
Genetic Approaches to Study Plant Responses to Environmental Stresses: An Overview

Abstract
:1. Introduction
2. Northern Blot
2.1. Setup and Use
2.2. Principle
2.3. Advantages
2.4. Disadvantages
3. RNase Protection Assay (RPA)
3.1. Setup and Use
3.2. Principle
3.3. Advantages
3.4. Disadvantages
4. Differential Display of mRNA by PCR (DD-PCR)
4.1. Setup and Use
4.2. Principle
4.3. Advantages
4.4. Disadvantages
5. cDNA Amplified Fragment Length Polymorphism (cDNA AFLP)
5.1. Setup and Use
5.2. Principle
5.3. Advantages
5.4. Disadvantages
6. Serial Analysis of Gene Expression (SAGE)
6.1. Setup and Use
6.2. Principle
6.3. Advantages
6.4. Disadvantages
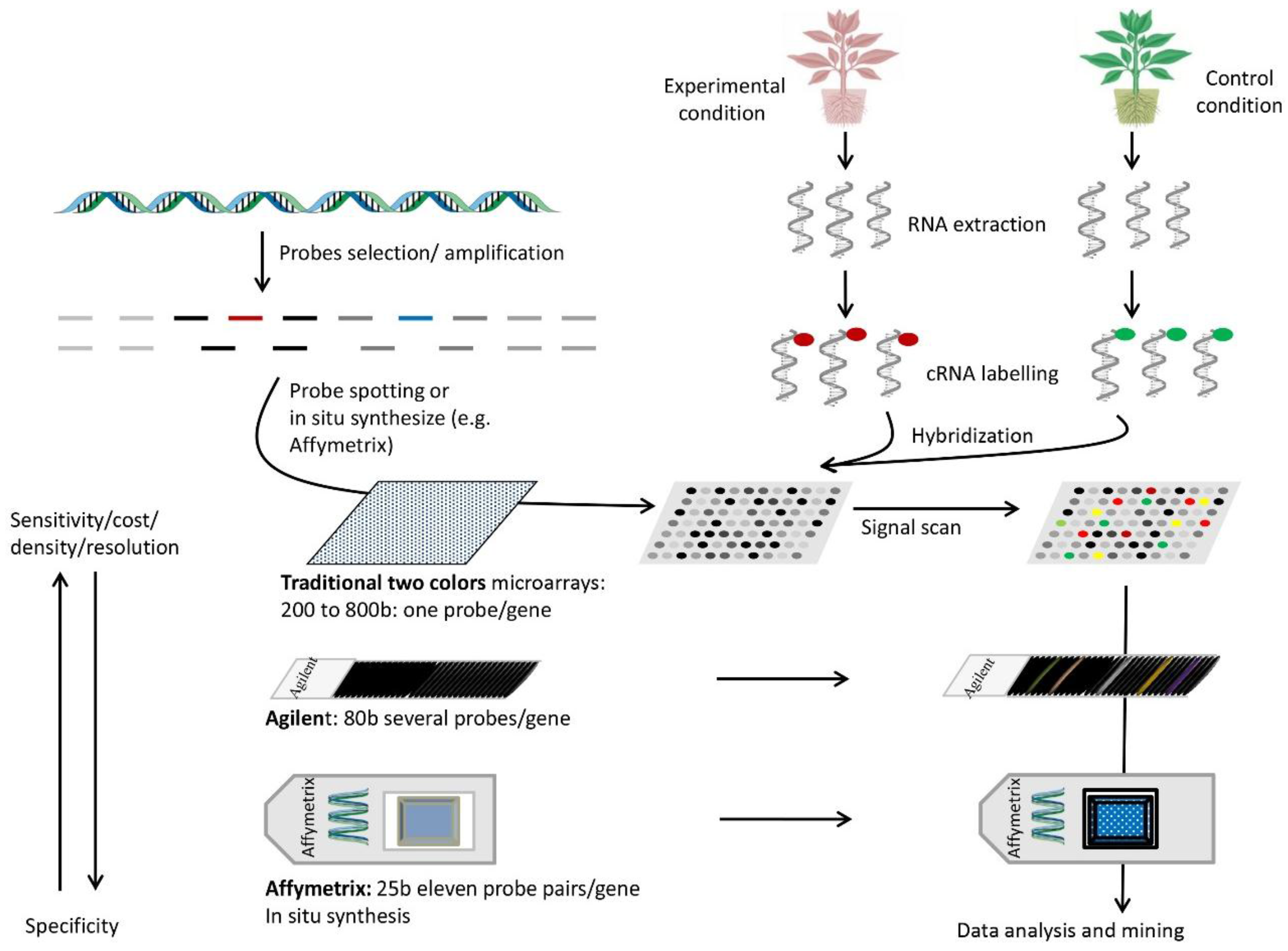
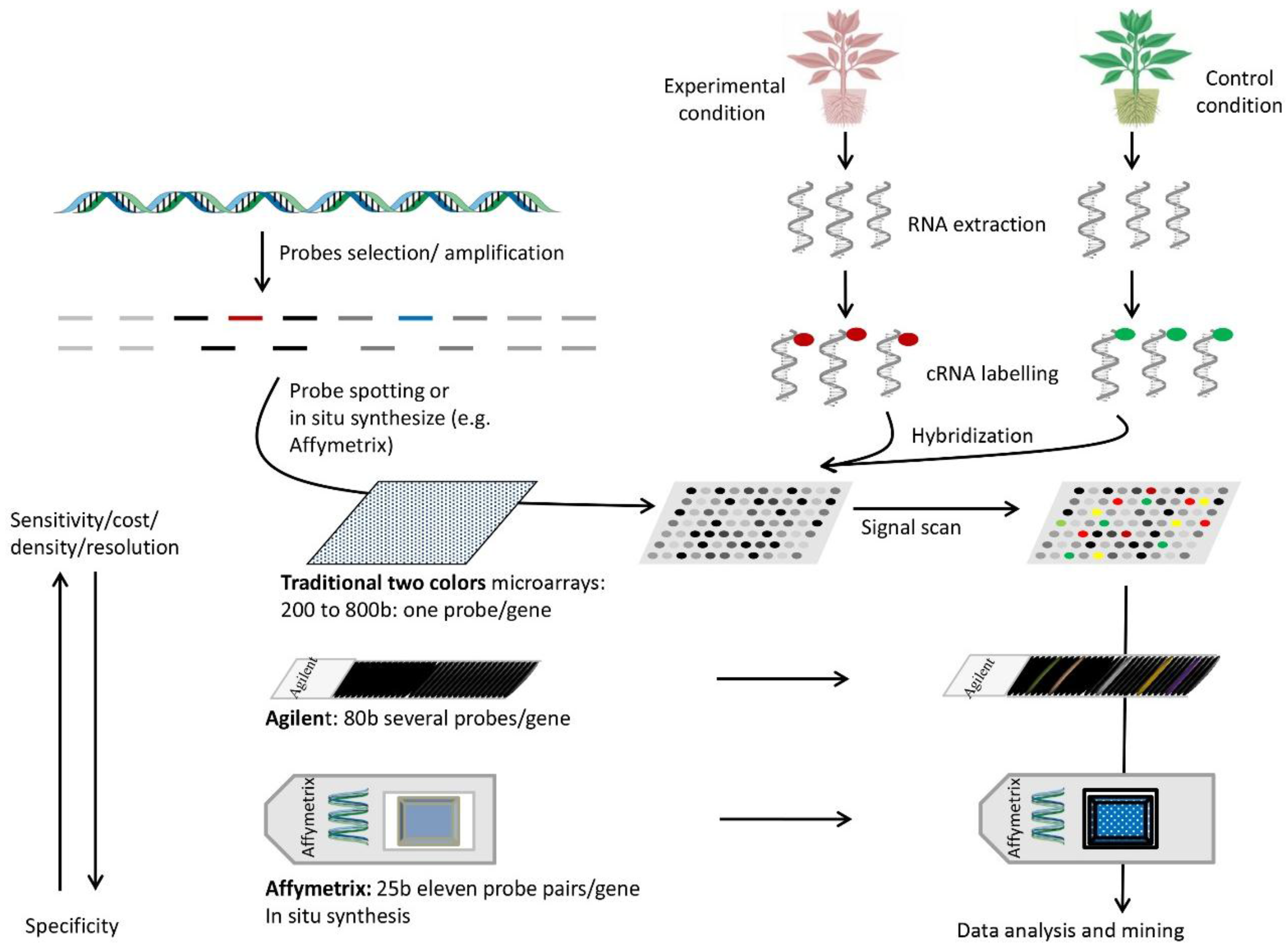
7. DNA Arrays
7.1. Setup and Use
7.2. Principle
7.3. Advantages
7.4. Disadvantages
8. Real-Time PCR (or Quantitative PCR)
8.1. Setup and Use
8.2. Principle
8.3. Advantages
8.4. Disadvantages
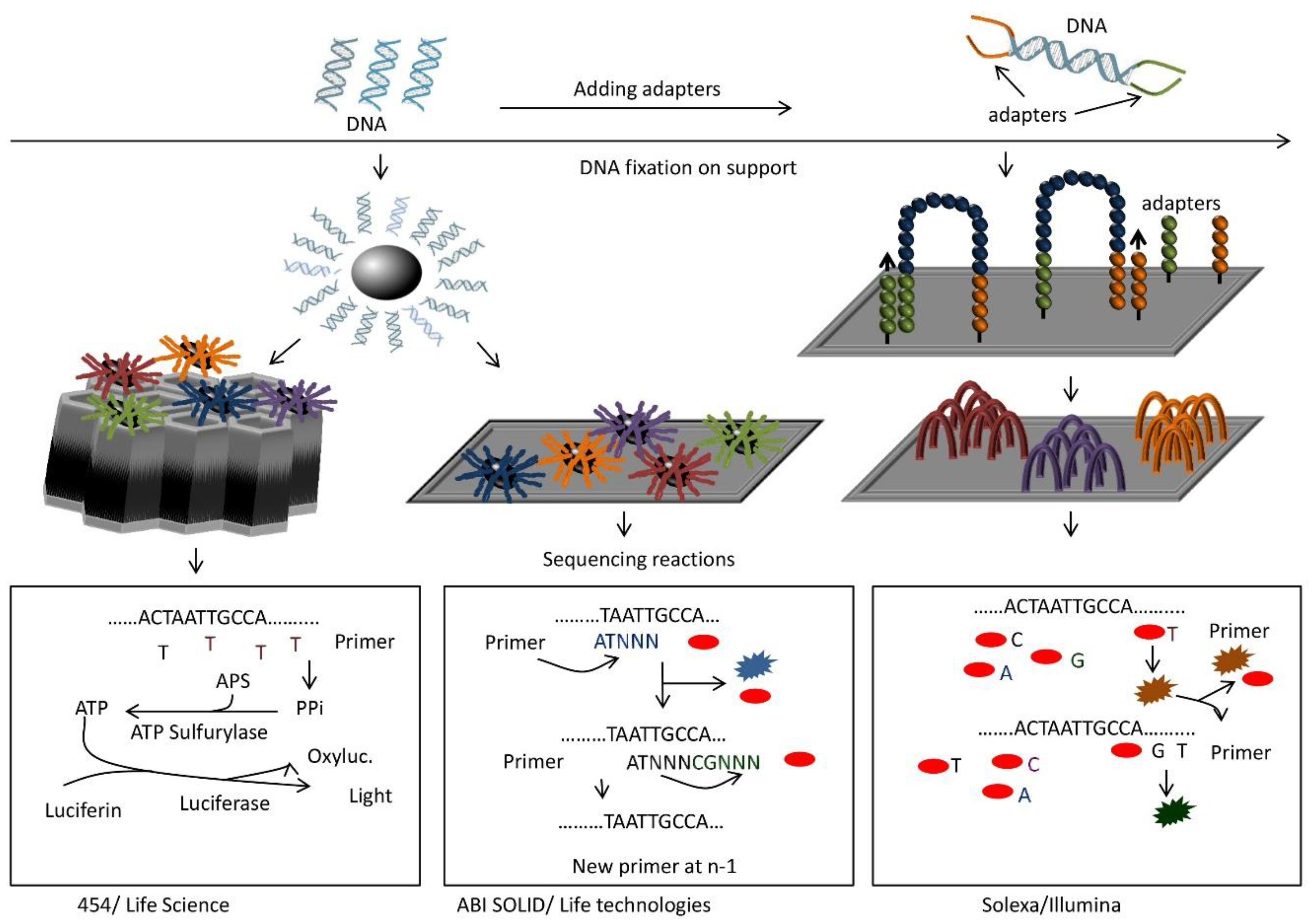
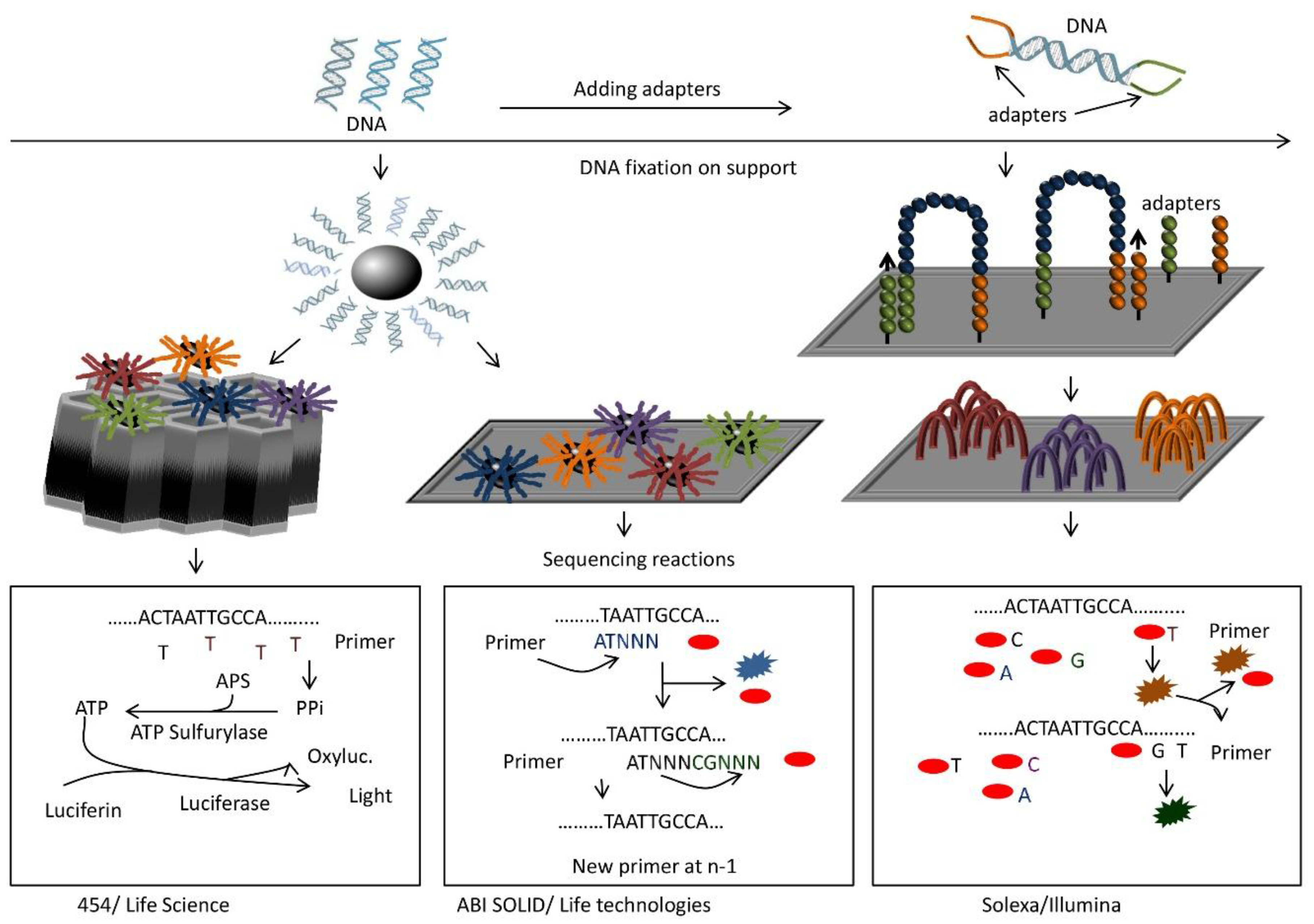
9. Next-Generation Sequencing (NGS)
9.1. Setup and Use
9.2. Principle
9.3. Advantages
9.4. Disadvantages
10. Conclusions
Conflicts of Interest
References
- Alwine, J.C.; Kemp, D.J.; Stark, G.R. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc. Natl. Acad. Sci. USA 1977, 74, 5350–5354. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Du, X.; Tu, L.; Tang, W.; Wang, P.; Wang, M.; Liu, Z.; Zhang, X. Fibre elongation requires normal redox homeostasis modulated by cytosolic ascorbate peroxidase in cotton (Gossypium hirsutum). J. Exp. Bot. 2016. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Li, X.; Zhu, W.; Wu, C.; Yang, G.; Zheng, C. Cotton metallothionein GhMT3a, a reactive oxygen species scavenger, increased tolerance against abiotic stress in transgenic tobacco and yeast. J. Exp. Bot. 2009, 60, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Wang, F.; Geng, X.; Zhang, L.; Yao, Y.; Ni, Z.; Peng, H.; Sun, Q. Overexpression of heat stress-responsive TaMBF1c, a wheat (Triticum aestivum L.) Multiprotein Bridging Factor, confers heat tolerance in both yeast and rice. Plant Mol. Biol. 2015, 87, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yuan, H.; Yang, Y.; Fish, T.; Lyi, S.M.; Thannhauser, T.W.; Zhang, L.; Li, L. Plastid ribosomal protein S5 is involved in photosynthesis, plant development, and cold stress tolerance in Arabidopsis. J. Exp. Bot. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Back, K.; Lee, H.Y.; Lee, H.J.; Phung, T.H.; Grimm, B.; Jung, S. Increased expression of Fe-chelatase leads to increased metabolic flux into heme and confers protection against photodynamically induced oxidative stress. Plant Mol. Biol. 2014, 86, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Southern, E.M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 1975, 98, 503–517. [Google Scholar] [CrossRef]
- Löw, R. Nonradioactive Northern Blotting of RNA. In The Nucleic Acid Protocols Handbook; Rapley, R., Ed.; Humana Press: Totowa, NJ, USA, 2000; pp. 239–247. [Google Scholar]
- Kroczek, R.A.; Siebert, E. Optimization of Northern analysis by vacuum-blotting, RNA-transfer visualization, and ultraviolet fixation. Anal. Biochem. 1990, 184, 90–95. [Google Scholar] [CrossRef]
- Josefsen, K.; Nielsen, H. Northern blotting analysis. Methods Mol. Biol. 2011, 703, 87–105. [Google Scholar] [PubMed]
- Taniguchi, M.; Miura, K.; Iwao, H.; Yamanaka, S. Quantitative assessment of DNA microarrays—Comparison with Northern blot analyses. Genomics 2001, 71, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Memelink, J.; Swords, K.M.; Staehelin, L.A.; Hoge, J.H. Southern, Northern and Western Blot Analysis. In Plant Molecular Biology Manual; Gelvin, S., Schilperoort, R., Eds.; Kluwer Academic Publishers: The Netherlands, 1994; pp. 273–295. [Google Scholar]
- Huang, Q.; Mao, Z.; Li, S.; Hu, J.; Zhu, Y. A non-radioactive method for small RNA detection by Northern blotting. Rice 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Azrolan, N.; Breslow, J.L. A solution hybridization/RNase protection assay with riboprobes to determine absolute levels of apoB, A-I, and E mRNA in human hepatoma cell lines. J. Lipid Res. 1990, 31, 1141–1146. [Google Scholar] [PubMed]
- Friedberg, T.; Grassow, M.A.; Oesch, F. Selective detection of mRNA forms encoding the major phenobarbital inducible cytochromes P450 and other members of the P450IIB family by the RNAse A protection assay. Arch. Biochem. Biophys 1990, 279, 167–173. [Google Scholar] [CrossRef]
- Finer, M.H. The RNase protection assay. Methods Mol. Biol. 1991, 7, 283–296. [Google Scholar] [PubMed]
- Emery, P. RNase protection assay. Methods Mol. Biol. 2007, 362, 343–348. [Google Scholar] [PubMed]
- Junk, D.J.; Mourad, G.S. Isolation and expression analysis of the isopropylmalate synthase gene family of Arabidopsis thaliana. J. Exp. Bot. 2002, 53, 2453–2454. [Google Scholar] [CrossRef] [PubMed]
- Zubo, Y.O.; Kusnetsov, V.V.; Borner, T.; Liere, K. Reverse protection assay: A tool to analyze transcriptional rates from individual promoters. Plant Methods 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Meisinger, C.; Grothe, C. A sensitive RNase protection assay using 33P labeled antisense riboprobes. Mol. Biotechnol. 1996, 5, 289–291. [Google Scholar] [PubMed]
- Rosenau, C.; Kaboord, B.; Qoronfleh, M.W. Development of a chemiluminescence-based ribonuclease protection assay. Biotechniques 2002, 33, 1354–1358. [Google Scholar] [PubMed]
- Sambrook, J.; Russell, D.W. Purification of nucleic acids by extraction with phenol:chloroform. Cold Spring Harb. Protoc. 2006. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Dissen, G.A.; Rage, F.; Ojeda, S.R. RNase protection assay. Methods 1996, 10, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.M. RNase protection assay analysis of mRNA for TGFbeta1(-3) in Ovarian Tumors. Methods Mol. Med. 2001, 39, 431–437. [Google Scholar] [PubMed]
- Stalder, A.K.; Pagenstecher, A.; Kincaid, C.L.; Campbell, I.L. Analysis of gene expression by multiprobe RNase protection assay. Methods Mol. Med. 1999, 22, 53–66. [Google Scholar] [PubMed]
- Newman, C.S.; Krieg, P.A. Ribonuclease protection analysis of gene expression in Xenopus. Methods Mol. Biol. 1999, 127, 29–40. [Google Scholar] [PubMed]
- Qu, Y.; Boutjdir, M. RNase protection assay for quantifying gene expression levels. Methods Mol. Biol. 2007, 366, 145–158. [Google Scholar] [PubMed]
- Ilyin, S.E.; Gayle, D.; Plata-Salaman, C.R. Modifications of RNase protection assay for neuroscience applications. J. Neurosci. Methods 1998, 84, 139–141. [Google Scholar] [CrossRef]
- Liang, P.; Pardee, A.B. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 1992, 257, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Pasentsis, K.; Falara, V.; Pateraki, I.; Gerasopoulos, D.; Kanellis, A.K. Identification and expression profiling of low oxygen regulated genes from Citrus flavedo tissues using RT-PCR differential display. J. Exp. Bot. 2007, 58, 2203–2216. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; He, X.-H.; Hu, Y.; Yu, H.-X.; Ou, S.-J.; Fang, Z.-B. Oligo-dT anchored cDNA–SCoT: A novel differential display method for analyzing differential gene expression in response to several stress treatments in mango (Mangifera indica L.). Gene 2014, 548, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Berke, J.D. Analysis of mRNA expression in striatal tissue by differential display polymerase chain reaction. Methods Mol. Med. 2003, 79, 193–209. [Google Scholar] [PubMed]
- Sturtevant, J. Applications of differential-display reverse transcription-PCR to molecular pathogenesis and medical mycology. Clin. Microbiol. Rev. 2000, 13, 408–427. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Schlichter, U.H.A.; Adams, M.J.; Burrow, P.R.; Steinbib, H.-H.; Antoniw, J.F. An analysis of differential display shows a strong bias towards high copy number mRNAs. Nucleic Acids Res. 1995, 23, 4520–4523. [Google Scholar] [CrossRef] [PubMed]
- Irian, S. Large-scale tag/PCR-based gene expression profiling. World J. Microbiol. Biotechnol. 2014, 30, 2125–2139. [Google Scholar] [CrossRef] [PubMed]
- Bachem, C.W.; van der Hoeven, R.S.; de Bruijn, S.M.; Vreugdenhil, D.; Zabeau, M.; Visser, R.G. Visualization of differential gene expression using a novel method of RNA fingerprinting based on AFLP: Analysis of gene expression during potato tuber development. Plant J. 1996, 9, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; van de Lee, T.; Hornes, M.; Frijters, A.; Pot, J.; Peleman, J.; Kuiper, M.; et al. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef] [PubMed]
- Shirani Bidabadi, M.; Shiran, B.; Fallahi, H.; Rafiei, F.; Esmaeili, F.; Ebrahimie, E. Identification of differential expressed transcripts of almond (Prunus dulcis ‘Sefied’) in response to water-deficit stress by cDNA-AFLP. J. For. Res. 2015, 20, 403–410. [Google Scholar] [CrossRef]
- Xue, R.; Wu, J.; Zhu, Z.; Wang, L.; Wang, X.; Wang, S.; Blair, M.W. Differentially expressed genes in resistant and susceptible common bean (Phaseolus vulgaris L.) genotypes in response to Fusarium oxysporum f. sp. phaseoli. PLoS ONE 2015, 10, e0127698. [Google Scholar] [CrossRef] [PubMed]
- Armoniené, R.; Brazauskas, G. Nonsense-mediated decay of sucrose synthase 1 mRNA with induced premature chain termination codon during cold acclimation in winter wheat. Turk. J. Bot. 2014, 38, 1147–1156. [Google Scholar] [CrossRef]
- Ren, L.; Zhang, D.; Jiang, X.-N.; Gai, Y.; Wang, W.-M.; Reed, B.M.; Shen, X.-H. Peroxidation due to cryoprotectant treatment is a vital factor for cell survival in Arabidopsis cryopreservation. Plant Sci. 2013, 212, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Decorosi, F.; Viti, C.; Mengoni, A.; Bazzicalupo, M.; Giovannetti, L. Improvement of the cDNA-AFLP method using fluorescent primers for transcription analysis in bacteria. J. Microbiol. Methods 2005, 63, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, H.; Tang, C. A silver-staining cDNA-AFLP protocol suitable for transcript profiling in the latex of Hevea brasiliensis (Para rubber tree). Mol. Biotechnol. 2009, 42, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Breyne, P.; Zabeau, M. Genome-wide expression analysis of plant cell cycle modulated genes. Curr. Opin. Plant Biol. 2001, 4, 136–142. [Google Scholar] [CrossRef]
- Fukumura, R.; Takahashi, H.; Saito, T.; Tsutsumi, Y.; Fujimori, A.; Sato, S.; Tatsumi, K.; Araki, R.; Abe, M. A sensitive transcriptome analysis method that can detect unknown transcripts. Nucleic Acids Res. 2003, 31. [Google Scholar] [CrossRef]
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial analysis of gene expression. Science 1995, 270, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, S.V. Serial Analysis of Gene Expression (SAGE): 13 Years of application in research. Curr. Pharm. Biotechnol. 2008, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, H.; Reich, S.; Ito, A.; Saitoh, H.; Kamoun, S.; Winter, P.; Kahl, G.; Reuter, M.; Kruger, D.H.; Terauchi, R. Gene expression analysis of plant host-pathogen interactions by SuperSAGE. Proc. Natl. Acad. Sci. USA 2003, 100, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Fregene, M.; Matsumura, H.; Akano, A.; Dixon, A.; Terauchi, R. Serial analysis of gene expression (SAGE) of host-plant resistance to the cassava mosaic disease (CMD). Plant Mol. Biol. 2004, 56, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Lee, J.Y.; Lee, D.H. Use of SAGE technology to reveal changes in gene expression in Arabidopsis leaves undergoing cold stress. Plant Mol. Biol. 2003, 52, 553–567. [Google Scholar] [CrossRef] [PubMed]
- El Kelish, A.; Zhao, F.; Heller, W.; Durner, J.; Winkler, J.; Behrendt, H.; Traidl-Hoffmann, C.; Horres, R.; Pfeifer, M.; Frank, U.; et al. Ragweed (Ambrosia artemisiifolia) pollen allergenicity: SuperSAGE transcriptomic analysis upon elevated CO2 and drought stress. BMC Plant Biol. 2014, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Doring, F.; Streubel, M.; Brautigam, A.; Gowik, U. Most photorespiratory genes are preferentially expressed in the bundle sheath cells of the C4 grass Sorghum bicolor. J. Exp. Bot. 2016. [Google Scholar] [CrossRef] [PubMed]
- Van Ruissen, F.; Ruijter, J.M.; Schaaf, G.J.; Asgharnegad, L.; Zwijnenburg, D.A.; Kool, M.; Baas, F. Evaluation of the similarity of gene expression data estimated with SAGE and Affymetrix GeneChips. BMC Genom. 2005. [Google Scholar] [CrossRef]
- Gowda, M.; Jantasuriyarat, C.; Dean, R.A.; Wang, G.L. Robust-LongSAGE (RL-SAGE): A substantially improved LongSAGE method for gene discovery and transcriptome analysis. Plant Physiol. 2004, 134, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, H.; Urasaki, N.; Yoshida, K.; Kruger, D.H.; Kahl, G.; Terauchi, R. SuperSAGE: Powerful serial analysis of gene expression. Methods Mol. Biol. 2012, 883, 1–17. [Google Scholar] [PubMed]
- Gowda, M.; Venu, R.C.; Jia, Y.; Stahlberg, E.; Pampanwar, V.; Soderlund, C.; Wang, G.L. Use of robust-long serial analysis of gene expression to identify novel fungal and plant genes involved in host-pathogen interactions. Methods Mol. Biol. 2007, 354, 131–144. [Google Scholar] [PubMed]
- Thomas, S.W.; Glaring, M.A.; Rasmussen, S.W.; Kinane, J.T.; Oliver, R.P. Transcript profiling in the barley mildew pathogen Blumeria graminis by serial analysis of gene expression (SAGE). Mol. Plant Microbe Interact. 2002, 15, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.J.; Wang, Z.X.; Qiao, Z.D. Modified PCR methods for 3’ end amplification from serial analysis of gene expression (SAGE) tags. FEBS J. 2009, 276, 2657–2668. [Google Scholar] [CrossRef] [PubMed]
- Datson, N.A.; van der Perk-de Jong, J.; van den Berg, M.P.; de Kloet, E.R.; Vreugdenhil, E. MicroSAGE: A modified procedure for serial analysis of gene expression in limited amounts of tissue. Nucleic Acids Res. 1999, 27, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Sham, A.; Moustafa, K.; Al-Ameri, S.; Al-Azzawi, A.; Iratni, R.; AbuQamar, S. Identification of Arabidopsis candidate genes in response to biotic and abiotic stresses using comparative microarrays. PLoS ONE 2015, 10, e0125666. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.N.; Moon, S.; Jung, K.H. Genome-wide expression analysis of rice ABC transporter family across spatio-temporal samples and in response to abiotic stresses. J. Plant Physiol. 2014, 171, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Leviatan, N.; Alkan, N.; Leshkowitz, D.; Fluhr, R. Genome-wide survey of cold stress regulated alternative splicing in Arabidopsis thaliana with tiling microarray. PLoS ONE 2013, 8, e66511. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Dong, X.; Park, J.I.; Thamilarasan, S.K.; Lee, S.S.; Kim, Y.K.; Lim, Y.P.; Nou, I.S.; Hur, Y. Genome-wide transcriptome analysis of two contrasting Brassica rapa doubled haploid lines under cold-stresses using Br135K oligomeric chip. PLoS ONE 2014, 9, e106069. [Google Scholar] [CrossRef] [PubMed]
- Yokota, H.; Iehisa, J.C.; Shimosaka, E.; Takumi, S. Line differences in Cor/Lea and fructan biosynthesis-related gene transcript accumulation are related to distinct freezing tolerance levels in synthetic wheat hexaploids. J. Plant Physiol. 2015, 176, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Kwasniewski, M.; Daszkowska-Golec, A.; Janiak, A.; Chwialkowska, K.; Nowakowska, U.; Sablok, G.; Szarejko, I. Transcriptome analysis reveals the role of the root hairs as environmental sensors to maintain plant functions under water-deficiency conditions. J. Exp. Bot. 2016, 67, 1079–1094. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.K.; Liu, S.; Rudd, J.C.; Xue, Q.; Payton, P.; Finlayson, S.A.; Mahan, J.; Akhunova, A.; Holalu, S.V.; Lu, N. Physiology and transcriptomics of water-deficit stress responses in wheat cultivars TAM 111 and TAM 112. J. Plant Physiol. 2014, 171, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Pasini, L.; Bergonti, M.; Fracasso, A.; Marocco, A.; Amaducci, S. Microarray analysis of differentially expressed mRNAs and miRNAs in young leaves of sorghum under dry-down conditions. J. Plant Physiol. 2014, 171, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Allu, A.D.; Soja, A.M.; Wu, A.; Szymanski, J.; Balazadeh, S. Salt stress and senescence: Identification of cross-talk regulatory components. J. Exp. Bot. 2014, 65, 3993–4008. [Google Scholar] [CrossRef] [PubMed]
- Buckley, B.A.; Gracey, A.Y.; Somero, G.N. The cellular response to heat stress in the goby Gillichthys mirabilis: A cDNA microarray and protein-level analysis. J. Exp. Biol. 2006, 209, 2660–2677. [Google Scholar] [CrossRef] [PubMed]
- Foley, M.E.; Chao, W.S.; Horvath, D.P.; Dogramaci, M.; Anderson, J.V. The transcriptomes of dormant leafy spurge seeds under alternating temperature are differentially affected by a germination-enhancing pretreatment. J. Plant Physiol. 2013, 170, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Ophir, R.; Pang, X.; Halaly, T.; Venkateswari, J.; Lavee, S.; Galbraith, D.; Or, E. Gene-expression profiling of grape bud response to two alternative dormancy-release stimuli expose possible links between impaired mitochondrial activity, hypoxia, ethylene-ABA interplay and cell enlargement. Plant Mol. Biol. 2009, 71, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Brazma, A.; Hingamp, P.; Quackenbush, J.; Sherlock, G.; Spellman, P.; Stoeckert, C.; Aach, J.; Ansorge, W.; Ball, C.A.; Causton, H.C.; et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat. Genet. 2001, 29, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Altman, N. Replication, variation and normalisation in microarray experiments. Appl. Bioinf. 2005, 4, 33–44. [Google Scholar] [CrossRef]
- Spruill, S.E.; Lu, J.; Hardy, S.; Weir, B. Assessing sources of variability in microarray gene expression data. Biotechniques 2002, 33, 916–920, 922–923. [Google Scholar] [PubMed]
- Bumgarner, R. DNA microarrays: Types, applications and their future. Curr. Protoc. Mol. Biol. 2013. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Biotechnology 1993, 11, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.J.; Xu, Z.S.; Sun, J.; Li, L.C.; Chen, M.; Yang, G.X.; He, G.Y.; Ma, Y.Z. Investigation of the ASR family in foxtail millet and the role of ASR1 in drought/oxidative stress tolerance. Plant Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.C.; Mei, C.; Liang, S.; Yu, Y.T.; Lu, K.; Wu, Z.; Wang, X.F.; Zhang, D.P. Crucial roles of the pentatricopeptide repeat protein SOAR1 in Arabidopsis response to drought, salt and cold stresses. Plant Mol. Biol. 2015, 88, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Li, Y.; Wu, L.; Zhou, H.; Zhang, G.; Ma, Z. Ectopic expression of a novel Ser/Thr protein kinase from cotton (Gossypium barbadense), enhances resistance to Verticillium dahliae infection and oxidative stress in Arabidopsis. Plant Cell Rep. 2013, 32, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Y.; Chang, L.; Zhang, T.; An, J.; Liu, Y.; Cao, Y.; Zhao, X.; Sha, X.; Hu, T.; et al. MsZEP, a novel zeaxanthin epoxidase gene from alfalfa (Medicago sativa), confers drought and salt tolerance in transgenic tobacco. Plant Cell Rep. 2015, 35, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Machnik, G.; Skudrzyk, E.; Buldak, L.; Labuzek, K.; Ruczynski, J.; Alenowicz, M.; Rekowski, P.; Nowak, P.J.; Okopien, B. A novel, highly selective RT-QPCR method for quantification of MSRV using PNA clamping syncytin-1 (ERVWE1). Mol. Biotechnol. 2015, 57, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Arya, M.; Shergill, I.S.; Williamson, M.; Gommersall, L.; Arya, N.; Patel, H.R. Basic principles of real-time quantitative PCR. Expert Rev. Mol. Diagn. 2005, 5, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.T.; Herrmann, M.G.; Moss, A.A.; Rasmussen, R.P. Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques 1997, 22, 130–131, 134–138. [Google Scholar] [PubMed]
- Gutierrez, L.; Mauriat, M.; Guenin, S.; Pelloux, J.; Lefebvre, J.F.; Louvet, R.; Rusterucci, C.; Moritz, T.; Guerineau, F.; Bellini, C.; et al. The lack of a systematic validation of reference genes: A serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol. J. 2008, 6, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Beaulieu, J.F.; Huggett, J.; Jaggi, R.; Kibenge, F.S.; Olsvik, P.A.; Penning, L.C.; Toegel, S. MIQE precis: Practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. BMC Mol. Biol. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Orntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Ronaghi, M.; Karamohamed, S.; Pettersson, B.; Uhlen, M.; Nyren, P. Real-time DNA sequencing using detection of pyrophosphate release. Anal. Biochem. 1996, 242, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xu, L.; Wang, Y.; Luo, X.; Zhu, X.; Kinuthia, K.B.; Nie, S.; Feng, H.; Li, C.; Liu, L. Transcriptome-based gene expression profiling identifies differentially expressed genes critical for salt stress response in radish (Raphanus sativus L.). Plant Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.R.; Goswami, S.; Sharma, S.K.; Kala, Y.K.; Rai, G.K.; Mishra, D.C.; Grover, M.; Singh, G.P.; Pathak, H.; Rai, A.; et al. Harnessing next generation sequencing in climate change: RNA-seq analysis of heat stress-responsive genes in wheat (Triticum aestivum L.). OMICS 2015, 19, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Moliterni, V.M.; Paris, R.; Onofri, C.; Orru, L.; Cattivelli, L.; Pacifico, D.; Avanzato, C.; Ferrarini, A.; Delledonne, M.; Mandolino, G. Early transcriptional changes in Beta vulgaris in response to low temperature. Planta 2015, 242, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Opitz, N.; Marcon, C.; Paschold, A.; Malik, W.A.; Lithio, A.; Brandt, R.; Piepho, H.-P.; Nettleton, D.; Hochholdinger, F. Extensive tissue-specific transcriptomic plasticity in maize primary roots upon water deficit. J. Exp. Bot. 2016, 67, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Tombuloglu, G.; Tombuloglu, H.; Sakcali, M.S.; Unver, T. High-throughput transcriptome analysis of barley (Hordeum vulgare) exposed to excessive boron. Gene 2015, 557, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Wang, Q.; Sun, R.; Zhang, B. Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J. Exp. Bot. 2015, 66, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Z.; Chen, S.; Ma, L.; Wang, H.; Dong, R.; Wang, Y.; Liu, Z. Global transcriptome profiling analysis reveals insight into saliva-responsive genes in alfalfa. Plant Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, S.; Llaca, V.; May, G.D. Genotyping-by-sequencing in plants. Biology 2012, 1, 460–483. [Google Scholar] [CrossRef] [PubMed]
- Hert, D.G.; Fredlake, C.P.; Barron, A.E. Advantages and limitations of next-generation sequencing technologies: A comparison of electrophoresis and non-electrophoresis methods. Electrophoresis 2008, 29, 4618–4626. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, J.M.; Buck, M.J. Key principles and clinical applications of “next-generation” DNA sequencing. Cancer Prev. Res. 2012, 5, 887–900. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Northern Blot | RPA | DD-PCR | SAGE | DNA Arrays | qPCR | NGS | |
|---|---|---|---|---|---|---|---|
| No. of genes | low | low | medium | high | high | medium | high |
| Specificity | high | high | high | medium | medium | high | high |
| Targeted | yes | yes | no | no | yes/no * | yes | no |
| Scalability | medium | medium | medium | medium | high | high | high |
| Difficulty | low | high | high | high | high | medium | high |
| Cost | low | low | low | medium | high | medium | high |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moustafa, K.; Cross, J.M. Genetic Approaches to Study Plant Responses to Environmental Stresses: An Overview. Biology 2016, 5, 20. https://doi.org/10.3390/biology5020020
Moustafa K, Cross JM. Genetic Approaches to Study Plant Responses to Environmental Stresses: An Overview. Biology. 2016; 5(2):20. https://doi.org/10.3390/biology5020020
Chicago/Turabian StyleMoustafa, Khaled, and Joanna M. Cross. 2016. "Genetic Approaches to Study Plant Responses to Environmental Stresses: An Overview" Biology 5, no. 2: 20. https://doi.org/10.3390/biology5020020
APA StyleMoustafa, K., & Cross, J. M. (2016). Genetic Approaches to Study Plant Responses to Environmental Stresses: An Overview. Biology, 5(2), 20. https://doi.org/10.3390/biology5020020