
Whole-Genome Sequencing and Biosynthetic Gene Cluster Analysis of Novel Entomopathogenic Bacteria Xenorhabdus thailandensis ALN 7.1 and ALN 11.5

 ,
,  and
and 
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Genome Sequencing
2.2. Genome Assembly and Annotation
2.3. Bioinfomatics Analyses
3. Results
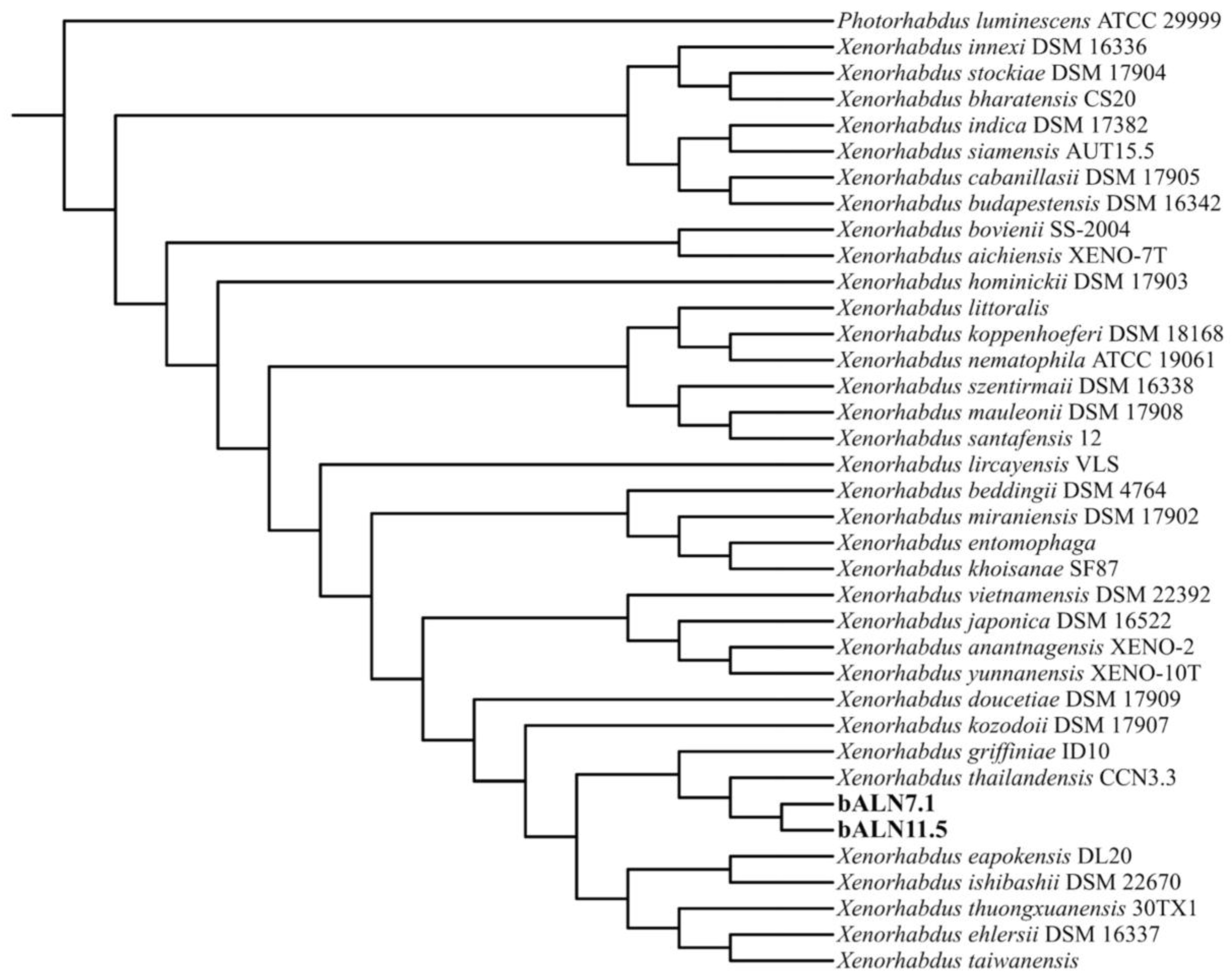
3.1. Bacterial Strain Identification
3.2. Genome Characterization and Comparison
3.3. Functional Genomics Analysis
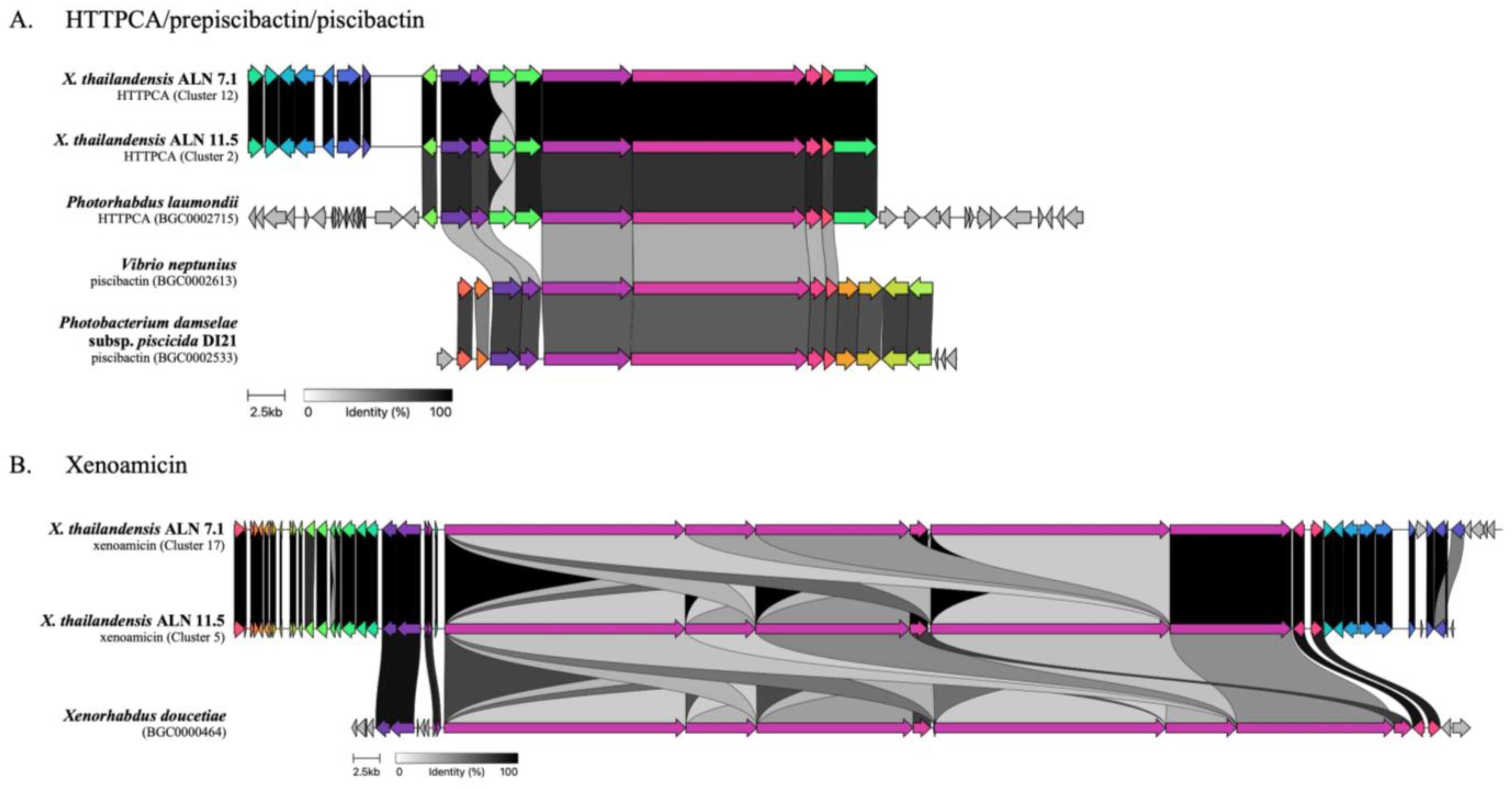
3.4. Elucidation of Biosynthetic Gene Clusters
3.5. Unknown BGCs in Xenorhabdus thailandensis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BGCs | Biosynthetic gene clusters |
| TYGS | The Type Strain Genome Server |
| dDDH | DNA–DNA hybridization |
| GGDC | Genome-to-Genome Distance Calculator |
| ANI | Average Nucleotide Identity |
| COG | Clusters of orthologous genes |
References
- Gokulan, K.; Khare, S.; Cerniglia, C. Metabolic Pathways: Production of secondary metabolites of bacteria. In Encyclopedia of Food Microbiology, 2nd ed.; Batt, C.A., Tortorello, M.L., Eds.; Academic Press: Oxford, UK, 2014; pp. 561–569. [Google Scholar]
- Korkina, L.; Mikhal’Chik, E.; Suprun, M.; Pastore, S.; Dal Toso, R. Molecular mechanisms underlying wound healing and anti-inflammatory properties of naturally occurring biotechnologically produced phenylpropanoid glycosides. Cell. Mol. Biol. 2007, 53, 84–91. [Google Scholar] [PubMed]
- Omojate Godstime, C.; Enwa Felix, O.; Jewo Augustina, O.; Eze Christopher, O. Mechanisms of antimicrobial actions of phytochemicals against enteric pathogens—A review. J. Pharm. Chem. Biol. Sci. 2014, 2, 77–85. [Google Scholar]
- Shukla, S.; Habbu, P.; Kulkarni, V.; Jagadish, K.; Pandey, A.; Sutariya, V. Endophytic microbes: A novel source for biologically/pharmacologically active secondary metabolites. Asian J. Pharmacol. Toxicol. 2014, 2, 1–6. [Google Scholar]
- Jensen, P.R.; Chavarria, K.L.; Fenical, W.; Moore, B.S.; Ziemert, N. Challenges and triumphs to genomics-based natural product discovery. J. Ind. Microbiol. Biotechnol. 2014, 41, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Qiao, Y.; Ang, E.L.; Zhao, H. Using natural products for drug discovery: The impact of the genomics era. Expert Opin. Drug Discov. 2017, 12, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Albarano, L.; Esposito, R.; Ruocco, N.; Costantini, M. Genome mining as new challenge in natural products discovery. Mar. Drugs 2020, 18, 199. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, B.O.; Van Lanen, S.G.; Baltz, R.H. Microbial genome mining for accelerated natural products discovery: Is a renaissance in the making? J. Ind. Microbiol. Biotechnol. 2014, 41, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Dutky, S.R. Insect microbiology. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 1959; Volume 1, pp. 175–200. [Google Scholar]
- Poinaer Jr, G.; Thomas, G. Significance of Achromobacter nematophilus (Achromobacteriaceae: Eubacteriales) in the development of the nematode, DD-136 (Neoaplectana sp.: Steinernematidae). Parasitology 1966, 56, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Chaston, J.M.; Suen, G.; Tucker, S.L.; Andersen, A.W.; Bhasin, A.; Bode, E.; Bode, H.B.; Brachmann, A.O.; Cowles, C.E.; Cowles, K.N. The entomopathogenic bacterial endosymbionts Xenorhabdus and Photorhabdus: Convergent lifestyles from divergent genomes. PLoS ONE 2011, 6, e27909. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.W.; Chen, G.; Webster, J.M.; Dunphy, G.B. Stability and activities of antibiotics produced during infection of the insect Galleria mellonella by two isolates of Xenorhabdus nematophilus. Appl. Environ. Microbiol. 1994, 60, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Tobias, N.J.; Shi, Y.-M.; Bode, H.B. Refining the natural product repertoire in entomopathogenic bacteria. Trends Microbiol. 2018, 26, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Pantel, L.; Florin, T.; Dobosz-Bartoszek, M.; Racine, E.; Sarciaux, M.; Serri, M.; Houard, J.; Campagne, J.M.; de Figueiredo, R.M.; Midrier, C.; et al. Odilorhabdins, Antibacterial Agents that Cause Miscoding by Binding at a New Ribosomal Site. Mol. Cell 2018, 70, 83–94.e7. [Google Scholar] [CrossRef] [PubMed]
- Nosopharm. Press Release: Nosopharm and Evotec Enter into Collaboration to Accelerate Development of Novel Antibiotics. Available online: https://www.life-sciences-germany.com/news/nosopharm-evotec-partner-advance-biodd-sas-fse-evt-2001-115234.html (accessed on 8 July 2025).
- McInerney, B.V.; Taylor, W.C.; Lacey, M.J.; Akhurst, R.J.; Gregson, R.P. Biologically active metabolites from Xenorhabdus spp., Part 2. Benzopyran-1-one derivatives with gastroprotective activity. J. Nat. Prod. 1991, 54, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Huber, E.M.; Westphalen, M.; Gellner, J.; Bode, E.; Köbel, T.; Grün, P.; Alanjary, M.M.; Glatter, T.; Schindler, D.; et al. Identification, structure and function of the methyltransferase involved in the biosynthesis of the dithiolopyrrolone antibiotic xenorhabdin. bioRxiv 2024. [Google Scholar] [CrossRef]
- Nollmann, F.I.; Dauth, C.; Mulley, G.; Kegler, C.; Kaiser, M.; Waterfield, N.R.; Bode, H.B. Insect-specific production of new GameXPeptides in Photorhabdus luminescens TTO1, widespread natural products in entomopathogenic bacteria. ChemBioChem 2015, 16, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Tobias, N.J.; Wolff, H.; Djahanschiri, B.; Grundmann, F.; Kronenwerth, M.; Shi, Y.M.; Simonyi, S.; Grün, P.; Shapiro-Ilan, D.; Pidot, S.J.; et al. Natural product diversity associated with the nematode symbionts Photorhabdus and Xenorhabdus. Nat. Microbiol. 2017, 2, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-M.; Hirschmann, M.; Shi, Y.-N.; Ahmed, S.; Abebew, D.; Tobias, N.J.; Grün, P.; Crames, J.J.; Pöschel, L.; Kuttenlochner, W. Global analysis of biosynthetic gene clusters reveals conserved and unique natural products in entomopathogenic nematode-symbiotic bacteria. Nat. Chem. 2022, 14, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Bisch, G.; Ogier, J.C.; Médigue, C.; Rouy, Z.; Vincent, S.; Tailliez, P.; Givaudan, A.; Gaudriault, S. Comparative genomics between two Xenorhabdus bovienii strains highlights differential evolutionary scenarios within an entomopathogenic bacterial species. Genome Biol. Evol. 2016, 8, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Thanwisai, A.; Machado, R.A.; Bhat, A.H.; Pidot, S.J.; Tandhavanant, S.; Subkrasae, C.; Meesil, W.; Ardpairin, J.; Pansri, S.; Vitta, A. Xenorhabdus bharatensis sp. nov., Xenorhabdus entomophaga sp. nov., Xenorhabdus siamensis sp. nov., and Xenorhabdus thailandensis sp. nov. isolated from Steinernema Entomopathogenic Nematodes. Curr. Microbiol. 2025, 82, 10. [Google Scholar] [CrossRef] [PubMed]
- Ardpairin, J.; Subkrasae, C.; Dumidae, A.; Pansri, S.; Homkaew, C.; Meesil, W.; Kumchantuek, T.; Phoungpetchara, I.; Dillman, A.R.; Pavesi, C. Symbiotic bacteria associated with entomopathogenic nematodes showed molluscicidal activity against Biomphalaria glabrata, an intermediate host of Schistosoma mansoni. Parasites Vectors 2024, 17, 529. [Google Scholar] [CrossRef] [PubMed]
- Sherathiya, V.N.; Schaid, M.D.; Seiler, J.L.; Lopez, G.C.; Lerner, T.N. GuPPy, a Python toolbox for the analysis of fiber photometry data. Sci. Rep. 2021, 11, 24212. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 May 2025).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.; Menzel, P. Filtlong: Quality Filtering Tool for Long Reads. Github 2019. Available online: https://github.com/rrwick/Filtlong (accessed on 19 May 2025).
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Vader, L.; Szenei, J.; Reitz, Z.L.; Augustijn, H.E.; Cediel-Becerra, J.D.; de Crécy-Lagard, V.; Koetsier, R.A.; Williams, S.E. antiSMASH 8.0: Extended gene cluster detection capabilities and analyses of chemistry, enzymology, and regulation. Nucleic Acids Res. 2025, 53, W32–W38. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.; Chooi, Y.-H. Clinker & clustermap. js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef] [PubMed]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Palma, L.; Frizzo, L.; Kaiser, S.; Berry, C.; Caballero, P.; Bode, H.B.; Del Valle, E.E. Genome sequence analysis of native Xenorhabdus strains isolated from entomopathogenic nematodes in Argentina. Toxins 2024, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.; Conrad, K.; Ahuja, B.; Göker, M.; Hahnke, R.L.; Spunde, A.; Ivanova, N.N.; Seshadri, R.; Stephens, C. Genome sequences of key bacterial symbionts of entomopathogenic nematodes: Xenorhabdus cabanillasii DSM17905, Xenorhabdus ehlersii DSM16337, Xenorhabdus japonica DSM16522, Xenorhabdus koppenhoeferii DSM18168, and Xenorhabdus mauleonii DSM17908. Microbiol. Resour. Announc. 2023, 12, e00548-23. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yang, C.; Zhang, G.; Wu, D. GCI: A continuity inspector for complete genome assembly. Bioinformatics 2024, 40, btae633. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, Z.; Ke, J.; Engel, Y.; Shi, Y.-M.; Robinson, D.; Bingol, K.; Zhang, Z.; Bowen, B.; Louie, K. CRAGE enables rapid activation of biosynthetic gene clusters in undomesticated bacteria. Nat. Microbiol. 2019, 4, 2498–2510. [Google Scholar] [CrossRef] [PubMed]
- Galvis, F.; Ageitos, L.; Rodríguez, J.; Jiménez, C.; Barja, J.L.; Lemos, M.L.; Balado, M. Vibrio neptunius produces piscibactin and amphibactin and both siderophores contribute significantly to virulence for clams. Front. Cell. Infect. Microbiol. 2021, 11, 750567. [Google Scholar] [CrossRef] [PubMed]
- Osorio, C.R.; Rivas, A.J.; Balado, M.; Fuentes-Monteverde, J.C.; Rodríguez, J.; Jiménez, C.; Lemos, M.L.; Waldor, M.K. A transmissible plasmid-borne pathogenicity island confers piscibactin biosynthesis in the fish pathogen Photobacterium damselae subsp. piscicida. Appl. Environ. Microbiol. 2015, 81, 5867–5879. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.; Sze, S.-H.; Thon, M.R. Identifying clusters of functionally related genes in genomes. Bioinformatics 2007, 23, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Kohler, P.R.; Zheng, J.Y.; Schoffers, E.; Rossbach, S. Inositol catabolism, a key pathway in Sinorhizobium meliloti for competitive host nodulation. Appl. Environ. Microbiol. 2010, 76, 7972–7980. [Google Scholar] [CrossRef] [PubMed]
- Mootz, H.D.; Marahiel, M.A. The tyrocidine biosynthesis operon of Bacillus brevis: Complete nucleotide sequence and biochemical characterization of functional internal adenylation domains. J. Bacteriol. 1997, 179, 6843–6850. [Google Scholar] [CrossRef] [PubMed]
- Díaz, E.; Ferrández, A.; Prieto María, A.; García José, L. Biodegradation of aromatic compounds by Escherichia coli. Microbiol. Mol. Biol. Rev. 2001, 65, 523–569. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Anantharaman, V.; Balaji, S.; Babu, M.M.; Iyer, L.M. The many faces of the helix-turn-helix domain: Transcription regulation and beyond. FEMS Microbiol. Rev. 2005, 29, 231–262. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-Ya, K.; Omura, S.; Cane, D.E.; Ikeda, H. Terpene synthases are widely distributed in bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, J.D.; Alsup, T.A.; Xu, B.; Li, Z. Bacterial terpenome. Nat. Prod. Rep. 2021, 38, 905–980. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Ma, K. Characterization of a zinc-containing alcohol dehydrogenase with stereoselectivity from the hyperthermophilic archaeon Thermococcus guaymasensis. J. Bacteriol. 2011, 193, 3009–3019. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-F.; Lin, Y.-Y.; Lan, C.-Y. Contribution of EmrAB efflux pumps to colistin resistance in Acinetobacter baumannii. J. Microbiol. 2017, 55, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gil, J.; Behrendorff, J.; Douw, A.; Vickers, C.E. The methylerythritol phosphate pathway as an oxidative stress sense and response system. Nat. Commun. 2024, 15, 5303. [Google Scholar] [CrossRef] [PubMed]
- Huvet, M.; Toni, T.; Sheng, X.; Thorne, T.; Jovanovic, G.; Engl, C.; Buck, M.; Pinney, J.W.; Stumpf, M.P. The evolution of the phage shock protein response system: Interplay between protein function, genomic organization, and system function. Mol. Biol. Evol. 2011, 28, 1141–1155. [Google Scholar] [CrossRef] [PubMed]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, B.J.; Schwalen, C.J.; Mann, G.; Naismith, J.H.; Mitchell, D.A. YcaO-dependent posttranslational amide activation: Biosynthesis, structure, and function. Chem. Rev. 2017, 117, 5389–5456. [Google Scholar] [CrossRef] [PubMed]
- Blankenfeldt, W.; Parsons, J.F. The structural biology of phenazine biosynthesis. Curr. Opin. Struct. Biol. 2014, 29, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Diederich, C.; Leypold, M.; Culka, M.; Weber, H.; Breinbauer, R.; Ullmann, G.M.; Blankenfeldt, W. Mechanisms and specificity of phenazine biosynthesis protein PhzF. Sci. Rep. 2017, 7, 6272. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pan, H.-X.; Tang, G.-L. Newly Discovered mechanisms of antibiotic self-resistance with multiple enzymes acting at different locations and stages. Antibiotics 2023, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.L.; Wackett, L.P. Rings of Power: Enzymatic Routes to β-Lactones. Compr. Nat. Prod. III 2020, 4, 323–345. [Google Scholar] [CrossRef]
- Ogier, J.-C.; Pagès, S.; Bisch, G.; Chiapello, H.; Médigue, C.; Rouy, Z.; Teyssier, C.; Vincent, S.; Tailliez, P.; Givaudan, A. Attenuated virulence and genomic reductive evolution in the entomopathogenic bacterial symbiont species, Xenorhabdus poinarii. Genome Biol. Evol. 2014, 6, 1495–1513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-D.; Isbrandt, T.; Lindqvist, L.L.; Larsen, T.O.; Gram, L. Holomycin, an antibiotic secondary metabolite, is required for biofilm formation by the native producer Photobacterium galatheae S2753. Appl. Environ. Microbiol. 2021, 87, e00169-21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wever, W.J.; Walsh, C.T.; Bowers, A.A. Correction: Dithiolopyrrolones: Biosynthesis, synthesis, and activity of a unique class of disulfide-containing antibiotics. Nat. Prod. Rep. 2015, 32, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Merrouche, R.; Yekkour, A.; Coppel, Y.; Bouras, N.; Zitouni, A.; Mathieu, F.; Sabaou, N. Saccharothrix algeriensis NRRL B-24137, the first non-Streptomyces actinobacterium, produces holomycin after cystine feeding. Arch. Microbiol. 2020, 202, 2509–2516. [Google Scholar] [CrossRef] [PubMed]
- Schimming, O.; Challinor, V.L.; Tobias, N.J.; Adihou, H.; Grün, P.; Pöschel, L.; Richter, C.; Schwalbe, H.; Bode, H.B. Structure, biosynthesis, and occurrence of bacterial pyrrolizidine alkaloids. Angew. Chem. 2015, 54, 12702–12705. [Google Scholar] [CrossRef] [PubMed]
- Effert, J.; Westphalen, M.; Calderari, A.; Shi, Y.M.; Elamri, I.; Najah, S.; Grün, P.; Li, Y.; Gruez, A.; Weissman, K.J.; et al. Pyrrolizwilline, a unique bacterial alkaloid assembled by a nonribosomal peptide synthetase and non-enzymatic dimerization. Angew. Chem. 2024, 63, e202411258. [Google Scholar] [CrossRef] [PubMed]
- Ryall, B.; Mitchell, H.; Mossialos, D.; Williams, H.D. Cyanogenesis by the entomopathogenic bacterium Pseudomonas entomophila. Lett. Appl. Microbiol. 2009, 49, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Blumer, C.; Haas, D. Mechanism, regulation, and ecological role of bacterial cyanide biosynthesis. Arch. Microbiol. 2000, 173, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Moreno, A.; Stefanato, F.L.; Ford, J.J.; Trippel, C.; Uszkoreit, S.; Ferrafiat, L.; Grenga, L.; Dickens, R.; Kelly, N.; Kingdon, A.D.H.; et al. Pan-genome analysis identifies intersecting roles for Pseudomonas specialized metabolites in potato pathogen inhibition. eLife 2021, 10, e71900. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-H.; Cho, W.; Hamchand, R.; Oh, J.; Crawford, J.M. A conserved nonribosomal peptide synthetase in Xenorhabdus bovienii produces citrulline-functionalized lipopeptides. J. Nat. Prod. 2021, 84, 2692–2699. [Google Scholar] [CrossRef] [PubMed]
- Engel, Y.; Windhorst, C.; Lu, X.; Goodrich-Blair, H.; Bode, H.B. The global regulators Lrp, LeuO, and HexA control secondary metabolism in entomopathogenic bacteria. Front. Microbiol. 2017, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; De Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C. Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Bode, H.B. Entomopathogenic bacteria as a source of secondary metabolites. Curr. Opin. Struct. Biol. 2009, 13, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Fortinez, C.M.; Bloudoff, K.; Harrigan, C.; Sharon, I.; Strauss, M.; Schmeing, T.M. Structures and function of a tailoring oxidase in complex with a nonribosomal peptide synthetase module. Nat. Commun. 2022, 13, 548. [Google Scholar] [CrossRef] [PubMed]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Brown, L.C.W.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics | X. thailandensis Strain ALN7.1 | X. thailandensis Strain ALN11.5 |
|---|---|---|
| Genome size (bp) | 4,021,996 | 4,024,527 |
| Number of contigs | 49 | 19 |
| Largest contig | 1,262,586 | 1,303,736 |
| CDS | 3448 | 3471 |
| rRNA | 24 | 22 |
| repeat_region | 3 | 3 |
| tRNA | 79 | 81 |
| tmRNA | 1 | 1 |
| N50 | 1,245,011 | 1,078,348 |
| N90 | 216,926 | 406,632 |
| auN | 987,176 | 985,771 |
| L50 | 2 | 2 |
| L90 | 5 | 4 |
| GC content (%) | 43.33 | 43.36 |
| NCBI accession number | SAMN48884708 | SAMN48884709 |
| Cluster | Type | Similarity Confidence | Most Similar Known Cluster | Gene Cluster from Organisms | MiBiG Accession/Reference |
|---|---|---|---|---|---|
| Cluster 1 Region 1.1 | NRPS | unknown | |||
| Cluster 2 Region 1.2 | Terpene-precursor | unknown | |||
| Cluster 3 Region 1.3 | NRPS | High | holomycin | Photobacterium galatheae | BGC0002412 |
| Cluster 4 Region 1.4 | NRPS | High | pyrrolizixenamide A | Xenorhabdus szentirmaii DSM 16338 | BGC0001873 |
| Cluster 5 Region 1.5 | Other: furanz | unknown | |||
| Cluster 6 Region 1.6 | Other: butyrolactone | unknown | |||
| Cluster 7 Region 2.1 | Other: hydrogen-cyanide | High | hydrogen cyanide | Pseudomonas fluorescens | BGC0002345 |
| Cluster 8 Region 2.2 | NRPS | High | bovienimide A | Xenorhabdus bovienii SS-2004 | BGC0002135 |
| Cluster 9 Region 2.3 | NRPS | unknown | |||
| Cluster 10 Region 2.4 | Other: betalactone | IOC | |||
| Cluster 11 Region 2.5 | NRPS | High | gamexpeptide C | Photorhabdus laumondii subsp. laumondii TTO1 | BGC0001128 |
| Cluster 12 Region 3.1 | Hybrid: NRPS, T1PKS | Low | HTTPCA/prepiscibactin/piscibactin | Photorhabdus laumondii subsp. laumondii | BGC0002715 |
| Cluster 13 Region 3.1 | unknown | ||||
| Cluster 14 Region 3.2 | Azole-containing-RiPP | unknown | |||
| Cluster 15 Region 4.1 | Phenazine | unknown | |||
| Cluster 16 Region 4.2 | Hybrid: NRPS, T1PKS, NRPS-like | unknown | |||
| Cluster 17 Region 5.1 | NRPS | Medium | xenoamicin A/xenoamicin B | Xenorhabdus doucetiae | BGC0000464 |
| Cluster 18 Region 5.2 | Hybrid: T1PKS, NRPS | unknown | |||
| Cluster 19 Region 8.1 | Hybrid: betalactone, NRPKS-like | unknown |
| Cluster | Type | Similarity Confidence | Most Similar Known Cluster | Gene Cluster from Organisms | MiBiG Accession/Reference |
|---|---|---|---|---|---|
| Cluster 1 Region 1.1 | Azole-containing-RiPP | unknown | |||
| Cluster 2 Region 1.2 | Hybrid: NRP-metallophore, NRPS, T1PKS | Low | HTTPCA/prepiscibactin/piscibactin | Photorhabdus laumondii subsp. laumondii | BGC0002715 |
| Cluster 3 Region 1.2 | unknown | ||||
| Cluster 4 Region 1.3 | Hybrid: NRPS, T1PKS | unknown | |||
| Cluster 5 Region 1.4 | NRPS | Medium | xenoamicin A/xenoamicin B | Xenorhabdus doucetiae | BGC0000464 |
| Cluster 6 Region 1.5 | Hybrid: betalactone, NRPKS-like | unknown | |||
| Cluster 7 Region 1.6 | Other: hydrogen-cyanide | High | hydrogen cyanide | Pseudomonas fluorescens | BGC0002345 |
| Cluster 8 Region 1.7 | NRPS | High | bovienimide A | Xenorhabdus bovienii SS-2004 | BGC0002135 |
| Cluster 9 Region 2.1 | NRPS | High | gamexpeptide C | Photorhabdus laumondii subsp. laumondii TTO1 | BGC0001128 |
| Cluster 10 Region 2.2 | Other: betalactone | IOC | |||
| Cluster 11 Region 2.3 | NRPS | unknown | |||
| Cluster 12 Region 3.1 | NRPS | High | pyrrolizixenamide A | Xenorhabdus szentirmaii DSM 16338 | BGC0001873 |
| Cluster 13 Region 3.2 | Other: furan | unknown | |||
| Cluster 14 Region 3.3 | Other: butyrolactone | unknown | |||
| Cluster 15 Region 4.1 | NRPS | unknown | |||
| Cluster 16 Region 4.2 | Terpene-precursor | unknown | |||
| Cluster 17 Region 5.1 | Phenazine | unknown | |||
| Cluster 18 Region 5.2 | Hybrid: NRPS, T1PKS, NRPS-like | unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meesil, W.; Ardpairin, J.; Sharkey, L.K.R.; Pidot, S.J.; Vitta, A.; Thanwisai, A. Whole-Genome Sequencing and Biosynthetic Gene Cluster Analysis of Novel Entomopathogenic Bacteria Xenorhabdus thailandensis ALN 7.1 and ALN 11.5. Biology 2025, 14, 905. https://doi.org/10.3390/biology14080905
Meesil W, Ardpairin J, Sharkey LKR, Pidot SJ, Vitta A, Thanwisai A. Whole-Genome Sequencing and Biosynthetic Gene Cluster Analysis of Novel Entomopathogenic Bacteria Xenorhabdus thailandensis ALN 7.1 and ALN 11.5. Biology. 2025; 14(8):905. https://doi.org/10.3390/biology14080905
Chicago/Turabian StyleMeesil, Wipanee, Jiranun Ardpairin, Liam K. R. Sharkey, Sacha J. Pidot, Apichat Vitta, and Aunchalee Thanwisai. 2025. "Whole-Genome Sequencing and Biosynthetic Gene Cluster Analysis of Novel Entomopathogenic Bacteria Xenorhabdus thailandensis ALN 7.1 and ALN 11.5" Biology 14, no. 8: 905. https://doi.org/10.3390/biology14080905
APA StyleMeesil, W., Ardpairin, J., Sharkey, L. K. R., Pidot, S. J., Vitta, A., & Thanwisai, A. (2025). Whole-Genome Sequencing and Biosynthetic Gene Cluster Analysis of Novel Entomopathogenic Bacteria Xenorhabdus thailandensis ALN 7.1 and ALN 11.5. Biology, 14(8), 905. https://doi.org/10.3390/biology14080905