

HSP70-Mediated Autophagy-Apoptosis-Inflammation Network and Neuroprotection Induced by Heat Acclimatization


{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
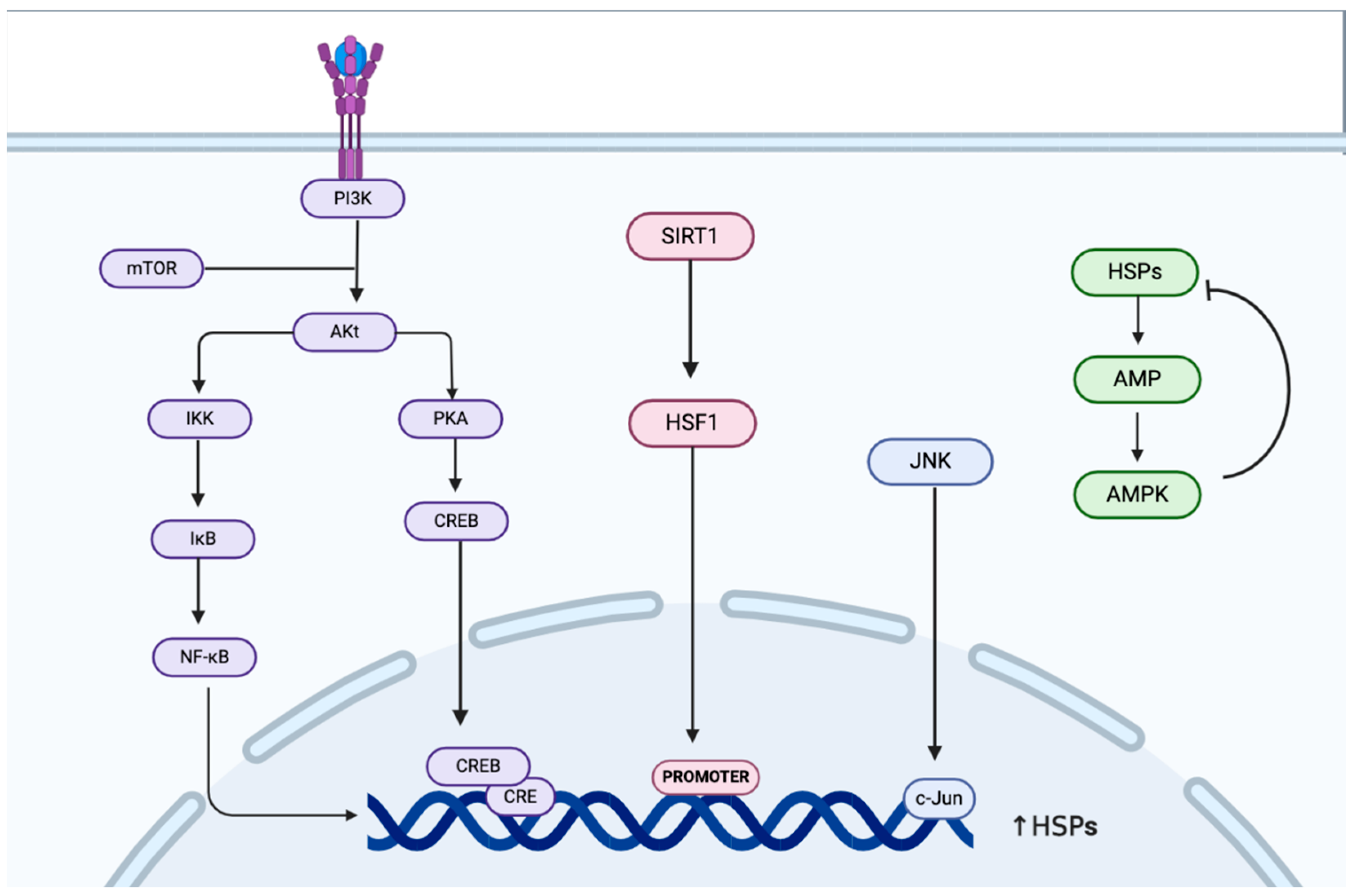
2. Regulation of HSP70 Expression and Activity During Heat Acclimatization
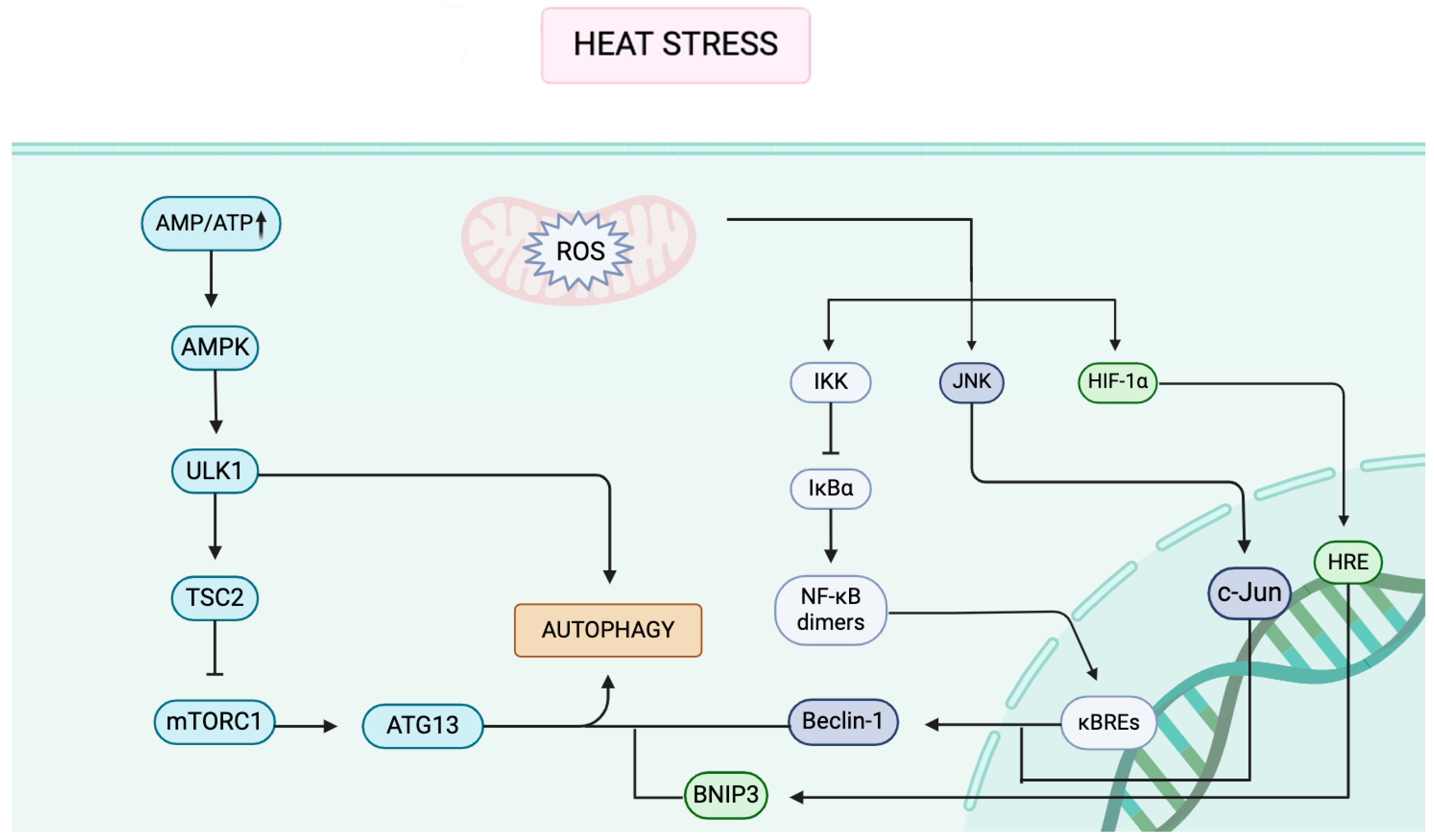
3. Autophagy Signaling Pathways Under Heat Stress
4. Expression of HSP70 in Brain Autophagy
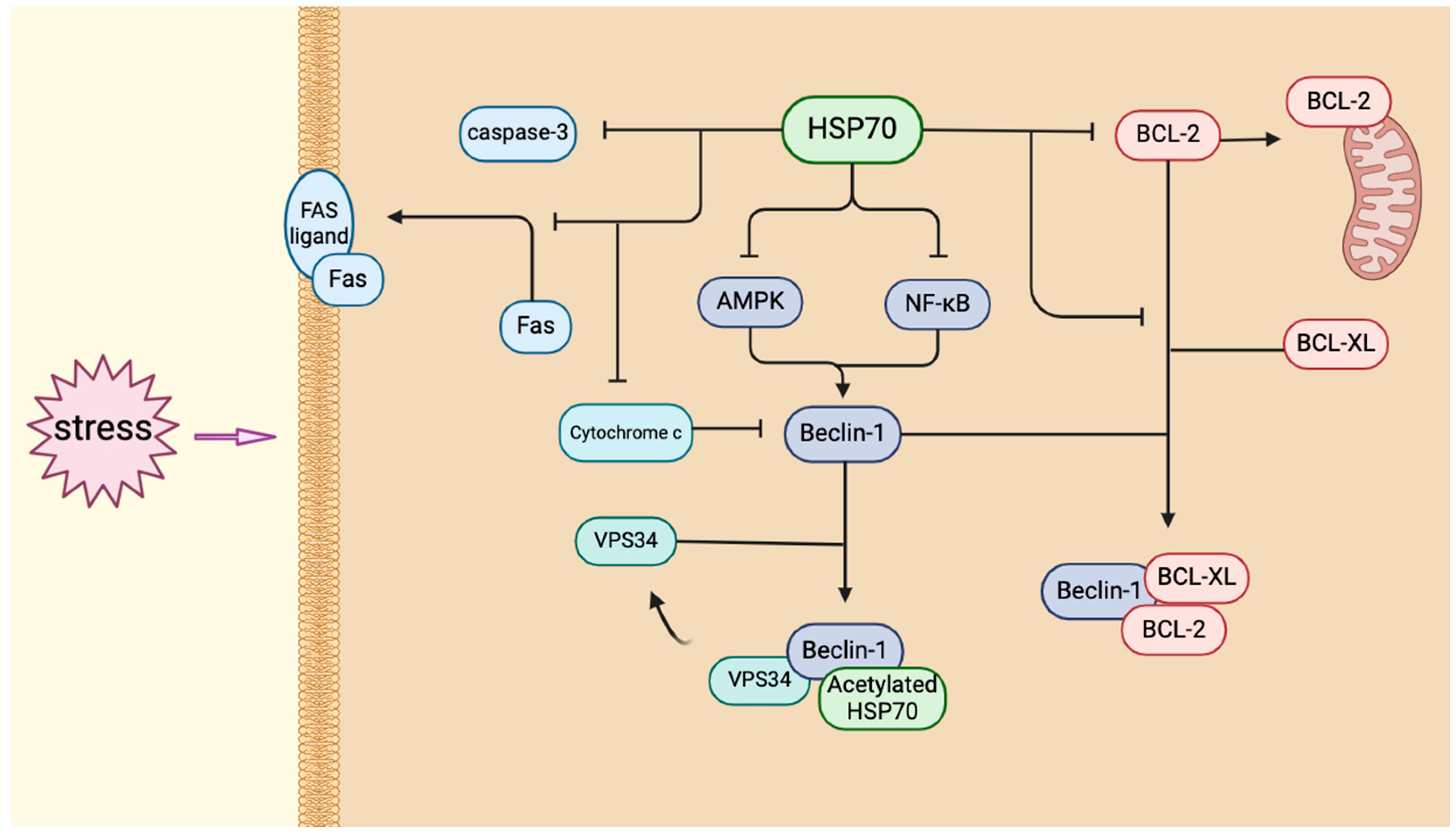
5. HSP70 and Autophagy in HA
6. Conclusions
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iba, T.; Kondo, Y.; Maier, C.L.; Helms, J.; Ferrer, R.; Levy, J.H. Impact of hyper- and hypothermia on cellular and whole-body physiology. J. Intensive Care 2025, 13, 4. [Google Scholar] [CrossRef]
- Tong, S.; Prior, J.; McGregor, G.; Shi, X.; Kinney, P. Urban heat: An increasing threat to global health. BMJ 2021, 375, n2467. [Google Scholar] [CrossRef]
- Kuo, W.Y.; Huang, C.C.; Chen, C.A.; Ho, C.H.; Tang, L.Y.; Lin, H.J.; Su, S.B.; Wang, J.J.; Hsu, C.C.; Chang, C.P.; et al. Heat-related illness and dementia: A study integrating epidemiological and experimental evidence. Alzheimer’s Res. Ther. 2024, 16, 145. [Google Scholar] [CrossRef] [PubMed]
- Romanello, M.; di Napoli, C.; Green, C.; Kennard, H.; Lampard, P.; Scamman, D.; Walawender, M.; Ali, Z.; Ameli, N.; Ayeb-Karlsson, S.; et al. The 2023 report of the Lancet Countdown on health and climate change: The imperative for a health-centred response in a world facing irreversible harms. Lancet 2023, 402, 2346–2394. [Google Scholar] [CrossRef] [PubMed]
- Kenny, G.P.; Jay, O. Thermometry, calorimetry, and mean body temperature during heat stress. Compr. Physiol. 2013, 3, 1689–1719. [Google Scholar] [CrossRef] [PubMed]
- Fealey, R.D. Interoception and autonomic nervous system reflexes thermoregulation. Handb. Clin. Neurol. 2013, 117, 79–88. [Google Scholar] [CrossRef]
- Crandall, C.G.; González-Alonso, J. Cardiovascular function in the heat-stressed human. Acta Physiol. 2010, 199, 407–423. [Google Scholar] [CrossRef]
- Misset, B.; De Jonghe, B.; Bastuji-Garin, S.; Gattolliat, O.; Boughrara, E.; Annane, D.; Hausfater, P.; Garrouste-Orgeas, M.; Carlet, J. Mortality of patients with heatstroke admitted to intensive care units during the 2003 heat wave in France: A national multiple-center risk-factor study. Crit. Care Med. 2006, 34, 1087–1092. [Google Scholar] [CrossRef]
- Khan, A.A. Heat related illnesses. Review of an ongoing challenge. Saudi Med. J. 2019, 40, 1195–1201. [Google Scholar] [CrossRef]
- Adnan Bukhari, H. A Systematic Review on Outcomes of Patients with Heatstroke and Heat Exhaustion. Open Access Emerg. Med. 2023, 15, 343–354. [Google Scholar] [CrossRef]
- Périard, J.D.; Travers, G.J.S.; Racinais, S.; Sawka, M.N. Cardiovascular adaptations supporting human exercise-heat acclimation. Auton. Neurosci. 2016, 196, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.; Ganio, M.S.; Schlader, Z.J.; Lucas, R.A.I.; Gagnon, D.; Rivas, E.; Davis, S.L.; Kowalske, K.J.; Crandall, C.G. Post Junctional Sudomotor and Cutaneous Vascular Responses in Noninjured Skin Following Heat Acclimation in Burn Survivors. J. Burn Care Res. 2017, 38, e284–e292. [Google Scholar] [CrossRef]
- Taylor, N.A.S. Human heat adaptation. Compr. Physiol. 2014, 4, 325–365. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, S.; Minson, C.T. Heat acclimation improves cutaneous vascular function and sweating in trained cyclists. J. Appl. Physiol. 2010, 109, 1736–1743. [Google Scholar] [CrossRef]
- Hodge, D.; Jones, D.; Martinez, R.; Buono, M.J. Time course of the attenuation of sympathetic nervous activity during active heat acclimation. Auton. Neurosci. 2013, 177, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Barua, S.; Huang, M.Y.; Park, J.; Yenari, M.A.; Lee, J.E. Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury. Cells 2020, 9, 2020. [Google Scholar] [CrossRef]
- Gotoh, T.; Terada, K.; Oyadomari, S.; Mori, M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004, 11, 390–402. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef]
- Neuer, A.; Spandorfer, S.D.; Giraldo, P.; Dieterle, S.; Rosenwaks, Z.; Witkin, S.S. The role of heat shock proteins in reproduction. Hum. Reprod. Update 2000, 6, 149–159. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.R. Induction of heat shock (stress) genes in the mammalian brain by hyperthermia and other traumatic events: A current perspective. J. Neurosci. Res. 1990, 27, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Gao, Y.; Chen, R.; Liu, W.; He, C.; Hao, J.; Zhou, M.; Kan, H. Temperature-related death burden of various neurodegenerative diseases under climate warming: A nationwide modelling study. Nat. Commun. 2023, 14, 8236. [Google Scholar] [CrossRef]
- Turturici, G.; Sconzo, G.; Geraci, F. Hsp70 and its molecular role in nervous system diseases. Biochem. Res. Int. 2011, 2011, 618127. [Google Scholar] [CrossRef]
- Zatsepina, O.G.; Evgen’ev, M.B.; Garbuz, D.G. Role of a Heat Shock Transcription Factor and the Major Heat Shock Protein Hsp70 in Memory Formation and Neuroprotection. Cells 2021, 10, 1638. [Google Scholar] [CrossRef]
- Kim, J.Y.; Han, Y.; Lee, J.E.; Yenari, M.A. The 70-kDa heat shock protein (Hsp70) as a therapeutic target for stroke. Expert Opin. Ther. Targets 2018, 22, 191–199. [Google Scholar] [CrossRef]
- Beckmann, R.P.; Lovett, M.; Welch, W.J. Examining the function and regulation of hsp70 in cells subjected to metabolic stress. J. Cell Biol. 1992, 117, 1137–1150. [Google Scholar] [CrossRef]
- Bittencourt, A.; Porto, R.R. eHSP70/iHSP70 and divergent functions on the challenge: Effect of exercise and tissue specificity in response to stress. Clin. Physiol. Funct. Imaging 2017, 37, 99–105. [Google Scholar] [CrossRef]
- Horowitz, M.; Eli-Berchoer, L.; Wapinski, I.; Friedman, N.; Kodesh, E. Stress-related genomic responses during the course of heat acclimation and its association with ischemic-reperfusion cross-tolerance. J. Appl. Physiol. 2004, 97, 1496–1507. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Sharp, F.R.; Zhan, X.; Liu, D.Z. Heat Shock Proteins in the Brain: Role of Hsp70, Hsp 27, and HO-1 (Hsp32) and Their Therapeutic Potential. Transl. Stroke Res. 2013, 4, 685–692. [Google Scholar] [CrossRef]
- De Graff, A.M.; Mosedale, D.E.; Sharp, T.; Dill, K.A.; Grainger, D.J. Proteostasis is adaptive: Balancing chaperone holdases against foldases. PLoS Comput. Biol. 2020, 16, e1008460. [Google Scholar] [CrossRef]
- Venediktov, A.A.; Bushueva, O.Y.; Kudryavtseva, V.A.; Kuzmin, E.A.; Moiseeva, A.V.; Baldycheva, A.; Meglinski, I.; Piavchenko, G.A. Closest horizons of Hsp70 engagement to manage neurodegeneration. Front. Mol. Neurosci. 2023, 16, 1230436. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Koukoulas, I. HSP72 gene expression progressively increases in human skeletal muscle during prolonged, exhaustive exercise. J. Appl. Physiol. 2000, 89, 1055–1060. [Google Scholar] [CrossRef]
- Puntschart, A.; Vogt, M.; Widmer, H.R.; Hoppeler, H.; Billeter, R. Hsp70 expression in human skeletal muscle after exercise. Acta Physiol. Scand. 1996, 157, 411–417. [Google Scholar] [CrossRef]
- Liu, Y.; Mayr, S.; Opitz-Gress, A.; Zeller, C.; Lormes, W.; Baur, S.; Lehmann, M.; Steinacker, J.M. Human skeletal muscle HSP70 response to training in highly trained rowers. J. Appl. Physiol. 1999, 86, 101–104. [Google Scholar] [CrossRef]
- Fehrenbach, E.; Passek, F.; Niess, A.M.; Pohla, H.; Weinstock, C.; Dickhuth, H.H.; Northoff, H. HSP expression in human leukocytes is modulated by endurance exercise. Med. Sci. Sports Exerc. 2000, 32, 592–600. [Google Scholar] [CrossRef]
- Kilgore, J.L.; Musch, T.I.; Ross, C.R. Physical Activity, Muscle, and the HSP70 Response. Can. J. Appl. Physiol. 1998, 23, 245–260. [Google Scholar] [CrossRef]
- Kumar, Y.; Tatu, U. Stress protein flux during recovery from simulated ischemia: Induced heat shock protein 70 confers cytoprotection by suppressing JNK activation and inhibiting apoptotic cell death. Proteomics 2003, 3, 513–526. [Google Scholar] [CrossRef]
- Nava, R.; Zuhl, M.N. Heat acclimation-induced intracellular HSP70 in humans: A meta-analysis. Cell Stress Chaperones 2020, 25, 35–45. [Google Scholar] [CrossRef]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2018, 69, 227–237.e4. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. Med. Comm. 2022, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Minoia, M.; Boncoraglio, A.; Vinet, J.; Morelli, F.F.; Brunsting, J.F.; Poletti, A.; Krom, S.; Reits, E.; Kampinga, H.H.; Carra, S. BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: Implications for a proteasome-to-autophagy switch. Autophagy 2014, 10, 1603–1621. [Google Scholar] [CrossRef] [PubMed]
- Behl, C. BAG3 and friends: Co-chaperones in selective autophagy during aging and disease. Autophagy 2011, 7, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Misselwitz, B.; Staeck, O.; Rapoport, T.A. J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol. Cell 1998, 2, 593–603. [Google Scholar] [CrossRef]
- Zhao, H.; Michaelis, M.L.; Blagg, B.S.J. Hsp90 modulation for the treatment of Alzheimer’s disease. Adv. Pharmacol. 2012, 64, 1–25. [Google Scholar] [CrossRef]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, X.; Zhang, W.; He, J.; Xu, B.; Lei, B.; Wang, Z.; Cates, C.; Rousselle, T.; Li, J. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism 2018, 83, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol. Int. 2018, 42, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Schmitz, K.; Cox, J.; Esser, L.M.; Voss, M.; Sander, K.; Löffler, A.; Hillebrand, F.; Erkelenz, S.; Schaal, H.; Kähne, T.; et al. An essential role of the autophagy activating kinase ULK1 in snRNP biogenesis. Nucleic Acids Res. 2021, 49, 6437–6455. [Google Scholar] [CrossRef]
- Huang, Y.; Li, S.; Jia, Z.; Zhao, W.; Zhou, C.; Zhang, R.; Ali, D.W.; Michalak, M.; Chen, X.Z.; Tang, J. Transient Receptor Potential Melastatin 8 (TRPM8) Channel Regulates Proliferation and Migration of Breast Cancer Cells by Activating the AMPK-ULK1 Pathway to Enhance Basal Autophagy. Front. Oncol. 2020, 10, 573127. [Google Scholar] [CrossRef]
- Takebe, K. 148 Chondrocyte Apoptosis with Heat Stress Is Induced by P53 Pathway. Osteoarthr. Cartil. 2008, 16, S77. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; De Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The Regulation of AMPK β1, TSC2, and PTEN Expression by p53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the IGF-1-AKT-mTOR Pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Chen, S.; Wang, Z.; Yao, Y.; Chen, T.; Ye, Z.; Lin, P. Andrographolide induces apoptosis in human osteosarcoma cells via the ROS/JNK pathway. Int. J. Oncol. 2020, 56, 1417–1428. [Google Scholar] [CrossRef]
- Pereira, L.; Igea, A.; Canovas, B.; Dolado, I.; Nebreda, A.R. Inhibition of p38 MAPK sensitizes tumour cells to cisplatin-induced apoptosis mediated by reactive oxygen species and JNK. EMBO Mol. Med. 2013, 5, 1759–1774. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.R.; Harris, A.L. The role of the hypoxia-inducible BH3-only proteins BNIP3 and BNIP3L in cancer. Cancer Metastasis Rev. 2007, 26, 553. [Google Scholar] [CrossRef] [PubMed]
- Greijer, A.E.; Van Der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Maini, R.; Bai, X.; Nangreave, R.C.; Dedkova, L.M.; Hecht, S.M. Incorporation of Phosphorylated Tyrosine into Proteins: In Vitro Translation and Study of Phosphorylated IκB-α and Its Interaction with NF-κB. J. Am. Chem. Soc. 2017, 139, 14098–14108. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Trocoli, A.; Djavaheri-Mergny, M. The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. Am. J. Cancer Res. 2011, 1, 629–649. [Google Scholar]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Blomgren, K.; Kroemer, G. Autophagy in acute brain injury. Nat. Rev. Neurosci. 2016, 17, 467–484. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Bento, C.F.; Deretic, V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J. Exp. Med. 2015, 212, 979–990. [Google Scholar] [CrossRef]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, G.D.S.; Quaas, C.E.; Bertolini, I.; Zuccotti, A.; Saatci, O.; Kashatus, J.A.; Sharmin, S.; Lu, D.Y.; Poli, A.N.R.; Quesnelle, A.F.; et al. HSP70-mediated mitochondrial dynamics and autophagy represent a novel vulnerability in pancreatic cancer. Cell Death Differ. 2024, 31, 881–896. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef]
- Fuchs, M.; Poirier, D.J.; Seguin, S.J.; Lambert, H.; Carra, S.; Charette, S.J.; Landry, J. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem. J. 2009, 425, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Rizza, S.; Muniyappa, R.; Iantorno, M.; Kim, J.-a.; Chen, H.; Pullikotil, P.; Senese, N.; Tesauro, M.; Lauro, D.; Cardillo, C.; et al. Citrus polyphenol hesperidin stimulates production of nitric oxide in endothelial cells while improving endothelial function and reducing inflammatory markers in patients with metabolic syndrome. J. Clin. Endocrinol. Metab. 2011, 96, E782–E792. [Google Scholar] [CrossRef]
- Jung, J.H.; Lee, J.O.; Kim, J.H.; Lee, S.K.; You, G.Y.; Park, S.H.; Park, J.M.; Kim, E.K.; Suh, P.G.; An, J.K.; et al. Quercetin suppresses HeLa cell viability via AMPK-induced HSP70 and EGFR down-regulation. J. Cell. Physiol. 2010, 223, 408–414. [Google Scholar] [CrossRef]
- Wang, T.; Yu, Q.; Chen, J.; Deng, B.; Qian, L.; Le, Y. PP2A Mediated AMPK Inhibition Promotes HSP70 Expression in Heat Shock Response. PLoS ONE 2010, 5, e13096. [Google Scholar] [CrossRef] [PubMed]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Bao, H.; Xie, H.; Tian, G.; Jiang, T. Heat shock protein expression and autophagy after incomplete thermal ablation and their correlation. Int. J. Hyperth. 2019, 36, 95–103. [Google Scholar] [CrossRef]
- Stokoe, D.; Engel, K.; Campbell, D.G.; Cohen, P.; Gaestel, M. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett. 1992, 313, 307–313. [Google Scholar] [CrossRef]
- Avni, D.; Glucksam, Y.; Zor, T. The phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 modulates cytokine expression in macrophages via p50 nuclear factor κB inhibition, in a PI3K-independent mechanism. Biochem. Pharmacol. 2012, 83, 106–114. [Google Scholar] [CrossRef]
- Schulze-Osthoff, K.; Ferrari, D.; Riehemann, K.; Wesselborg, S. Regulation of NF-kappa B activation by MAP kinase cascades. Immunobiology 1997, 198, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C.; Clarke, B.A.; Zimmermann, S.; Nuñez, L.; Rossman, R.; Reid, K.; Moelling, K.; Yancopoulos, G.D.; Glass, D.J. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 1999, 286, 1738–1741. [Google Scholar] [CrossRef] [PubMed]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Cattelan, A.; Ceolotto, G.; Bova, S.; Albiero, M.; Kuppusamy, M.; De Martin, S.; Semplicini, A.; Fadini, G.P.; De Kreutzenberg, S.V.; Avogaro, A. NAD+-dependent SIRT1 deactivation has a key role on ischemia–reperfusion-induced apoptosis. Vasc. Pharmacol. 2015, 70, 35–44. [Google Scholar] [CrossRef]
- Yao, M.; Zhao, Z.; Wei, L.; Zhou, D.; Xue, Z.; Ge, S. HSF1/HSP pathway in the hippocampus is involved in SIRT1-mediated caloric restriction-induced neuroprotection after surgery in aged mice. Exp. Gerontol. 2019, 119, 184–192. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Pooya, S.; Lorentz, S.; Gauchotte, G.; Arnold, C.; Gueant, J.L.; Battaglia-Hsu, S.F. Decreased vitamin B12 availability induces ER stress through impaired SIRT1-deacetylation of HSF1. Cell Death Dis. 2013, 4, e553. [Google Scholar] [CrossRef]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef]
- Zhang, C.; Qu, S.; Wei, X.; Feng, Y.; Zhu, H.; Deng, J.; Wang, K.; Liu, K.; Liu, M.; Zhang, H.; et al. HSP25 down-regulation enhanced p53 acetylation by dissociation of SIRT1 from p53 in doxorubicin-induced H9c2 cell apoptosis. Cell Stress Chaperones 2016, 21, 251–260. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Ghislat, G.; Cho, D.H.; Rubinsztein, D.C. Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci. 2016, 129, 3059–3066. [Google Scholar] [CrossRef]
- Kregel, K.C. Invited Review: Heat shock proteins: Modifying factors in physiological stress responses and acquired thermotolerance. J. Appl. Physiol. 2002, 92, 2177–2186. [Google Scholar] [CrossRef] [PubMed]
- Barbe, M.F.; Tytell, M.; Gower, D.J.; Welch, W.J. Hyperthermia protects against light damage in the rat retina. Science 1988, 241, 1817–1820. [Google Scholar] [CrossRef]
- Daugaard, M.; Rohde, M.; Jäättelä, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar] [CrossRef]
- Lou, S.L.; Zhang, X.Y.; Wang, D.H. HSP70 plays a role in the defense of acute and chronic heat stress in Mongolian gerbils (Meriones unguiculatus). J. Therm. Biol. 2019, 86, 102452. [Google Scholar] [CrossRef]
- Nishizawa, M.; Giviziez, P.E.N.; Ferro, J.A.; Ferro, M.I.T.; Macari, M. Effect of heat stress or lipopolysaccharide (E. coli) injection on HSP70 levels in the liver and brain of adrenalectomized rats. J. Therm. Biol. 1999, 24, 429–432. [Google Scholar] [CrossRef]
- Rout, P.K.; Kaushik, R.; Ramachandran, N. Differential expression pattern of heat shock protein 70 gene in tissues and heat stress phenotypes in goats during peak heat stress period. Cell Stress Chaperones 2016, 21, 645–651. [Google Scholar] [CrossRef]
- Mahmoud, K.Z.; Edens, F.W.; Eisen, E.J.; Havenstein, G.B. The effect of dietary phosphorus on heat shock protein mRNAs during acute heat stress in male broiler chickens (Gallus gallus). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2004, 137, 11–18. [Google Scholar] [CrossRef]
- Walters, T.J.; Ryan, K.L.; Tehrany, M.R.; Jones, M.B.; Paulus, L.A.; Mason, P.A. HSP70 expression in the CNS in response to exercise and heat stress in rats. J. Appl. Physiol. 1998, 84, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Walters, T.J.; Ryan, K.L.; Mason, P.A. Regional distribution of Hsp70 in the CNS of young and old food-restricted rats following hyperthermia. Brain Res. Bull. 2001, 55, 367–374. [Google Scholar] [CrossRef]
- Maroni, P.; Bendinelli, P.; Tiberio, L.; Rovetta, F.; Piccoletti, R.; Schiaffonati, L. In vivo heat-shock response in the brain: Signalling pathway and transcription factor activation. Mol. Brain Res. 2003, 119, 90–99. [Google Scholar] [CrossRef]
- Sprang, G.K.; Brown, I.R. Selective induction of a heat shock gene in fibre tracts and cerebellar neurons of the rabbit brain detected by in situ hybridization. Brain Res. 1987, 427, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Vass, K.; Welch, W.J.; Nowak, T.S. Localization of 70-kDa stress protein induction in gerbil brain after ischemia. Acta Neuropathol. 1988, 77, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, V.L.; Hayward, N.J.; Poat, J.A.; Woodruff, G.N.; Hughes, J. The effect of dizocilpine and enadoline on immediate early gene expression in the gerbil global ischaemia model. Neuropharmacology 1993, 32, 1047–1059. [Google Scholar] [CrossRef]
- Wagstaff, M.J.D.; Collaço-Moraes, Y.; Aspey, B.S.; Coffin, R.S.; Harrison, M.J.G.; Latchman, D.S.; De Belleroche, J.S. Focal cerebral ischaemia increases the levels of several classes of heat shock proteins and their corresponding mRNAs. Mol. Brain Res. 1996, 42, 236–244. [Google Scholar] [CrossRef]
- Lyon, M.S.; Milligan, C. Extracellular heat shock proteins in neurodegenerative diseases: New perspectives. Neurosci. Lett. 2019, 711, 134462. [Google Scholar] [CrossRef]
- Blake, M.J.; Gershon, D.; Fargnoli, J.; Holbrook, N.J. Discordant expression of heat shock protein mRNAs in tissues of heat-stressed rats. J. Biol. Chem. 1990, 265, 15275–15279. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Han, D.; Sun, B.; Qiu, J.; Li, Y.; Li, M.; Zhang, T.; Yang, Z. Heat stress preconditioning improves cognitive outcome after diffuse axonal injury in rats. J. Neurotrauma 2009, 26, 1695–1706. [Google Scholar] [CrossRef] [PubMed]
- Porto, R.R.; Dutra, F.D.; Crestani, A.P.; Holsinger, R.M.D.; Quillfeldt, J.A.; Homem de Bittencourt, P.I.; de Oliveira Alvares, L. HSP70 Facilitates Memory Consolidation of Fear Conditioning through MAPK Pathway in the Hippocampus. Neuroscience 2018, 375, 108–118. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef]
- Shan, R.; Liu, N.; Yan, Y.; Liu, B. Apoptosis, autophagy and atherosclerosis: Relationships and the role of Hsp27. Pharmacol. Res. 2021, 166, 105169. [Google Scholar] [CrossRef]
- Cui, D.; Xiong, X.; Zhao, Y. Cullin-RING ligases in regulation of autophagy. Cell Div. 2016, 11, 8. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-i.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, W.; Sun, X.; Xu, D.; Wang, C.; Zhang, Q.; Wang, H.; Luo, W.; Chen, Y.; Chen, H.; et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy 2016, 12, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, D.; Gu, S.; Chen, X.; Bi, Y.; Xiong, X.; Zhao, Y. Autophagy regulates apoptosis by targeting NOXA for degradation. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1105–1113. [Google Scholar] [CrossRef]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Thorburn, A. FOXO3 links autophagy to apoptosis. Autophagy 2018, 14, 1467–1468. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Galea, E.; Aquino, D.A.; Li, G.C.; Xu, H.; Reis, D.J. Heat shock protein 70 suppresses astroglial-inducible nitric-oxide synthase expression by decreasing NFkappaB activation. J. Biol. Chem. 1996, 271, 17724–17732. [Google Scholar] [CrossRef]
- Zimmermann, M.; Reichert, A.S. How to get rid of mitochondria: Crosstalk and regulation of multiple mitophagy pathways. Biol. Chem. 2017, 399, 29–45. [Google Scholar] [CrossRef]
- Kim, J.Y.; Yenari, M.A.; Lee, J.E. Regulation of inflammatory transcription factors by heat shock protein 70 in primary cultured astrocytes exposed to oxygen-glucose deprivation. Neuroscience 2015, 286, 272–280. [Google Scholar] [CrossRef]
- Mignotte, B.; Vayssiere, J.L. Mitochondria and apoptosis. Eur. J. Biochem. 1998, 252, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.M.; Morishima, Y.; Pratt, W.B.; Osawa, Y. Modulation of heme/substrate binding cleft of neuronal nitric-oxide synthase (nNOS) regulates binding of Hsp90 and Hsp70 proteins and nNOS ubiquitination. J. Biol. Chem. 2012, 287, 1556–1565. [Google Scholar] [CrossRef]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Livieri, T.; Cuttaia, C.; Vetrini, R.; Concato, M.; Peruch, M.; Neri, M.; Radaelli, D.; D’Errico, S. Old and Promising Markers Related to Autophagy in Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 24, 72. [Google Scholar] [CrossRef]
- Yang, Y.; Fiskus, W.; Yong, B.; Atadja, P.; Takahashi, Y.; Pandita, T.K.; Wang, H.G.; Bhalla, K.N. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 6841–6846. [Google Scholar] [CrossRef]
- Yang, Y.; Xing, D.; Zhou, F.; Chen, Q. Mitochondrial autophagy protects against heat shock-induced apoptosis through reducing cytosolic cytochrome c release and downstream caspase-3 activation. Biochem. Biophys. Res. Commun. 2010, 395, 190–195. [Google Scholar] [CrossRef]
- Li, H.; Wang, P.; Sun, Q.; Ding, W.X.; Yin, X.M.; Sobol, R.W.; Stolz, D.B.; Yu, J.; Zhang, L. Following cytochrome c release, autophagy is inhibited during chemotherapy-induced apoptosis by caspase 8-mediated cleavage of Beclin 1. Cancer Res. 2011, 71, 3625–3634. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, N.; Zheng, Z.; Lee, J.E.; Yenari, M.A. 70-kDa Heat Shock Protein Downregulates Dynamin in Experimental Stroke: A New Therapeutic Target? Stroke 2016, 47, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, V.N.; Ronai, Z.; Hei, T.K. Opposite roles of FAP-1 and dynamin in the regulation of Fas (CD95) translocation to the cell surface and susceptibility to Fas ligand-mediated apoptosis. J. Biol. Chem. 2006, 281, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Y.; Zheng, X. HSP70-Mediated Autophagy-Apoptosis-Inflammation Network and Neuroprotection Induced by Heat Acclimatization. Biology 2025, 14, 774. https://doi.org/10.3390/biology14070774
Su Y, Zheng X. HSP70-Mediated Autophagy-Apoptosis-Inflammation Network and Neuroprotection Induced by Heat Acclimatization. Biology. 2025; 14(7):774. https://doi.org/10.3390/biology14070774
Chicago/Turabian StyleSu, Yuchen, and Xinyan Zheng. 2025. "HSP70-Mediated Autophagy-Apoptosis-Inflammation Network and Neuroprotection Induced by Heat Acclimatization" Biology 14, no. 7: 774. https://doi.org/10.3390/biology14070774
APA StyleSu, Y., & Zheng, X. (2025). HSP70-Mediated Autophagy-Apoptosis-Inflammation Network and Neuroprotection Induced by Heat Acclimatization. Biology, 14(7), 774. https://doi.org/10.3390/biology14070774