberts, B.T. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell 1991, 66, 507–517. [Google Scholar] [CrossRef]
- Spencer, F.; Hieter, P. Centromere DNA mutations induce a mitotic delay in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1992, 89, 8908–8912. [Google Scholar] [CrossRef] [PubMed]
- Bernat, R.L.; Delannoy, M.R.; Rothfield, N.F.; Earnshaw, W.C. Disruption of centromere assembly during interphase inhibits kinetochore morphogenesis and function in mitosis. Cell 1991, 66, 1229–1238. [Google Scholar] [CrossRef]
- Rieder, C.L.; Schultz, A.; Cole, R.; Sluder, G. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J. Cell Biol. 1994, 127, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Rieder, C.L.; Cole, R.W.; Khodjakov, A.; Sluder, G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 1995, 130, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Benezra, R. Identification of a human mitotic checkpoint gene: hsMAD2. Science 1996, 274, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Waters, J.C.; Salmon, E.D.; Murray, A.W. Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science 1996, 274, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, K.G.; Murray, A.W. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J. Cell Biol. 1995, 131, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.J.; Hoffman, D.B.; Fang, G.; Murray, A.W.; Salmon, E.D. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J. Cell Biol. 2000, 150, 1233–1250. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.J.; Moree, B.; Farrar, E.M.; Stewart, S.; Fang, G.; Salmon, E.D. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr. Biol. 2004, 14, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Holloway, S.L.; Glotzer, M.; King, R.W.; Murray, A.W. Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell 1993, 73, 1393–1402. [Google Scholar] [CrossRef]
- Glotzer, M.; Murray, A.W.; Kirschner, M.W. Cyclin is degraded by the ubiquitin pathway. Nature 1991, 349, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Surana, U.; Robitsch, H.; Price, C.; Schuster, T.; Fitch, I.; Futcher, A.B.; Nasmyth, K. The role of CDC28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell 1991, 65, 145–161. [Google Scholar] [CrossRef]
- Sudakin, V.; Ganoth, D.; Dahan, A.; Heller, H.; Hershko, J.; Luca, F.C.; Ruderman, J.V.; Hershko, A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell 1995, 6, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Irniger, S.; Piatti, S.; Michaelis, C.; Nasmyth, K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell 1995, 81, 269–278. [Google Scholar] [CrossRef]
- Schwab, M.; Lutum, A.S.; Seufert, W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 1997, 90, 683–693. [Google Scholar] [CrossRef]
- Hwang, L.H.; Lau, L.F.; Smith, D.L.; Mistrot, C.A.; Hardwick, K.G.; Hwang, E.S.; Amon, A.; Murray, A.W. Budding yeast Cdc20: A target of the spindle checkpoint. Science 1998, 279, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, K.G.; Johnston, R.C.; Smith, D.L.; Murray, A.W. MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J. Cell Biol. 2000, 148, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Sudakin, V.; Chan, G.K.; Yen, T.J. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 2001, 154, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Izawa, D.; Pines, J. The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 2015, 517, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Guacci, V.; Koshland, D. Pds1p is required for faithful execution of anaphase in the yeast, Saccharomyces cerevisiae. J. Cell Biol. 1996, 133, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Guacci, V.; Koshland, D. Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s). J. Cell Biol. 1996, 133, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Surana, U.; Amon, A.; Dowzer, C.; McGrew, J.; Byers, B.; Nasmyth, K. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J. 1993, 12, 1969–1978. [Google Scholar] [PubMed]
- Michaelis, C.; Ciosk, R.; Nasmyth, K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997, 91, 35–45. [Google Scholar] [CrossRef]
- Guacci, V.; Koshland, D.; Strunnikov, A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 1997, 91, 47–57. [Google Scholar] [CrossRef]
- Zou, H.; McGarry, T.J.; Bernal, T.; Kirschner, M.W. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science 1999, 285, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F.; Lottspeich, F.; Nasmyth, K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 1999, 400, 37–42. [Google Scholar] [PubMed]
- Uhlmann, F.; Wernic, D.; Poupart, M.A.; Koonin, E.V.; Nasmyth, K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell 2000, 103, 375–386. [Google Scholar] [CrossRef]
- Ciosk, R.; Zachariae, W.; Michaelis, C.; Shevchenko, A.; Mann, M.; Nasmyth, K. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell 1998, 93, 1067–1076. [Google Scholar] [CrossRef]
- Wigge, P.A.; Jensen, O.N.; Holmes, S.; Soues, S.; Mann, M.; Kilmartin, J.V. Analysis of the Saccharomyces spindle pole by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. J. Cell Biol. 1998, 141, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, K.G.; Weiss, E.; Luca, F.C.; Winey, M.; Murray, A.W. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science 1996, 273, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Rieder, C.L. The Formation, structure, and composition of the mammalian kinetochore and kinetochore fiber. Int. Rev. Cytol. 1982, 79, 1–58. [Google Scholar] [PubMed]
- McIntosh, J.R.; O’Toole, E.; Zhudenkov, K.; Morphew, M.; Schwartz, C.; Ataullakhanov, F.I.; Grishchuk, E.L. Conserved and divergent features of kinetochores and spindle microtubule ends from five species. J. Cell Biol. 2013, 200, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.J.; McEwen, B.F.; Canman, J.C.; Hoffman, D.B.; Farrar, E.M.; Rieder, C.L.; Salmon, E.D. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J. Cell Biol. 2001, 155, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Dick, A.E.; Gerlich, D.W. Kinetic framework of spindle assembly checkpoint signalling. Nat. Cell Biol. 2013, 15, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Hiruma, Y.; Sacristan, C.; Pachis, S.T.; Adamopoulos, A.; Kuijt, T.; Ubbink, M.; von Castelmur, E.; Perrakis, A.; Kops, G.J. CELL DIVISION CYCLE. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science 2015, 348, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Gao, H.; Yu, H. CELL DIVISION CYCLE. Kinetochore attachment sensed by competitive Mps1 and microtubule binding to Ndc80C. Science 2015, 348, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Liu, X.; Wang, W.; Zhu, T.; Wang, X.; Xu, L.; Abrieu, A.; Fu, C.; Hill, D.L.; Yao, X. Dynamic localization of Mps1 kinase to kinetochores is essential for accurate spindle microtubule attachment. Proc. Natl. Acad. Sci. USA 2015, 112, E4546–E4555. [Google Scholar] [CrossRef] [PubMed]
- Kemmler, S.; Stach, M.; Knapp, M.; Ortiz, J.; Pfannstiel, J.; Ruppert, T.; Lechner, J. Mimicking Ndc80 phosphorylation triggers spindle assembly checkpoint signalling. EMBO J. 2009, 28, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, J.G.; Moree, B.; Hickey, J.M.; Kilmartin, J.V.; Salmon, E.D. hNuf2 inhibition blocks stable kinetochore-microtubule attachment and induces mitotic cell death in HeLa cells. J. Cell Biol. 2002, 159, 549–555. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, J.G.; Howell, B.J.; Canman, J.C.; Hickey, J.M.; Fang, G.; Salmon, E.D. Nuf2 and Hec1 are required for retention of the checkpoint proteins Mad1 and Mad2 to kinetochores. Curr. Biol. 2003, 13, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Novelle, M.D.; Petronczki, M. Relocation of the Chromosomal Passenger Complex Prevents Mitotic Checkpoint Engagement at Anaphase. Curr. Biol. 2010, 20, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.A.; Hamilton, R.S.; Pauli, A.; Davis, I.; Nasmyth, K. Cohesin cleavage and Cdk inhibition trigger formation of daughter nuclei. Nat. Cell Biol. 2010, 12, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Kuijt, T.E.; Omerzu, M.; Saurin, A.T.; Kops, G.J. Conditional targeting of MAD1 to kinetochores is sufficient to reactivate the spindle assembly checkpoint in metaphase. Chromosoma 2014, 123, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Ballister, E.R.; Riegman, M.; Lampson, M.A. Recruitment of Mad1 to metaphase kinetochores is sufficient to reactivate the mitotic checkpoint. J. Cell Biol. 2014, 204, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.; Kapoor, T.M. Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat. Cell Biol. 2011, 13, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Aravamudhan, P.; Goldfarb, A.A.; Joglekar, A.P. The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling. Nat. Cell Biol. 2015, 17, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, A.P.; Bloom, K.; Salmon, E.D. In vivo protein architecture of the eukaryotic kinetochore with nanometer scale accuracy. Curr. Biol. 2009, 19, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; O’Quinn, R.P.; Pierce, H.L.; Joglekar, A.P.; Gall, W.E.; DeLuca, J.G.; Carroll, C.W.; Liu, S.T.; Yen, T.J.; McEwen, B.F.; et al. Protein architecture of the human kinetochore microtubule attachment site. Cell 2009, 137, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, R.B.; Chaleckis, R.; Lehner, C.F. Intrakinetochore localization and essential functional domains of Drosophila Spc105. EMBO J. 2009, 28, 2374–2386. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Long, S.; Ciferri, C.; Westermann, S.; Drubin, D.; Barnes, G.; Nogales, E. Architecture and flexibility of the yeast Ndc80 kinetochore complex. J. Mol. Biol. 2008, 383, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Espeut, J.; Cheerambathur, D.K.; Krenning, L.; Oegema, K.; Desai, A. Microtubule binding by KNL-1 contributes to spindle checkpoint silencing at the kinetochore. J. Cell Biol. 2012, 196, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Wignall, S.M.; Villeneuve, A.M. Lateral microtubule bundles promote chromosome alignment during acentrosomal oocyte meiosis. Nat. Cell Biol. 2009, 11, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; O’Connell, C.B.; Khodjakov, A.; Walczak, C.E. Chromosome congression in the absence of kinetochore fibres. Nat. Cell Biol. 2009, 11, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, A.P.; Aravamudhan, P. How the kinetochore switches off the spindle assembly checkpoint. Cell Cycle 2016, 15, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Aravamudhan, P.; Felzer-Kim, I.; Gurunathan, K.; Joglekar, A.P. Assembling the protein architecture of the budding yeast kinetochore-microtubule attachment using FRET. Curr. Biol. 2014, 24, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Shepperd, L.A.; Meadows, J.C.; Sochaj, A.M.; Lancaster, T.C.; Zou, J.; Buttrick, G.J.; Rappsilber, J.; Hardwick, K.G.; Millar, J.B. Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr. Biol. 2012, 22, 891–899. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Ceto, S.; Ranish, J.A.; Biggins, S. Phosphoregulation of Spc105 by Mps1 and PP1 regulates Bub1 localization to kinetochores. Curr. Biol. 2012, 22, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Vleugel, M.; Tromer, E.; Omerzu, M.; Groenewold, V.; Nijenhuis, W.; Snel, B.; Kops, G.J. Arrayed BUB recruitment modules in the kinetochore scaffold KNL1 promote accurate chromosome segregation. J. Cell Biol. 2013, 203, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Primorac, I.; Weir, J.R.; Chiroli, E.; Gross, F.; Hoffmann, I.; van Gerwen, S.; Ciliberto, A.; Musacchio, A. Bub3 reads phosphorylated MELT repeats to promote spindle assembly checkpoint signaling. Elife 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, B.; Yu, H. The Bub1-Plk1 kinase complex promotes spindle checkpoint signalling through Cdc20 phosphorylation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, B.; Davey, N.E.; Hagting, A.; Izawa, D.; Mansfeld, J.; Gibson, T.J.; Pines, J. The ABBA Motif Binds APC/C Activators and Is Shared by APC/C Substrates and Regulators. Dev. Cell 2015, 32, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Overlack, K.; Primorac, I.; van Gerwen, S.; Musacchio, A. KI motifs of human Knl1 enhance assembly of comprehensive spindle checkpoint complexes around MELT repeats. Curr. Biol. 2014, 24, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kiyomitsu, T.; Murakami, H.; Yanagida, M. Protein interaction domain mapping of human kinetochore protein Blinkin reveals a consensus motif for binding of spindle assembly checkpoint proteins Bub1 and BubR1. Mol. Cell Biol. 2011, 31, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Wehenkel, A.; Li, X.; Santaguida, S.; Musacchio, A. Structural analysis reveals features of the spindle checkpoint kinase Bub1-kinetochore subunit Knl1 interaction. J. Cell Biol. 2012, 196, 451–467. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Biggins, S. Mad1 kinetochore recruitment by Mps1-mediated phosphorylation of Bub1 signals the spindle checkpoint. Genes Dev. 2014, 28, 140–152. [Google Scholar] [CrossRef] [PubMed]
- De Antoni, A.; Pearson, C.G.; Cimini, D.; Canman, J.C.; Sala, V.; Nezi, L.; Mapelli, M.; Sironi, L.; Faretta, M.; Salmon, E.D.; Musacchio, A. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr. Biol. 2005, 15, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, M.; Massimiliano, L.; Santaguida, S.; Musacchio, A. The MAD2 conformational dimer: Structure and implications for the Spindle Assembly Checkpoint. Cell 2007, 131, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Tipton, A.R.; Ji, W.; Sturt-Gillespie, B.; Bekier, M.E., 2nd; Wang, K.; Taylor, W.R.; Liu, S.T. Monopolar spindle 1 (MPS1) kinase promotes production of closed MAD2 (C-MAD2) conformer and assembly of the mitotic checkpoint complex. J. Biol. Chem. 2013, 288, 35149–35158. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, L.; Tighe, A.; Santaguida, S.; White, A.M.; Jones, C.D.; Musacchio, A.; Green, S.; Taylor, S.S. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J. Cell Biol. 2010, 190, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Musacchio, A. The Aurora B Kinase in Chromosome Bi-Orientation and Spindle Checkpoint Signaling. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, A.; Maffini, S.; Rainey, M.D.; Kaczmarczyk, A.; Gaboriau, D.; Musacchio, A.; Santocanale, C. Requirement for PLK1 kinase activity in the maintenance of a robust spindle assembly checkpoint. Biol. Open 2015, 5, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Yekezare, M.; Minshull, J.; Pines, J. The APC/C maintains the spindle assembly checkpoint by targeting Cdc20 for destruction. Nat. Cell Biol. 2008, 10, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Aravamudhan, P.; Chen, R.; Roy, B.; Sim, J.; Joglekar, A.P. Dual mechanisms regulate the recruitment of spindle assembly checkpoint proteins to the budding yeast kinetochore. Mol. Biol. Cell 2016, 27, 3405–3417. [Google Scholar] [CrossRef] [PubMed]
- Brito, D.A.; Rieder, C.L. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Rieder, C.L.; Maiato, H. Stuck in division or passing through: What happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 2004, 7, 637–651. [Google Scholar] [CrossRef] [PubMed]
- He, E.; Kapuy, O.; Oliveira, R.A.; Uhlmann, F.; Tyson, J.J.; Novák, B. System-level feedbacks make the anaphase switch irreversible. Proc. Natl. Acad. Sci. USA 2011, 108, 10016–10021. [Google Scholar] [CrossRef] [PubMed]
- Collin, P.; Nashchekina, O.; Walker, R.; Pines, J. The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat. Cell Biol. 2013, 15, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, S.; Geissen, E.M.; Kamenz, J.; Trautmann, S.; Widmer, C.; Drewe, P.; Knop, M.; Radde, N.; Hasenauer, J.; Hauf, S. Determinants of robustness in spindle assembly checkpoint signalling. Nat. Cell Biol. 2013, 15, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Bravo, V.; Maciejowski, J.; Corona, J.; Buch, H.K.; Collin, P.; Kanemaki, M.T.; Shah, J.V.; Jallepalli, P.V. Nuclear pores protect genome integrity by assembling a premitotic and Mad1-dependent anaphase inhibitor. Cell 2014, 156, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Wynne, D.J.; Funabiki, H. Kinetochore function is controlled by a phospho-dependent coexpansion of inner and outer components. J. Cell Biol. 2015, 210, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Vleugel, M.; Backer, C.B.; Hori, T.; Fukagawa, T.; Cheeseman, I.M.; Lampson, M.A. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J. Cell Biol. 2010, 188, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.S.; Cross, F.R.; Funabiki, H. KNL1/Spc105 recruits PP1 to silence the spindle assembly checkpoint. Curr. Biol. 2011, 21, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Espert, A.; Uluocak, P.; Bastos, R.N.; Mangat, D.; Graab, P.; Gruneberg, U. PP2A-B56 opposes Mps1 phosphorylation of Knl1 and thereby promotes spindle assembly checkpoint silencing. J. Cell Biol. 2014, 206, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, S.; Janczyk, P.L.; Qu, Q.; Brautigam, C.A.; Stukenberg, P.T.; Yu, H.; Gorbsky, G.J. The human SKA complex drives the metaphase-anaphase cell cycle transition by recruiting protein phosphatase 1 to kinetochores. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, B.; Tomchick, D.R.; Machius, M.; Rizo, J.; Yu, H.; Luo, X. p31comet blocks Mad2 activation through structural mimicry. Cell 2007, 131, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Eytan, E.; Wang, K.; Miniowitz-Shemtov, S.; Sitry-Shevah, D.; Kaisari, S.; Yen, T.J.; Liu, S.T.; Hershko, A. Disassembly of mitotic checkpoint complexes by the joint action of the AAA-ATPase TRIP13 and p31(comet). Proc. Natl. Acad. Sci. USA 2014, 111, 12019–12024. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Rosenberg, S.C.; Moeller, A.; Speir, J.A.; Su, T.Y.; Corbett, K.D. TRIP13 is a protein-remodeling AAA+ATPase that catalyzes MAD2 conformation switching. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Miniowitz-Shemtov, S.; Eytan, E.; Kaisari, S.; Sitry-Shevah, D.; Hershko, A. Mode of interaction of TRIP13 AAA-ATPase with the Mad2-binding protein p31comet and with mitotic checkpoint complexes. Proc. Natl. Acad. Sci. USA 2015, 112, 11536–11540. [Google Scholar] [CrossRef] [PubMed]
- Gorbsky, G.J. Cohesion fatigue. Curr. Biol. 2013, 23, R986–R988. [Google Scholar] [CrossRef] [PubMed]
- Daum, J.R.; Potapova, T.A.; Sivakumar, S.; Daniel, J.J.; Flynn, J.N.; Rankin, S.; Gorbsky, G.J. Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr. Biol. 2011, 21, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Meitinger, F.; Anzola, J.V.; Kaulich, M.; Richardson, A.; Stender, J.D.; Benner, C.; Glass, C.K.; Dowdy, S.F.; Desai, A.; Shiau, A.K.; Oegema, K. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J. Cell Biol. 2016, 214, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.; Gassmann, R.; Oegema, K.; Desai, A. Uncoordinated loss of chromatid cohesion is a common outcome of extended metaphase arrest. PLoS ONE 2011, 6, e22969. [Google Scholar] [CrossRef] [PubMed]
- Pilaz, L.J.; McMahon, J.J.; Miller, E.E.; Lennox, A.L.; Suzuki, A.; Salmon, E.; Silver, D.L. Prolonged Mitosis of Neural Progenitors Alters Cell Fate in the Developing Brain. Neuron 2016, 89, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.V.; Cleveland, D.W. Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell 2000, 103, 997–1000. [Google Scholar] [CrossRef]
- Etemad, B.; Kops, G.J. Attachment issues: Kinetochore transformations and spindle checkpoint silencing. Curr. Opin. Cell Biol. 2016, 39, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Magidson, V.; He, J.; Ault, J.G.; O'Connell, C.B.; Yang, N.; Tikhonenko, I.; McEwen, B.F.; Sui, H.; Khodjakov, A. Unattached kinetochores rather than intrakinetochore tension arrest mitosis in taxol-treated cells. J. Cell Biol. 2016, 212, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Tauchman, E.C.; Boehm, F.J.; DeLuca, J.G. Stable kinetochore-microtubule attachment is sufficient to silence the spindle assembly checkpoint in human cells. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Etemad, B.; Kuijt, T.E.; Kops, G.J. Kinetochore-microtubule attachment is sufficient to satisfy the human spindle assembly checkpoint. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Badger, B.L.; Salmon, E.D. A quantitative description of Ndc80 complex linkage to human kinetochores. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Aravamudhan, P.; Felzer-Kim, I.; Joglekar, A.P. The budding yeast point centromere associates with two Cse4 molecules during mitosis. Curr. Biol. 2013, 23, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, A.P.; Bouck, D.C.; Molk, J.N.; Bloom, K.S.; Salmon, E.D. Molecular architecture of a kinetochore-microtubule attachment site. Nat. Cell Biol. 2006, 8, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Vleugel, M.; Omerzu, M.; Groenewold, V.; Hadders, M.A.; Lens, S.M.; Kops, G.J. Sequential multisite phospho-regulation of KNL1-BUB3 interfaces at mitotic kinetochores. Mol. Cell 2015, 57, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Westhorpe, F.G.; Tighe, A.; Lara-Gonzalez, P.; Taylor, S.S. p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J. Cell Sci. 2011, 124, 3905–3916. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Weaver, B.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Schvartzman, J.M.; Sotillo, R.; Benezra, R. Mitotic chromosomal instability and cancer: Mouse modelling of the human disease. Nat. Rev. Cancer 2010, 10, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Tighe, A.; Johnson, V.L.; Albertella, M.; Taylor, S.S. Aneuploid colon cancer cells have a robust spindle checkpoint. EMBO Rep. 2001, 2, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Vleugel, M.; Hoogendoorn, E.; Snel, B.; Kops, G.J. Evolution and function of the mitotic checkpoint. Dev. Cell 2012, 23, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Tromer, E.; Snel, B.; Kops, G.J. Widespread Recurrent Patterns of Rapid Repeat Evolution in the Kinetochore Scaffold KNL1. Genome Biol. Evol. 2015, 7, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Morgan, D.O. Cell Size Determines the Strength of the Spindle Assembly Checkpoint during Embryonic Development. Dev. Cell 2016, 36, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Sear, R.P.; Howard, M. Modeling dual pathways for the metazoan spindle assembly checkpoint. Proc. Natl. Acad. Sci. USA 2006, 103, 16758–16763. [Google Scholar] [CrossRef] [PubMed]
- Doncic, A.; Ben-Jacob, E.; Barkai, N. Evaluating putative mechanisms of the mitotic spindle checkpoint. Proc. Natl. Acad. Sci. USA 2005, 102, 6332–6337. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joglekar, A.P. A Cell Biological Perspective on Past, Present and Future Investigations of the Spindle Assembly Checkpoint. Biology 2016, 5, 44. https://doi.org/10.3390/biology5040044
Joglekar AP. A Cell Biological Perspective on Past, Present and Future Investigations of the Spindle Assembly Checkpoint. Biology. 2016; 5(4):44. https://doi.org/10.3390/biology5040044
Chicago/Turabian StyleJoglekar, Ajit P. 2016. "A Cell Biological Perspective on Past, Present and Future Investigations of the Spindle Assembly Checkpoint" Biology 5, no. 4: 44. https://doi.org/10.3390/biology5040044
APA StyleJoglekar, A. P. (2016). A Cell Biological Perspective on Past, Present and Future Investigations of the Spindle Assembly Checkpoint. Biology, 5(4), 44. https://doi.org/10.3390/biology5040044