

Genomic Evolution of White Spot Syndrome Virus in Shrimp: Insights from Transposon Dynamics


 , , and
, , and
Simple Summary
Abstract
1. Introduction
2. Methods and Materials
2.1. Sequence Data Collection
2.2. Reannotated ORFs of WSSV in Public Databases
2.3. Analysis of Variable Region
2.4. Recombination and Evolution Analyses
3. Results
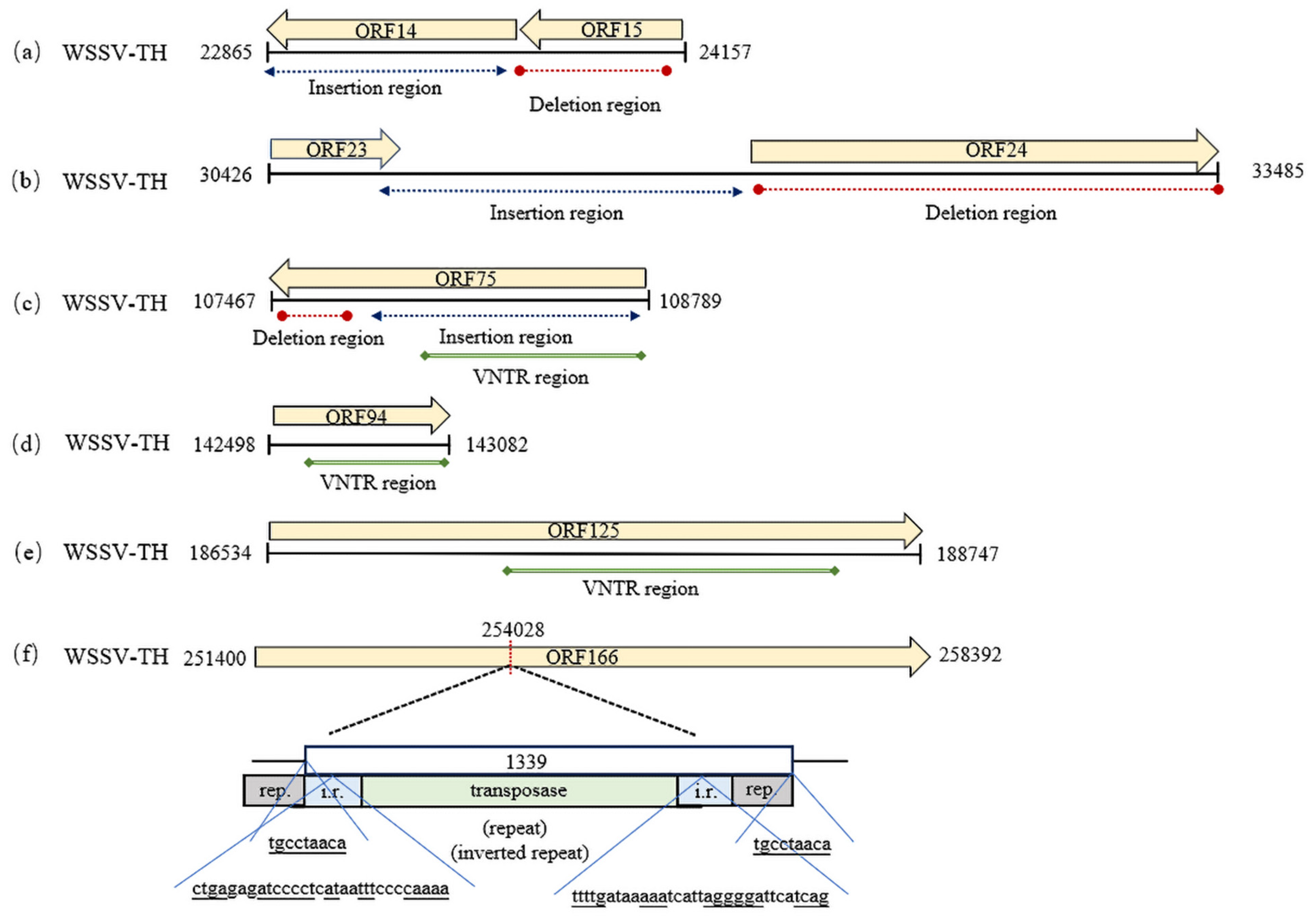
3.1. Variable Regions
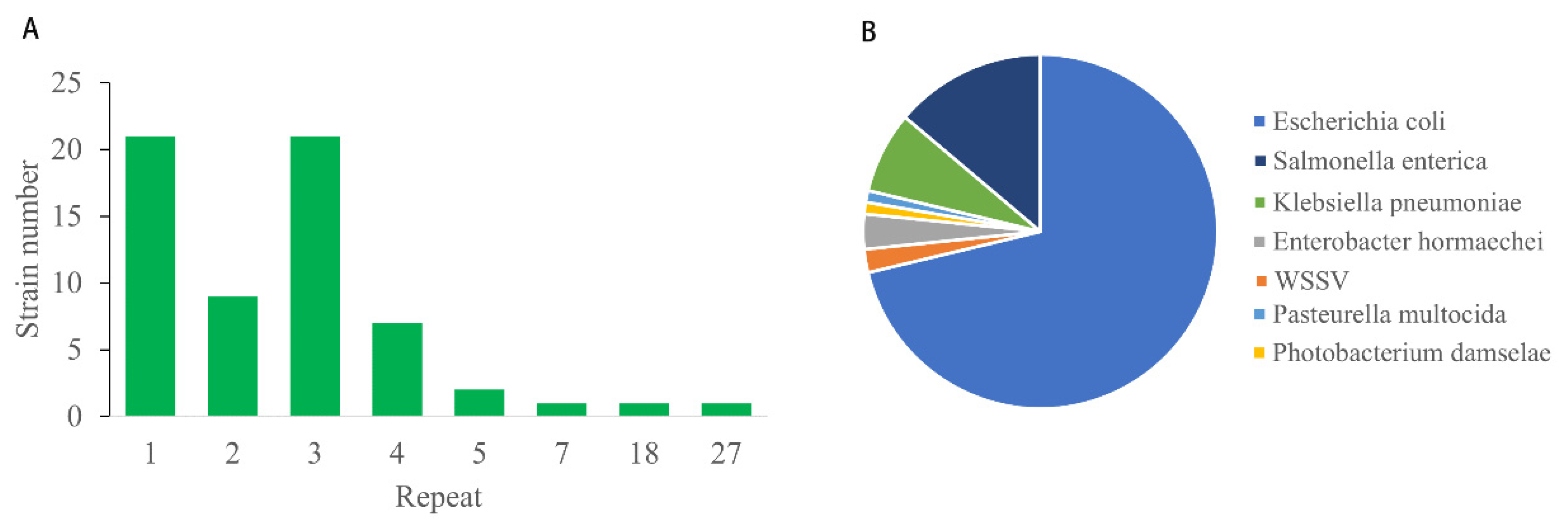
3.2. Variations in ORF Number and Sequences
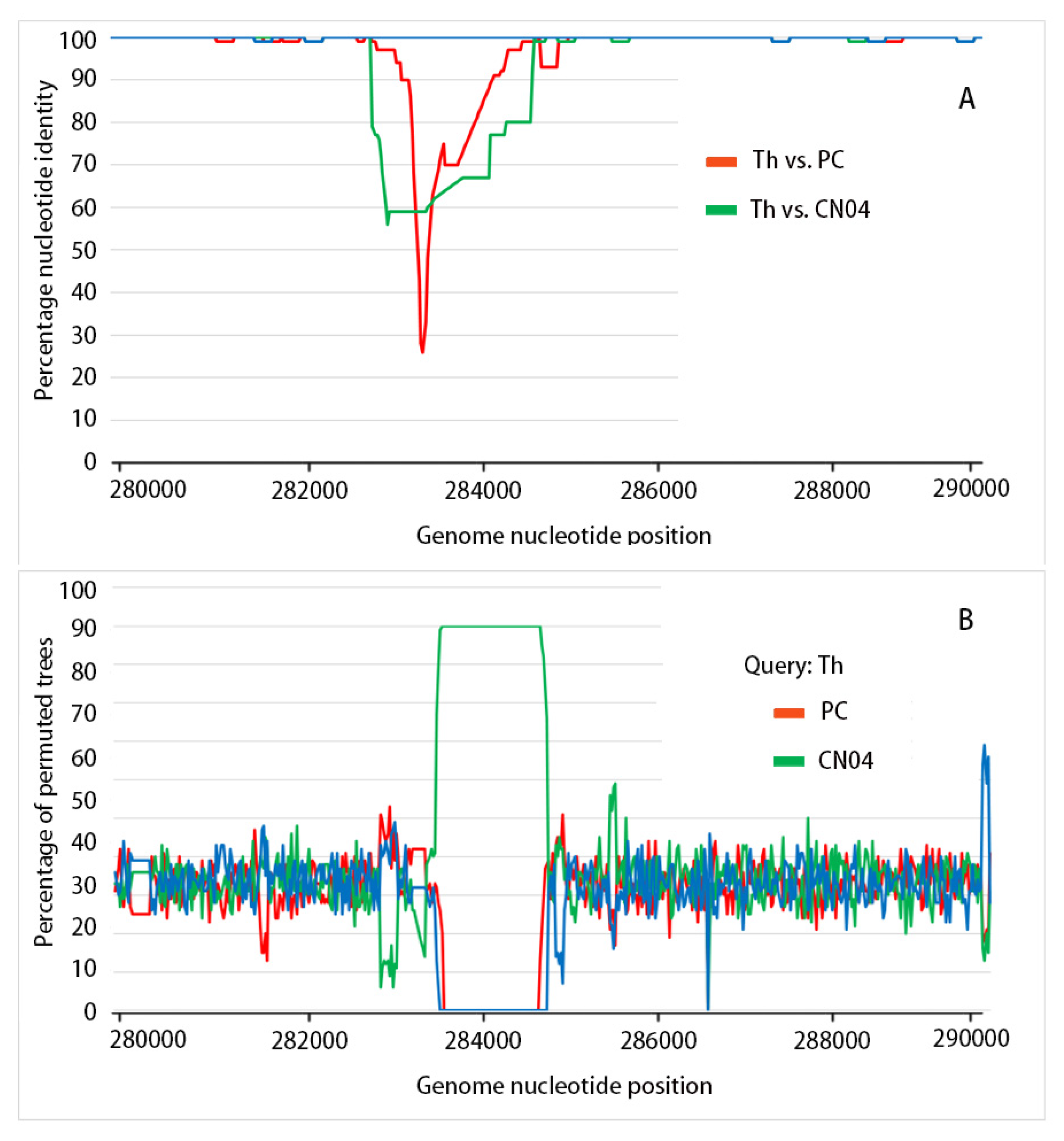
3.3. Recombination Analysis
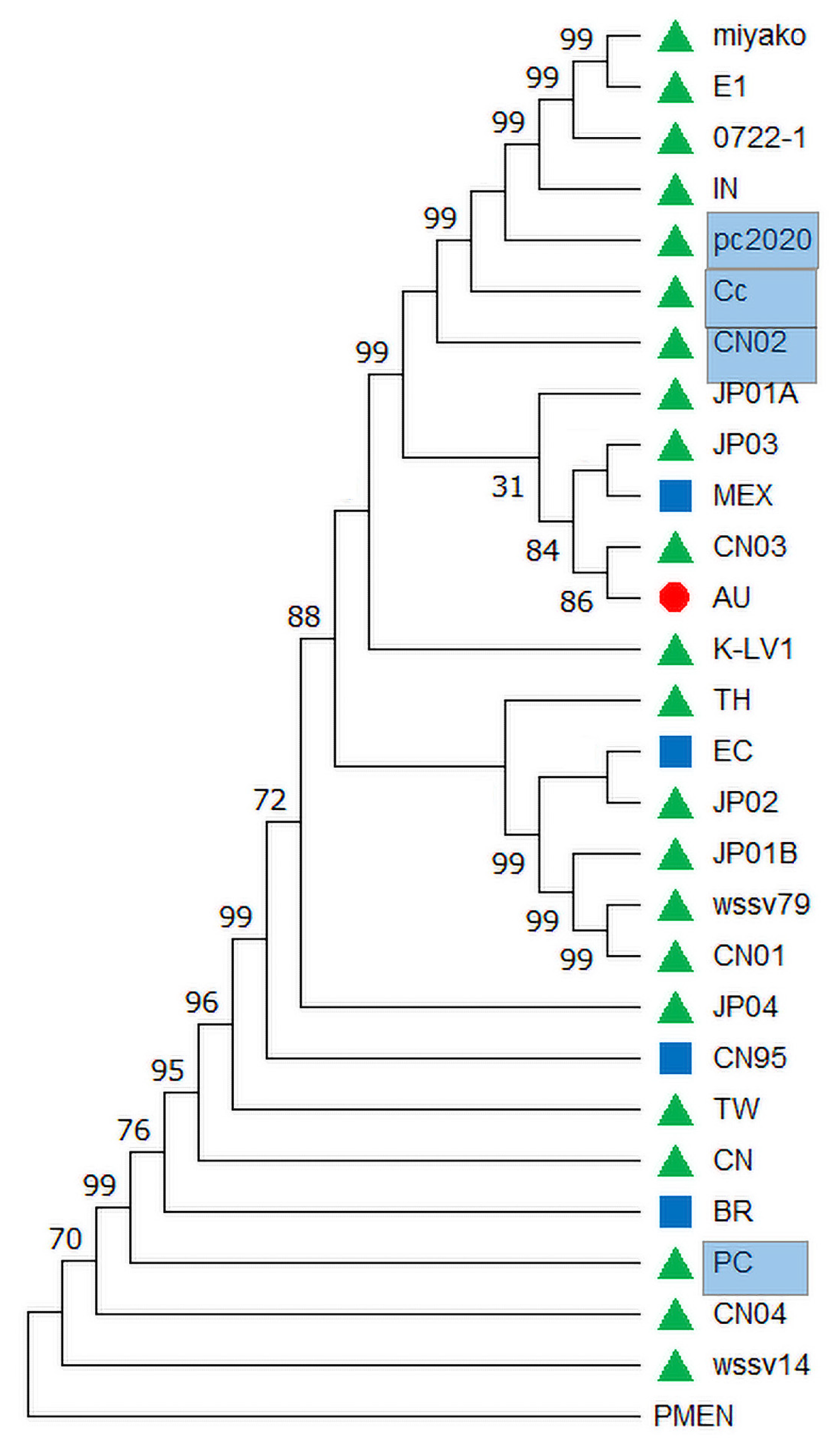
3.4. Evolutionary Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chou, H.Y.; Huang, C.Y.; Wang, C.H.; Chiang, H.C.; Lo, C.F. Pathogenicity of a baculovirus infection causing white spot syndrome in cultured penaeid shrimp in Taiwan. Dis. Aquat. Org. 1995, 23, 165–173. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, Y.S.; Lee, S.; Lee, Y. An infectious viral disease of penaeid shrimp newly found in Korea. Dis. Aquat. Org. 1998, 34, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Koube, H.; Umezawa, S.; Momoyama, K.; Oseko, N. Mass Mortalities of Cultured Kuruma Shrimp, Penaeus japonicus, in Japan in 1993: Epizootiological Survey and Infection Trials. Fish Pathol. 1994, 29, 135–139. [Google Scholar] [CrossRef]
- Mohan, C.V.; Shankar, K.M.; Kulkarni, S.; Sudha, P.M. Histopathology of cultured shrimp showing gross signs of yellow head syndrome and white spot syndrome during 1994 Indian epizootics. Dis. Aquat. Org. 1998, 34, 9–12. [Google Scholar] [CrossRef]
- van Hulten, M.C.W.; Witteveldt, J.; Peters, S.; Kloosterboer, N.; Tarchini, R.; Fiers, M.; Sandbrink, H.; Lankhorst, R.K.; Vlak, J.M. The white spot syndrome virus DNA genome sequence. Virology 2001, 286, 7–22. [Google Scholar] [CrossRef]
- Flegel, T.W.; Fegan, D.F. Strategies for preventing the spread of fish and shellfish diseases. Fish. Sci. 2002, 68, 776–788. [Google Scholar] [CrossRef]
- Oakey, J.; Smith, C.; Underwood, D.; Afsharnasab, M.; Alday-Sanz, V.; Dhar, A.; Sivakumar, S.; Sahul Hameed, A.S.; Beattie, K.; Crook, A. Global distribution of white spot syndrome virus genotypes determined using a novel genotyping assay. Arch. Virol. 2019, 164, 2061–2082. [Google Scholar] [CrossRef]
- Afsharnasab, M.; Kakoolaki, S.; Afazli, F. The Status of white spot syndrome virus (WSSV) in Islamic Republic of Iran. Iran. J. Fish. Sci. 2014, 13, 1021–1055. [Google Scholar]
- Shekar, M.; Pradeep, B.; Karunasagar, I. White spot syndrome virus: Genotypes, Epidemiology and Evolutionary Studies. Indian J. Virol. 2012, 23, 175–183. [Google Scholar] [CrossRef]
- Liu, L.K.; Liu, M.J.; Li, D.L.; Liu, H.P. Recent insights into anti-WSSV immunity in crayfish. Dev. Comp. Immunol. 2021, 116, 103947. [Google Scholar] [CrossRef]
- Marks, H.; Goldbach, R.W.; Vlak, J.M.; van Hulten, M.C.W. Genetic variation among isolates of White spot syndrome virus. Arch. Virol. 2004, 149, 673–697. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; He, J.; Lin, X.H.; Li, Q.; Pan, D.; Zhang, X.B.; Xu, X. Complete genome sequence of the shrimp white spot bacilliform virus. J. Virol. 2001, 75, 11811–11820. [Google Scholar] [CrossRef] [PubMed]
- Kawato, S.; Shitara, A.; Wang, Y.Y.; Nozaki, R.; Kondo, H.; Hirono, I. Crustacean Genome Exploration Reveals the Evolutionary Origin of White Spot Syndrome Virus. J. Virol. 2019, 93, e01144-18. [Google Scholar] [CrossRef]
- Kawato, S.; Omine, R.; Teruya, S.; Kubo, H.; Yasumoto, S.; Kondo, M.; Takahashi, Y.; Nozaki, R.; Kondo, H.; Hirono, I. Evolutionary genomics of white spot syndrome virus. Fish. Sci. 2023, 89, 769–783. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Samson, S.; Lord, É.; Makarenkov, V. SimPlot++: A Python application for representing sequence similarity and detecting recombination. Bioinformatics 2022, 38, 3118–3120. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Ke, F.; Gui, L.; Li, T.; Li, F.H.; Zhao, X.; Chen, Z.Y.; Lei, C.K.; Zhang, Q.Y. The features of an emerging whispovirus isolate from freshwater crayfish. Aquac. Rep. 2021, 20, 100728. [Google Scholar] [CrossRef]
- Zwart, M.P.; Dieu, B.T.M.; Hemerik, L.; Vlak, J.M. Evolutionary Trajectory of White Spot Syndrome Virus (WSSV) Genome Shrinkage during Spread in Asia. PLoS ONE 2010, 5, e13400. [Google Scholar] [CrossRef]
- Onihary, A.M.; Razanajatovo, I.M.; Rabetafika, L.; Bastaraud, A.; Heraud, J.M.; Rasolofo, V. Genotype Diversity and Spread of (WSSV) in Madagascar (2012–2016). Viruses 2021, 13, 1713. [Google Scholar] [CrossRef]
- Puttirungroj, P.; Kawato, S.; Mwamburi, S.M.; Furukawa, M.; Oomine, R.; Koiwai, K.; Kondo, H.; Hirono, I. Comparative genomics highlights the virulence and evolutionary trajectory of white spot syndrome virus. J. Gen. Virol. 2024, 105, 002042. [Google Scholar] [CrossRef]
- Alatorre-García, T.A.; Fonseca-Coronado, S.; González-Candelas, F. Homologous recombination as a mechanism of genetic changes in bovine parainfluenza-3 virus. Vet. Microbiol. 2021, 261, 109185. [Google Scholar] [CrossRef]
- Li, Z.P.; Li, F.; Han, Y.L.; Xu, L.M.; Yang, F. VP24 Is a Chitin-Binding Protein Involved in White Spot Syndrome Virus Infection. J. Virol. 2016, 90, 842–850. [Google Scholar] [CrossRef]
- Jeronimo, P.M.C.; Aksenen, C.F.; Duarte, I.O.; Lins, R.D.; Miyajima, F. Evolutionary deletions within the SARS-CoV-2 genome as signature trends for virus fitness and adaptation. J. Virol. 2024, 98, e0140423. [Google Scholar] [CrossRef]
- Reis, A.L.; Rathakrishnan, A.; Goulding, L.V.; Barber, C.; Goatley, L.C.; Dixon, L.K. Deletion of the gene for the African swine fever virus BCL-2 family member A179L increases virus uptake and apoptosis but decreases virus spread in macrophages and reduces virulence in pigs. J. Virol. 2023, 97, e0110623. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.S.; Nanda, H.; Aliferis, C.; Langlois, R.A. Characterization of influenza A virus induced transposons reveals a subgroup of transposons likely possessing the regulatory role as eRNAs. Sci. Rep. 2022, 12, 2188. [Google Scholar] [CrossRef] [PubMed]
- Macchietto, M.G.; Langlois, R.A.; Shen, S.S. Virus-induced transposable element expression up-regulation in human and mouse host cells. Life Sci. Alliance 2020, 3, e201900536. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, X.Y.; Guo, H.Y.; Zhang, X.G.; Zhang, M.; Tang, W.Q. Chromosome-level genome assembly of the endangered humphead wrasse: Insight into the expansion of opsin genes in fishes. Mol. Ecol. Resour. 2021, 21, 2388–2406. [Google Scholar] [CrossRef]
- Widen, S.A.; Bes, I.C.; Koreshova, A.; Pliota, P.; Krogull, D.; Burga, A. Virus-like transposons cross the species barrier and drive the evolution of genetic incompatibilities. Science 2023, 380, 1336. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Abbr. | Length (bp) | ORF Number | Host | Origin | Year |
|---|---|---|---|---|---|---|
| NC_003225 | CN01 | 309,286 | 184 | Penaeus japonicus | Mainland China | 1994 |
| AF440570 | TW | 307,287 | 184 | Penaeus monodon | Taiwan China | 1994 |
| AF369029 | TH | 292,967 | 184 | Penaeus monodon | Thailand | 1996 |
| AF332093 | CN | 305,119 | 184 | Penaeus japonicus | Mainland China | 1996 |
| AP027279 | JP01B | 293,923 | 184 | Penaeus japonicus | Japan | 2000 |
| AP027280 | JP02 | 311,562 | 184 | Penaeus japonicus | Japan | 2000 |
| KU216744 | MEX | 300,087 | 184 | Litopenaeus vannamei | Mexico | 2008 |
| KT995470 | CN02 | 294,261 | 183 | Procambarus clarkii | Mainland China | 2010 |
| KT995471 | CN03 | 284,148 | 179 | Litopenaeus vannamei | Mainland China | 2010 |
| JX515788 | K-LV1 | 295,884 | 184 | Litopenaeus vannamei | Korea | 2011 |
| KY827813 | CN04 | 281,054 | 179 | Marsupenaeus japonicus | Mainland China | 2012 |
| MG702567 | IN | 280,591 | 182 | Penaeus vannamei | India | 2013 |
| MH090824 | EC | 288,997 | 183 | Litopenaeus vannamei | El Guado | 2015 |
| KX686117 | PC | 300,223 | 184 | Procambarus clarkii | Mainland China | 2015 |
| MF784752 | BR | 292,912 | 184 | Litopenaeus vannamei | Brazil | 2015 |
| MF768985 | AU | 285,973 | 177 | Penaeus monodon | Australia | 2016 |
| MH663976 | Cc | 287,179 | 180 | Procambarus clarkii | Mainland China | 2016 |
| MN840357 | CN_95 | 305,094 | 184 | Penaeus vannamei | United States | 2017 |
| AP027286 | E1 | 289,353 | 174 | Penaeus japonicus | Japan | 2017 |
| AP027281 | JP03 | 301,236 | 182 | Metapenaeopsis lamellate | Japan | 2017 |
| AP027288 | 0722-1 | 288,252 | 176 | Penaeus japonicus | Japan | 2018 |
| AP027282 | JP04 | 301,054 | 181 | Trachysalambria curvirostris | Japan | 2018 |
| AP027289 | wssv14 | 288,494 | 175 | Penaeus japonicus | Japan | 2020 |
| AP027290 | Miyako | 288,190 | 172 | Penaeus japonicus | Japan | 2021 |
| AP027284 | pc2020 | 298,496 | 184 | Procambarus clarkii | Japan | 2023 |
| AP027278 | JP01A | 299,976 | 184 | Penaeus japonicus | Japan | 2023 |
| AP027283 | wssv79 | 295,104 | 181 | Penaeus japonicus | Japan | NA |
| Site | Allelic Frequency | Genotypic Frequency | Type of Mutation | Pi | Score |
|---|---|---|---|---|---|
| SNV1 (44276G > A) | G (0.0526) | GG (0.0526) | missense | 0.0997 | 0.142 |
| A (0.9474) | AA (0.9474) | ||||
| SNV2 (46674T > C) | T (0.4737) | TT (0.4737) | missense | 0.4986 | 0.281 |
| C (0.5263) | CC (0.5263) | ||||
| SNV3 (48301G > A) | G (0.2632) | GG (0.2632) | missense | 0.3879 | 0.232 |
| A (0.7368) | AA (0.7368) | ||||
| SNV4 (49066G > A) | G (0.0526) | GG (0.0526) | missense | 0.0997 | 0.102 |
| A (0.9474) | AA (0.9474) | ||||
| SNV5 (49357C > T) | C (0.0526) | CC (0.0526) | missense | 0.0997 | 0.117 |
| T (0.9474) | TT (0.9474) | ||||
| SNV6 (50153A > T) | A (0.8947) | AA (0.8947) | nonsense | 0.1884 | 0.083 |
| T (0.1053) | TT (0.1053) | ||||
| SNV7 (51086A > C) | A (0.0526) | AA (0.0526) | missense | 0.0997 | 0.154 |
| C (0.9474) | CC (0.9474) | ||||
| SNV8 (252108 C > T) | C (0.4737) | CC (0.4737) | missense | 0.4986 | 0.065 |
| T (0.5263) | TT (0.5263) | ||||
| SNV9 (252937 C > T) | C (0.8947) | CC (0.8947) | missense | 0.1884 | 0.095 |
| T (0.1053) | TT (0.1053) | ||||
| SNV10 (257897T > G) | T (0.8947) | TT (0.8947) | missense | 0.1884 | 0.161 |
| G (0.1053) | GG (0.1053) | ||||
| SNV11 (258889A > C) | A (0.4737) | AA (0.4737) | missense | 0.4986 | 0.253 |
| C (0.5263) | CC (0.5263) | ||||
| SNV12 (259660A > G) | A (0.4737) | AA (0.4737) | missense | 0.4986 | 0.153 |
| G (0.5263) | GG (0.5263) | ||||
| SNV13 (265557A > G) | A (0.4737) | AA (0.4737) | missense | 0.4986 | 0.129 |
| G (0.5263) | GG (0.5263) | ||||
| SNV14 (267494G > C) | G (0.0526) | GG (0.0526) | missense | 0.0997 | 0.203 |
| C (0.9474) | CC (0.9474) |
| Recombination Event | Start~End at WSSV-TH | Major Parent | Minor Parent | Analysis (** Denoted p < 0.01) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R | G | B | M | C | S | Q | ||||
| WSSV-BR | 283,000–284,840 | WSSV-PC | WSSV-CN04 | - | ** | - | ** | ** | ** | ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Huang, G.; Zhang, J.; Li, M.; Liu, Z.; Peng, S.; Wang, R.; Liu, D. Genomic Evolution of White Spot Syndrome Virus in Shrimp: Insights from Transposon Dynamics. Biology 2025, 14, 653. https://doi.org/10.3390/biology14060653
Li Z, Huang G, Zhang J, Li M, Liu Z, Peng S, Wang R, Liu D. Genomic Evolution of White Spot Syndrome Virus in Shrimp: Insights from Transposon Dynamics. Biology. 2025; 14(6):653. https://doi.org/10.3390/biology14060653
Chicago/Turabian StyleLi, Zhouquan, Guanghua Huang, Jingyi Zhang, Mingyou Li, Zhizhi Liu, Sihua Peng, Rui Wang, and Dong Liu. 2025. "Genomic Evolution of White Spot Syndrome Virus in Shrimp: Insights from Transposon Dynamics" Biology 14, no. 6: 653. https://doi.org/10.3390/biology14060653
APA StyleLi, Z., Huang, G., Zhang, J., Li, M., Liu, Z., Peng, S., Wang, R., & Liu, D. (2025). Genomic Evolution of White Spot Syndrome Virus in Shrimp: Insights from Transposon Dynamics. Biology, 14(6), 653. https://doi.org/10.3390/biology14060653