
Multi-Faceted Role of Histone Methyltransferase Enhancer of Zeste 2 (EZH2) in Neuroinflammation and Emerging Targeting Options


Simple Summary
Abstract
1. Introduction
2. Structural Aspects of EZH2
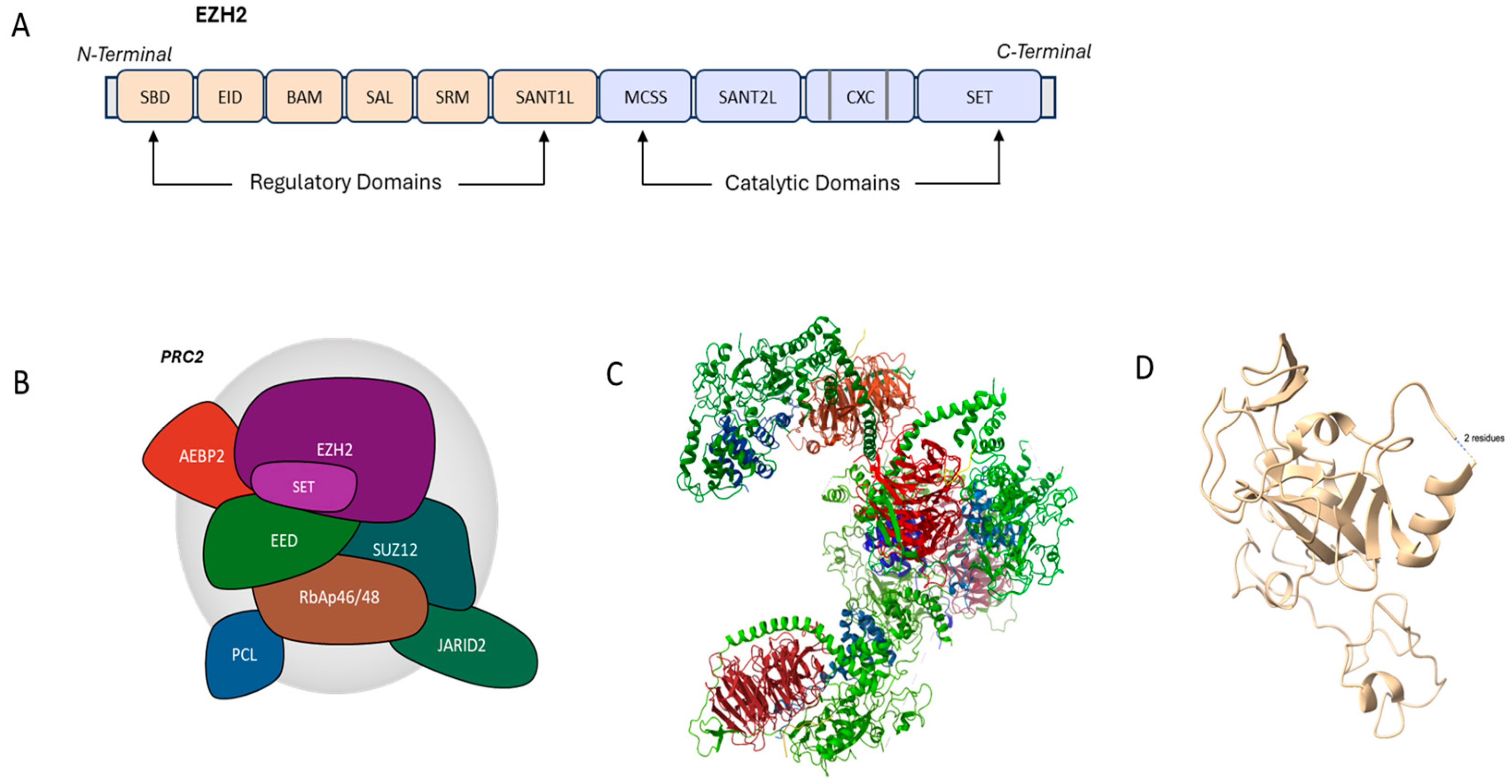
3. Physiological Functions of EZH2
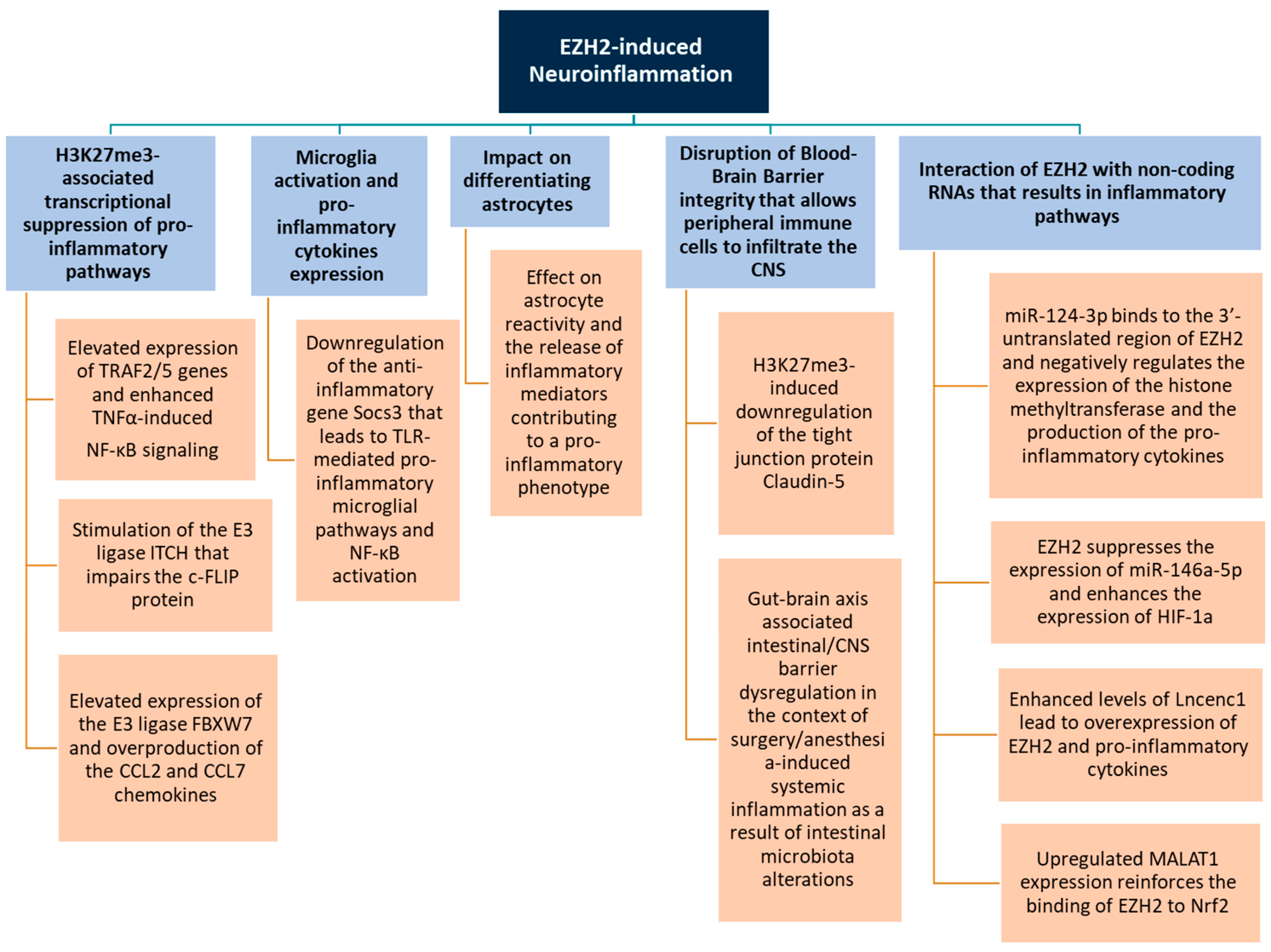
4. Role of EZH2 in Neuroinflammation
4.1. Regulation of Inflammatory Gene Expression
4.2. Microglia Activation
4.3. Astrocyte Function

4.4. Blood–Brain Barrier Integrity
4.5. Interaction with Non-Coding RNAs
5. EZH2 Role in Neuroinflammatory Diseases
5.1. Multiple Sclerosis (MS)
5.2. Alzheimer’s Disease (AD)
5.3. Parkinson’s Disease (PD)
6. Impact of EZH2 in Cholinergic Neurotransmission in Neuroinflammation
7. EZH2 Targeting Options in Treatment Strategies

{kind=link}
{kind=link}
{kind=link}
| EZH2 Inhibitor/Chemical Compound | Type of Inhibitor | Mechanism of Action/Effect | Disease | Type of Study | Side-Effects | Reference |
|---|---|---|---|---|---|---|
| DZNep/ 3-Deazaneplanocin A | Indirect | - Reduction in microglial pro-inflammatory activation (CD86+) and pro-inflammatory cytokines IL-6, IL-1β, TNF-α, and CXCL10 - Restriction of STAT3 phosphorylation - Stimulation of microglial anti-inflammatory polarization (CD206+) - Reduction in EZH2 activity and reversal of H3K27me3 accumulation | Ischemic stroke | In vitro and in vivo | [33] | |
| Neuropathic pain | In vivo | [22] | ||||
| - Potential neurotoxicity, impaired neurogenesis, BBB disruption - Anemia, immune suppression in animal models - Poor clinical viability due to off-target effects and global methylation interference | ||||||
| GSK-126 | Direct | - Inhibition of microglial, astrocytic, and pro-inflammatory mediators’ expression, - Standardization of EZH2 and H3K27me3 levels | Neuropathic pain | In vivo | - Nausea, diarrhea, reduced appetite - Mild anemia, neutropenia, lymphopenia - Changes in T cell function or inflammatory cytokines that can be beneficial or adverse due to disease context | [22] |
| EPZ-6438/Tazemetostat | Direct | - Inhibition of inflammatory mediators’ expression, including IRF1 and STAT1 - Socs3 activation - Reduction in TRAF6 proteins, NF-κΒ, p65, and pro-inflammatory cytokines production | Neuropathic pain | In vitro | Nausea, constipation, anorexia | [86] |
| Depressive behaviors | In vivo | Anemia, neutropenia, thrombocytopenia | [10] | |||
| Aging-related behavioral deficits | In vivo | Mild transaminase elevations | [10] | |||
| Subarachnoid hemorrhage brain injury | In vivo | Fatigue, pain, peripheral edema, weight loss | [63] | |||
| GSK-343 | Direct | Neuroprotective properties against dopaminergic degeneration through regulation of the NF-κB/IκBα pathway, the production of cytokines, and the activation of glia | Brain aging-related cognitive decline | In vitro | Cytopenias, impaired stem cell function | [7,92] |
| Parkinson’s disease | In vivo | Not characterized (poor oral bioavailability limits systemic exposure) - Altered T-cell activity, microglial cytokine imbalance | [75] | |||
| Curcumin | - | Anticancer, antioxidant, and anti-inflammatory properties, regulation of histone methylation | Neuroinflammation-related neurological disorders | In vitro | Nausea, diarrhea, bloating, indigestion (in high doses) - Reduced iron absorption - Elevated liver enzymes (rare) - Skin rash, itching (rare) | [7,93] |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mishra, A.; Bandopadhyay, R.; Singh, P.K.; Mishra, P.S.; Sharma, N.; Khurana, N. Neuroinflammation in Neurological Disorders: Pharmacotherapeutic Targets from Bench to Bedside. Metab. Brain Dis. 2021, 36, 1591–1626. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, W.; Liu, S.; Qiao, X.; Xing, Y.; Zhou, Q.; Zhang, Z. Epigenetic Regulation of Neuroinflammation in Alzheimer’s Disease. Cells 2023, 13, 79. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Monastero, R. Neuroinflammation and Neurodegenerative Diseases: How Much Do We Still Not Know? Brain Sci. 2023, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Komada, M.; Nishimura, Y. Epigenetics and Neuroinflammation Associated with Neurodevelopmental Disorders: A Microglial Perspective. Front. Cell Dev. Biol. 2022, 10, 852752. [Google Scholar] [CrossRef]
- Giallongo, S.; Longhitano, L.; Denaro, S.; D’Aprile, S.; Torrisi, F.; La Spina, E.; Giallongo, C.; Mannino, G.; Lo Furno, D.; Zappalà, A.; et al. The Role of Epigenetics in Neuroinflammatory-Driven Diseases. Int. J. Mol. Sci. 2022, 23, 15218. [Google Scholar] [CrossRef]
- Vitorakis, N.; Piperi, C. Insights into the Role of Histone Methylation in Brain Aging and Potential Therapeutic Interventions. Int. J. Mol. Sci. 2023, 24, 17339. [Google Scholar] [CrossRef]
- Li, L.; Chen, R.; Zhang, H.; Li, J.; Huang, H.; Weng, J.; Tan, H.; Guo, T.; Wang, M.; Xie, J. The epigenetic modification of DNA methylation in neurological diseases. Front. Immunol. 2024, 15, 1401962. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Piperi, C. Epigenetics: The missing link between environmental exposures and Parkinson’s disease? Epigenomics 2024, 16, 921–927. [Google Scholar] [CrossRef]
- Wang, W.; Qin, X.; Wang, R.; Xu, J.; Wu, H.; Khalid, A.; Jiang, H.; Liu, D.; Pan, F. EZH2 Is Involved in Vulnerability to Neuroinflammation and Depression-like Behaviors Induced by Chronic Stress in Different Aged Mice. J. Affect. Disord. 2020, 272, 452–464. [Google Scholar] [CrossRef]
- Yadav, R.; Weng, H.-R. EZH2 Regulates Spinal Neuroinflammation in Rats with Neuropathic Pain. Neuroscience 2017, 349, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Y.; Yuan, J.; Li, N.; Pei, S.; Xu, J.; Luo, X.; Mao, C.; Liu, J.; Yu, T.; et al. Macrophage/Microglial Ezh2 Facilitates Autoimmune Inflammation through Inhibition of Socs3. J. Exp. Med. 2018, 215, 1365–1382. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhu, Q.; Zhang, X.; Wen, Z.; Zhang, G.; Li, N.; Pei, Y.; Wang, Y.; Pei, S.; Xu, J.; et al. Ezh2 Competes with P53 to License lncRNA Neat1 Transcription for Inflammasome Activation. Cell Death Differ. 2022, 29, 2009–2023. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhang, J.; Sun, Y.; Wang, J.; Ren, C.; Banerjee, S.; Ouyang, L.; Wang, Y. Targeting EZH2 for Cancer Therapy: From Current Progress to Novel Strategies. Eur. J. Med. Chem. 2022, 238, 114419. [Google Scholar] [CrossRef]
- Wang, B.; Liu, Y.; Liao, Z.; Wu, H.; Zhang, B.; Zhang, L. EZH2 in Hepatocellular Carcinoma: Progression, Immunity, and Potential Targeting Therapies. Exp. Hematol. Oncol. 2023, 12, 52. [Google Scholar] [CrossRef]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 Histone Methyltransferase in Cancer Epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A Novel Target for Cancer Treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Q. The Roles of EZH2 in Cancer and Its Inhibitors. Med. Oncol. 2023, 40, 167. [Google Scholar] [CrossRef]
- Wen, Y.; Cai, J.; Hou, Y.; Huang, Z.; Wang, Z. Role of EZH2 in Cancer Stem Cells: From Biological Insight to a Therapeutic Target. Oncotarget 2017, 8, 37974–37990. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X. PRC2, Chromatin Regulation, and Human Disease: Insights from Molecular Structure and Function. Front. Oncol. 2022, 12, 894585. [Google Scholar] [CrossRef]
- Liu, K.-L.; Zhu, K.; Zhang, H. An Overview of the Development of EED Inhibitors to Disable the PRC2 Function. RSC Med. Chem. 2022, 13, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.-R.; Taing, K.; Chen, L.; Penney, A. EZH2 Methyltransferase Regulates Neuroinflammation and Neuropathic Pain. Cells 2023, 12, 1058. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zeng, H.; Dong, A.; Li, F.; He, H.; Senisterra, G.; Seitova, A.; Duan, S.; Brown, P.J.; Vedadi, M.; et al. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS ONE 2013, 8, e83737. [Google Scholar] [CrossRef]
- Stoccoro, A. Epigenetic Mechanisms Underlying Sex Differences in Neurodegenerative Diseases. Biology 2025, 14, 98. [Google Scholar] [CrossRef]
- Chlamydas, S.; Markouli, M.; Strepkos, D.; Piperi, C. Epigenetic Mechanisms Regulate Sex-Specific Bias in Disease Manifestations. J. Mol. Med. 2022, 100, 1111–1123. [Google Scholar] [CrossRef]
- Greathouse, K.L.; Bredfeldt, T.; Everitt, J.I.; Lin, K.; Berry, T.; Kannan, K.; Mittelstadt, M.L.; Ho, S.; Walker, C.L. Environmental Estrogens Differentially Engage the Histone Methyltransferase EZH2 to Increase Risk of Uterine Tumorigenesis. Mol. Cancer Res. 2012, 10, 546–557. [Google Scholar] [CrossRef]
- Takayama, K. Epigenetic Regulation by Androgen Receptor in Prostate Cancer. OBM Genet. 2018, 2, 1–25. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Hung, M.-C.; Li, L.-Y. EZH2: A Pivotal Regulator in Controlling Cell Differentiation. Am. J. Transl. Res. 2012, 4, 364–375. [Google Scholar]
- Sasaki, M.; Yamaguchi, J.; Itatsu, K.; Ikeda, H.; Nakanuma, Y. Over-Expression of Polycomb Group Protein EZH2 Relates to Decreased Expression of P16 INK4a in Cholangiocarcinogenesis in Hepatolithiasis. J. Pathol. 2008, 215, 175–183. [Google Scholar] [CrossRef]
- Chen, J.-F.; Luo, X.; Xiang, L.-S.; Li, H.-T.; Zha, L.; Li, N.; He, J.-M.; Xie, G.-F.; Xie, X.; Liang, H.-J. EZH2 Promotes Colorectal Cancer Stem-like Cell Expansion by Activating P21cip1-Wnt/β-Catenin Signaling. Oncotarget 2016, 7, 41540–41558. [Google Scholar] [CrossRef]
- Gonzalez, M.E.; DuPrie, M.L.; Krueger, H.; Merajver, S.D.; Ventura, A.C.; Toy, K.A.; Kleer, C.G. Histone Methyltransferase EZH2 Induces Akt-Dependent Genomic Instability and BRCA1 Inhibition in Breast Cancer. Cancer Res. 2011, 71, 2360–2370. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.-H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 Orchestrates Gene Expression for the Stepwise Differentiation of Tissue-Specific Stem Cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, M.; Zhang, X.; Fan, L.; Liu, P.; Yu, L.; Cao, X.; Qiu, S.; Xu, Y. EZH2 Inhibitor DZNep Modulates Microglial Activation and Protects against Ischaemic Brain Injury after Experimental Stroke. Eur. J. Pharmacol. 2019, 857, 172452. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb Complex PRC2 and Its Mark in Life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Kitchen, G.B.; Hopwood, T.; Gali Ramamoorthy, T.; Downton, P.; Begley, N.; Hussell, T.; Dockrell, D.H.; Gibbs, J.E.; Ray, D.W.; Loudon, A.S.I. The Histone Methyltransferase Ezh2 Restrains Macrophage Inflammatory Responses. FASEB J. 2021, 35, e21843. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, J.; Guo, Z.; Li, C.; Tan, Z.; Wang, J.; Yang, J.; Xue, L. Easy or Not-The Advances of EZH2 in Regulating T Cell Development, Differentiation, and Activation in Antitumor Immunity. Front. Immunol. 2021, 12, 741302. [Google Scholar] [CrossRef]
- Yang, X.-P.; Jiang, K.; Hirahara, K.; Vahedi, G.; Afzali, B.; Sciume, G.; Bonelli, M.; Sun, H.-W.; Jankovic, D.; Kanno, Y.; et al. EZH2 Is Crucial for Both Differentiation of Regulatory T Cells and T Effector Cell Expansion. Sci. Rep. 2015, 5, 10643. [Google Scholar] [CrossRef]
- Liu, Y.; Peng, J.; Sun, T.; Li, N.; Zhang, L.; Ren, J.; Yuan, H.; Kan, S.; Pan, Q.; Li, X.; et al. Epithelial EZH2 Serves as an Epigenetic Determinant in Experimental Colitis by Inhibiting TNFα-Mediated Inflammation and Apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E3796–E3805. [Google Scholar] [CrossRef]
- He, J.; Song, Y.; Li, G.; Xiao, P.; Liu, Y.; Xue, Y.; Cao, Q.; Tu, X.; Pan, T.; Jiang, Z.; et al. Fbxw7 Increases CCL2/7 in CX3CR1hi Macrophages to Promote Intestinal Inflammation. J. Clin. Investig. 2019, 129, 3877–3893. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef]
- Cheray, M.; Joseph, B. Epigenetics Control Microglia Plasticity. Front. Cell. Neurosci. 2018, 12, 243. [Google Scholar] [CrossRef] [PubMed]
- Neele, A.E.; de Winther, M.P.J. Repressing the Repressor: Ezh2 Mediates Macrophage Activation. J. Exp. Med. 2018, 215, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.E.; Lee, J.S. Mechanisms and Emerging Regulators of Neuroinflammation: Exploring New Therapeutic Strategies for Neurological Disorders. Curr. Issues Mol. Biol. 2025, 47, 8. [Google Scholar] [CrossRef]
- Lynch, M.A. Exploring Sex-Related Differences in Microglia May Be a Game-Changer in Precision Medicine. Front. Aging Neurosci. 2022, 14, 868448. [Google Scholar] [CrossRef]
- Gudkov, S.V.; Burmistrov, D.E.; Kondakova, E.V.; Sarimov, R.M.; Yarkov, R.S.; Franceschi, C.; Vedunova, M.V. An emerging role of astrocytes in aging/neuroinflammation and gut-brain axis with consequences on sleep and sleep disorders. Ageing Res. Rev. 2023, 83, 101775. [Google Scholar] [CrossRef]
- Pavlou, M.A.S.; Grandbarbe, L.; Buckley, N.J.; Niclou, S.P.; Michelucci, A. Transcriptional and epigenetic mechanisms underlying astrocyte identity. Prog. Neurobiol. 2019, 174, 36–52. [Google Scholar] [CrossRef]
- Hwang, W.W.; Salinas, R.D.; Siu, J.J.; Kelley, K.W.; Delgado, R.N.; Paredes, M.F.; Alvarez-Buylla, A.; Oldham, M.C.; Lim, D.A. Distinct and separable roles for EZH2 in neurogenic astroglia. eLife 2014, 3, e02439. [Google Scholar] [CrossRef]
- Neal, M.; Richardson, J.R. Epigenetic Regulation of Astrocyte Function in Neuroinflammation and Neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 432–443. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, M.; Zou, W.; Li, C.; Zhang, S.; Lv, Y.; Su, L.; Ji, F.; Jiao, J.; Gao, Y. Ezh2 Regulates Early Astrocyte Morphogenesis and Influences the Coverage of Astrocytic Endfeet on the Vasculature. Cell Prolif. 2025, 28, e70015. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef]
- Shafqat, A.; Albalkhi, I.; Magableh, H.M.; Saleh, T.; Alkattan, K.; Yaqinuddin, A. Tackling the glial scar in spinal cord regeneration: New discoveries and future directions. Front. Cell. Neurosci. 2023, 17, 1180825. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Wu, C.; Parker, E.; Liu, T.C.; Duan, R.; Yang, L. Microglia and Astrocytes in Alzheimer’s Disease: Significance and Summary of Recent Advances. Aging Dis. 2024, 15, 1537–1564. [Google Scholar] [CrossRef] [PubMed]
- Wever, I.; von Oerthel, L.; Wagemans, C.M.R.J.; Smidt, M.P. EZH2 Influences mdDA Neuronal Differentiation, Maintenance and Survival. Front. Mol. Neurosci. 2019, 11, 491. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-W.; Wang, X.; Zhao, Y.; Sun, Z.-X.; Wu, Y.-H.; Hu, H.; Zhang, L.; Wang, S.-D.; Li, F.; Wei, A.-J.; et al. Blood-Brain Barrier Dysfunction Mediated by the EZH2-Claudin-5 Axis Drives Stress-Induced TNF-α Infiltration and Depression-like Behaviors. Brain Behav. Immun. 2024, 115, 143–156. [Google Scholar] [CrossRef]
- Han, D.; Li, Z.; Liu, T.; Yang, N.; Li, Y.; He, J.; Qian, M.; Kuang, Z.; Zhang, W.; Ni, C.; et al. Prebiotics Regulation of Intestinal Microbiota Attenuates Cognitive Dysfunction Induced by Surgery Stimulation in APP/PS1 Mice. Aging Dis. 2020, 11, 1029–1045. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Suo, Z.; Yang, J.; Zhou, B.; Qu, Y.; Xu, W.; Li, M.; Xiao, T.; Zheng, H.; Ni, C. Whole-Transcriptome Sequencing Identifies Neuroinflammation, Metabolism and Blood-Brain Barrier Related Processes in the Hippocampus of Aged Mice during Perioperative Period. CNS Neurosci. Ther. 2022, 28, 1576–1595. [Google Scholar] [CrossRef]
- Neo, W.H.; Yap, K.; Lee, S.H.; Looi, L.S.; Khandelia, P.; Neo, S.X.; Makeyev, E.V.; Su, I.H. MicroRNA miR-124 controls the choice between neuronal and astrocyte differentiation by fine-tuning Ezh2 expression. J. Biol. Chem 2014, 289, 20788–20801. [Google Scholar] [CrossRef]
- Li, X.J.; Zhou, F.; Li, Y.J.; Xue, X.Y.; Qu, J.R.; Fan, G.F.; Liu, J.; Sun, R.; Wu, J.Z.; Zheng, Q.; et al. LncRNA H19-EZH2 interaction promotes liver fibrosis via reprogramming H3K27me3 profiles. Acta Pharmacol. Sin. 2023, 44, 2479–2491. [Google Scholar] [CrossRef]
- Tenchov, R.; Sasso, J.M.; Wang, X.; Zhou, Q.A. Aging Hallmarks and Progression and Age-Related Diseases: A Landscape View of Research Advancement. ACS Chem. Neurosci. 2024, 15, 1–30. [Google Scholar] [CrossRef]
- Huang, X.; Yang, Q.; Xie, L.; Lei, S. Histone Methyltransferase Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit Exacerbates Inflammation in Depression Rats by Modulating Microglia Polarization. Bioengineered 2022, 13, 5509–5524. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Huang, S.; Wang, Z.; Huang, J.; Xu, L.; Tang, X.; Wan, Y.Y.; Li, Q.-J.; Symonds, A.L.J.; Long, H.; et al. Targeting EZH2 Histone Methyltransferase Activity Alleviates Experimental Intestinal Inflammation. Nat. Commun. 2019, 10, 2427. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Fang, Y.; Kang, R.; Lenahan, C.; Gamdzyk, M.; Zhang, Z.; Okada, T.; Tang, J.; Chen, S.; Zhang, J.H. Inhibition of EZH2 (Enhancer of Zeste Homolog 2) Attenuates Neuroinflammation via H3k27me3/SOCS3/TRAF6/NF-κB (Trimethylation of Histone 3 Lysine 27/Suppressor of Cytokine Signaling 3/Tumor Necrosis Factor Receptor Family 6/Nuclear Factor-κB) in a Rat Model of Subarachnoid Hemorrhage. Stroke 2020, 51, 3320–3331. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Shang, J.; Gao, C.; Guan, X.; Chen, Y.; Zhu, L.; Zhang, L.; Zhang, C.; Zhang, J.; Pang, T. A Novel SIRT6 Activator Ameliorates Neuroinflammation and Ischemic Brain Injury via EZH2/FOXC1 Axis. Acta Pharm. Sin. B 2021, 11, 708–726. [Google Scholar] [CrossRef]
- Malhotra, S.; Villar, L.M.; Costa, C.; Midaglia, L.; Cubedo, M.; Medina, S.; Fissolo, N.; Río, J.; Castilló, J.; Álvarez-Cermeño, J.C.; et al. Circulating EZH2-Positive T Cells Are Decreased in Multiple Sclerosis Patients. J. Neuroinflamm. 2018, 15, 296. [Google Scholar] [CrossRef]
- Karantanos, T.; Chistofides, A.; Barhdan, K.; Li, L.; Boussiotis, V.A. Regulation of T Cell Differentiation and Function by EZH2. Front. Immunol. 2016, 7, 172. [Google Scholar] [CrossRef]
- Peeters, J.G.C.; Silveria, S.; Ozdemir, M.; Ramachandran, S.; DuPage, M. Hyperactivating EZH2 to Augment H3K27me3 Levels in Regulatory T Cells Enhances Immune Suppression by Driving Early Effector Differentiation. Cell Rep. 2024, 43, 114724. [Google Scholar] [CrossRef]
- El Waly, B.; Macchi, M.; Cayre, M.; Durbec, P. Oligodendrogenesis in the Normal and Pathological Central Nervous System. Front. Neurosci. 2014, 8, 145. [Google Scholar] [CrossRef]
- Yan, X.-W.; Liu, H.-J.; Hong, Y.-X.; Meng, T.; Du, J.; Chang, C. lncRNA XIST Induces Aβ Accumulation and Neuroinflammation by the Epigenetic Repression of NEP in Alzheimer’s Disease. J. Neurogenet. 2022, 36, 11–20. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Corpas, R.; Puigoriol-Illamola, D.; Palomera-Ávalos, V.; Sanfeliu, C.; Pallàs, M. Understanding Epigenetics in the Neurodegeneration of Alzheimer’s Disease: SAMP8 Mouse Model. J. Alzheimers Dis. 2018, 62, 943–963. [Google Scholar] [CrossRef]
- Li, C.; Ren, J.; Zhang, M.; Wang, H.; Yi, F.; Wu, J.; Tang, Y. The Heterogeneity of Microglial Activation and Its Epigenetic and Non-Coding RNA Regulations in the Immunopathogenesis of Neurodegenerative Diseases. Cell. Mol. Life Sci. 2022, 79, 511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, F.; Liu, Y.; Lei, X.; Li, H.; Ji, G.; Yuan, Z.; Jiao, J. Ezh2 Regulates Adult Hippocampal Neurogenesis and Memory. J. Neurosci. 2014, 34, 5184–5199. [Google Scholar] [CrossRef] [PubMed]
- m6A Regulates Neurogenesis and Neuronal Development by Modulating Histone Methyltransferase Ezh2—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/31154015/ (accessed on 8 May 2025).
- Kunoh, S.; Nakashima, H.; Nakashima, K. Epigenetic Regulation of Neural Stem Cells in Developmental and Adult Stages. Epigenomes 2024, 8, 22. [Google Scholar] [CrossRef]
- Mannino, D.; Scuderi, S.A.; Casili, G.; Bova, V.; Cucinotta, L.; Lanza, M.; Filippone, A.; Esposito, E.; Paterniti, I. Neuroprotective Effects of GSK-343 in an in Vivo Model of MPTP-Induced Nigrostriatal Degeneration. J. Neuroinflamm. 2023, 20, 155. [Google Scholar] [CrossRef]
- Cai, L.-J.; Tu, L.; Huang, X.-M.; Huang, J.; Qiu, N.; Xie, G.-H.; Liao, J.-X.; Du, W.; Zhang, Y.-Y.; Tian, J.-Y. LncRNA MALAT1 Facilitates Inflammasome Activation via Epigenetic Suppression of Nrf2 in Parkinson’s Disease. Mol. Brain 2020, 13, 130. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Meng, K.; Jia, H.; Hou, X.; Zhu, Z.; Lu, Y.; Feng, Y.; Feng, J.; Xia, Y.; Tan, R.; Cui, F.; et al. Mitochondrial Dysfunction in Neurodegenerative Diseases: Mechanisms and Corresponding Therapeutic Strategies. Biomedicines 2025, 13, 327. [Google Scholar] [CrossRef]
- Delgado-Olguín, P.; Dang, L.T.; He, D.; Thomas, S.; Chi, L.; Sukonnik, T.; Khyzha, N.; Dobenecker, M.W.; Fish, J.E.; Bruneau, B.G. Ezh2-mediated repression of a transcriptional pathway upstream of Mmp9 maintains integrity of the developing vasculature. Development 2014, 141, 4610–4617. [Google Scholar] [CrossRef]
- Del Moral-Morales, A.; González-Orozco, J.C.; Hernández-Vega, A.M.; Hernández-Ortega, K.; Peña-Gutiérrez, K.M.; Camacho-Arroyo, I. EZH2 Mediates Proliferation, Migration, and Invasion Promoted by Estradiol in Human Glioblastoma Cells. Front. Endocrinol. 2022, 13, 703733. [Google Scholar] [CrossRef]
- Rondeaux, J.; Groussard, D.; Renet, S.; Tardif, V.; Dumesnil, A.; Chu, A.; Di Maria, L.; Lemarcis, T.; Valet, M.; Henry, J.-P.; et al. Ezh2 Emerges as an Epigenetic Checkpoint Regulator during Monocyte Differentiation Limiting Cardiac Dysfunction Post-MI. Nat. Commun. 2023, 14, 4461. [Google Scholar] [CrossRef]
- Kempkes, R.W.M.; Prinjha, R.K.; de Winther, M.P.J.; Neele, A.E. Novel Insights into the Dynamic Function of PRC2 in Innate Immunity. Trends Immunol. 2024, 45, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Physiology and Immunology of the Cholinergic Antiinflammatory Pathway. J. Clin. Investig. 2007, 117, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Xiao, C.; Fan, T.; Deng, Z.; Wang, D.; Cai, W.; Li, J.; Liao, T.; Li, C.; He, J. The Epigenetic Hallmarks of Immune Cells in Cancer. Mol. Cancer 2025, 24, 66. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Wang, S.-R.; Ma, J.-X.; Yu, L.-H.; Jia, G.-W. Vagus Nerve Stimulation Is a Potential Treatment for Ischemic Stroke. Neural. Regen. Res. 2023, 18, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Penas, C.; Navarro, X. Epigenetic Modifications Associated to Neuroinflammation and Neuropathic Pain After Neural Trauma. Front. Cell. Neurosci. 2018, 12, 158. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, Y.; Xie, L.; Wang, K.; Wang, X. Pharmacological Inhibition of EZH2 by GSK126 Decreases Atherosclerosis by Modulating Foam Cell Formation and Monocyte Adhesion in Apolipoprotein E-Deficient Mice. Exp. Ther. Med. 2021, 22, 841. [Google Scholar] [CrossRef]
- Mitić, T.; Caporali, A.; Floris, I.; Meloni, M.; Marchetti, M.; Urrutia, R.; Angelini, G.D.; Emanueli, C. EZH2 modulates angiogenesis in vitro and in a mouse model of limb ischemia. Mol. Ther. 2015, 23, 32–42. [Google Scholar] [CrossRef]
- Julia, E.; Salles, G. EZH2 Inhibition by Tazemetostat: Mechanisms of Action, Safety and Efficacy in Relapsed/Refractory Follicular Lymphoma. Future Oncol. 2021, 17, 2127–2140. [Google Scholar] [CrossRef]
- Lhuissier, E.; Aury-Landas, J.; Bouet, V.; Bazille, C.; Repesse, Y.; Freret, T.; Boumédiene, K.; Baugé, C. Evaluation of the Impact of S-Adenosylmethionine-Dependent Methyltransferase Inhibitor, 3-Deazaneplanocin A, on Tissue Injury and Cognitive Function in Mice. Oncotarget 2018, 9, 20698–20708. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, X.; Hu, X.; Duan, X.; Wan, G.; Li, L.; Feng, Q.; Zhang, Y.; Wang, N.; Yu, L. Covalent Inhibitors of EZH2: Design, Synthesis and Evaluation. Biomed. Pharmacother. 2022, 147, 112617. [Google Scholar] [CrossRef]
- Verma, S.K.; Tian, X.; LaFrance, L.V.; Duquenne, C.; Suarez, D.P.; Newlander, K.A.; Romeril, S.P.; Burgess, J.L.; Grant, S.W.; Brackley, J.A.; et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med. Chem. Lett. 2012, 3, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Ali, S.; Banerjee, S.; Wang, Z.; Logna, F.; Azmi, A.S.; Kong, D.; Ahmad, A.; Li, Y.; Padhye, S.; et al. Curcumin Analogue CDF Inhibits Pancreatic Tumor Growth by Switching on Suppressor microRNAs and Attenuating EZH2 Expression. Cancer Res. 2012, 72, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Grinshtein, N.; Rioseco, C.C.; Marcellus, R.; Uehling, D.; Aman, A.; Lun, X.; Muto, O.; Podmore, L.; Lever, J.; Shen, Y.; et al. Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells. Oncotarget 2016, 7, 59360–59376. [Google Scholar] [CrossRef] [PubMed]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef]
- Corbin, J.; Yu, X.; Jin, J.; Cai, L.; Wang, G.G. EZH2 PROTACs target EZH2- and FOXM1-associated oncogenic nodes, suppressing breast cancer cell growth. Oncogene 2024, 43, 2722–2736. [Google Scholar] [CrossRef]
- Hong, S.S.; Oh, K.T.; Choi, H.G.; Lim, S.J. Liposomal Formulations for Nose-to-Brain Delivery: Recent Advances and Future Perspectives. Pharmaceutics 2019, 11, 540. [Google Scholar] [CrossRef]
| Disease | Pro-Inflammatory | Anti-Inflammatory/Repair | Reference |
|---|---|---|---|
| Multiple Sclerosis | EZH2 promotes Th17 differentiation, exacerbates autoimmunity | EZH2 promotes Treg stability; supports remyelination in oligodendrocytes | [65,66,67,68] |
| Alzheimer’s Disease | Loss of EZH2 enables chronic microglial activation; worsened neurodegeneration | EZH2 supports Aβ clearance; promotes microglial polarization to M2-like state | [41,47,54,55,72,73,74] |
| Parkinson’s Disease | Excessive EZH2 inhibition induces uncontrolled microglial activation | EZH2 induces neuroprotective effects in dopaminergic neurons; regulation of autophagy/mitophagy | [41,75,76,77,78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moraitis, S.; Piperi, C. Multi-Faceted Role of Histone Methyltransferase Enhancer of Zeste 2 (EZH2) in Neuroinflammation and Emerging Targeting Options. Biology 2025, 14, 749. https://doi.org/10.3390/biology14070749
Moraitis S, Piperi C. Multi-Faceted Role of Histone Methyltransferase Enhancer of Zeste 2 (EZH2) in Neuroinflammation and Emerging Targeting Options. Biology. 2025; 14(7):749. https://doi.org/10.3390/biology14070749
Chicago/Turabian StyleMoraitis, Sotirios, and Christina Piperi. 2025. "Multi-Faceted Role of Histone Methyltransferase Enhancer of Zeste 2 (EZH2) in Neuroinflammation and Emerging Targeting Options" Biology 14, no. 7: 749. https://doi.org/10.3390/biology14070749
APA StyleMoraitis, S., & Piperi, C. (2025). Multi-Faceted Role of Histone Methyltransferase Enhancer of Zeste 2 (EZH2) in Neuroinflammation and Emerging Targeting Options. Biology, 14(7), 749. https://doi.org/10.3390/biology14070749