


Integrating Proteomics and GWAS to Identify Key Tissues and Genes Underlying Human Complex Diseases

Simple Summary
Abstract
1. Introduction
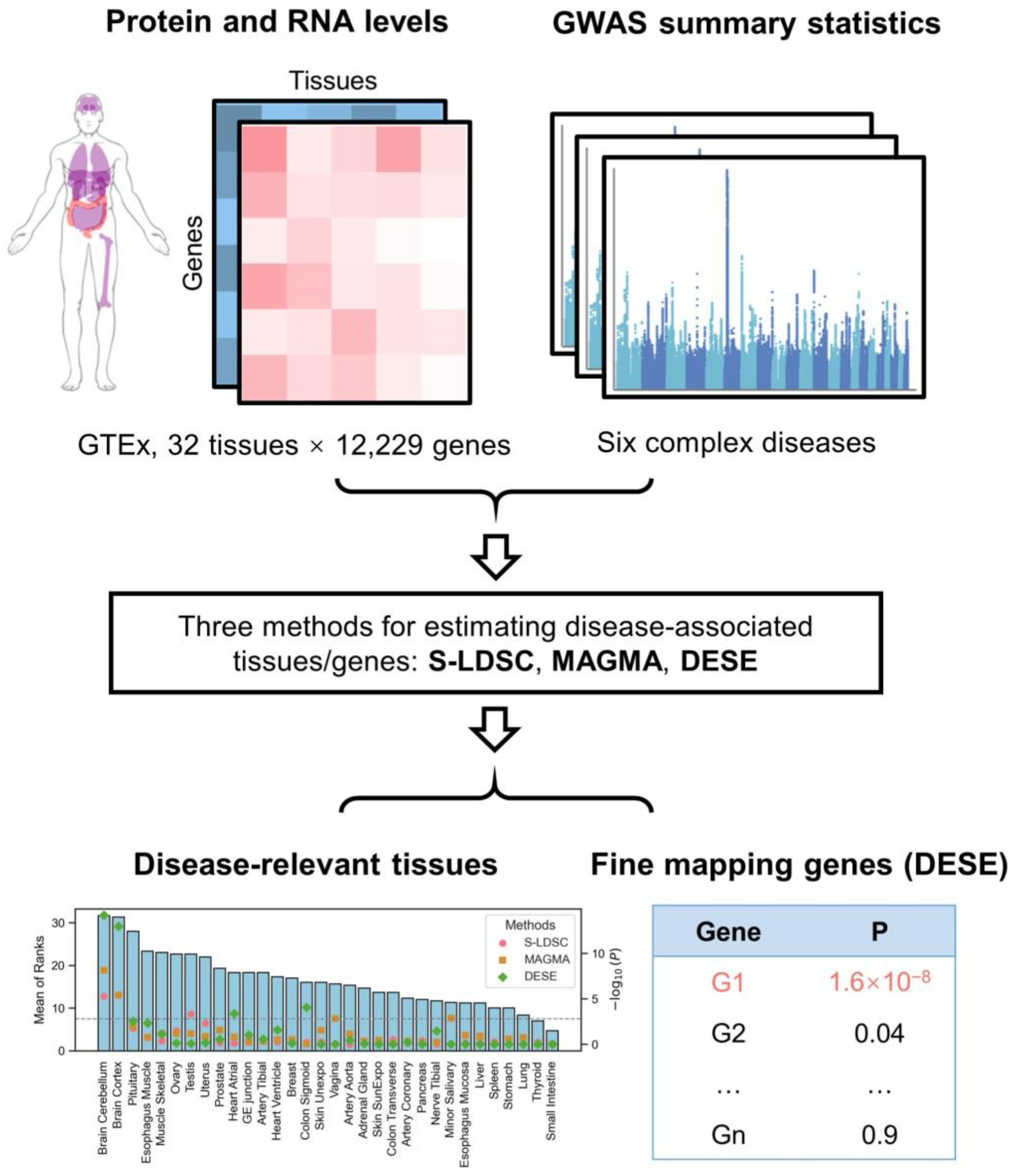
2. Materials and Methods
2.1. Paired Protein and RNA Expression Profiles
2.2. GWAS Summary Statistics
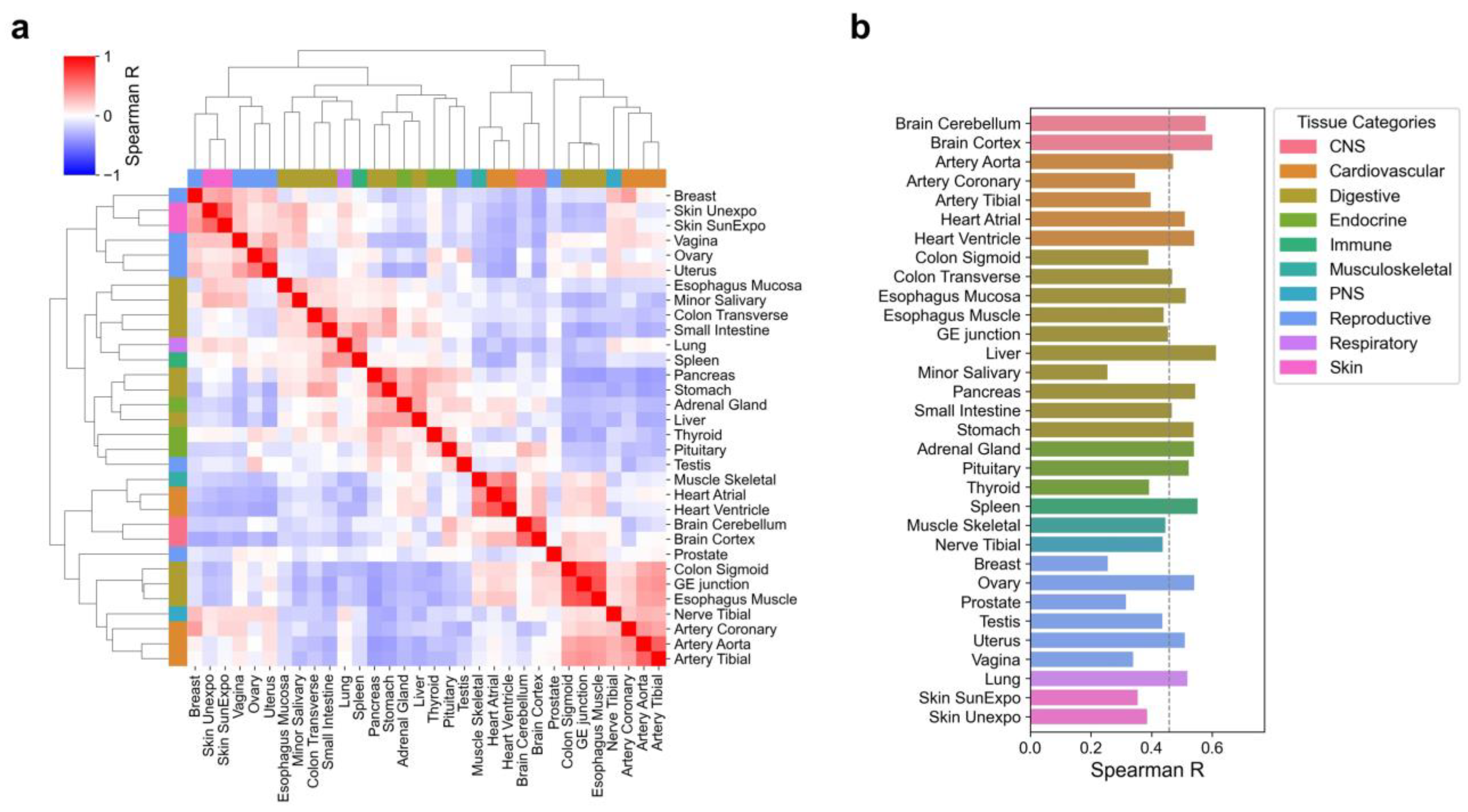
2.3. Tissue Correlation Analysis
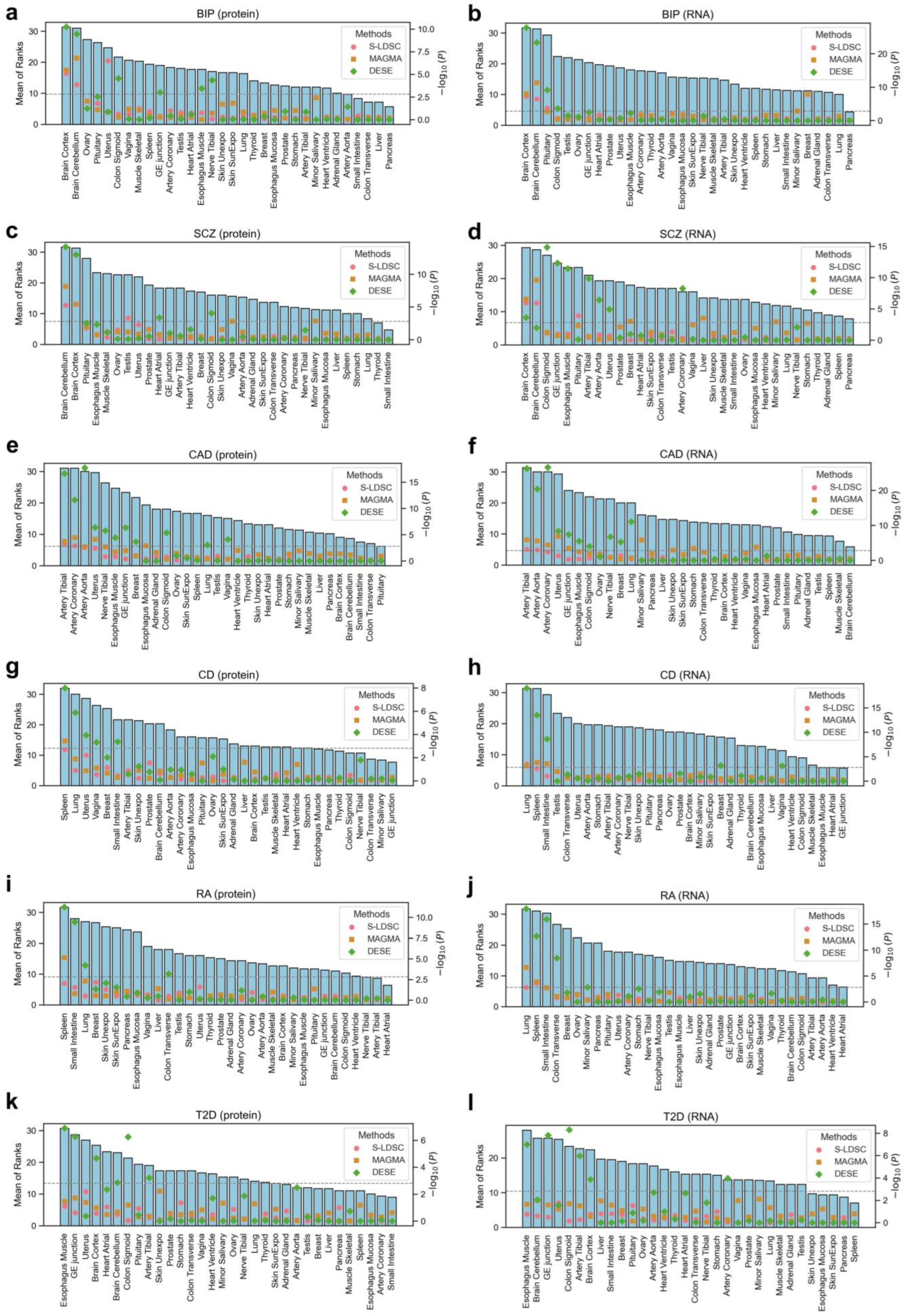
2.4. Identification of Disease-Associated Tissues
2.5. Fine-Mapping of Disease-Associated Genes
2.6. Evaluation of Disease-Associated Genes
2.7. Functional Enrichment Analysis
2.8. Protein-Specific Disease-Associated Gene Analysis
2.9. Analysis Code
3. Results
3.1. Characteristics of Tissue-Specific Protein Expression
3.2. Disease-Associated Tissues
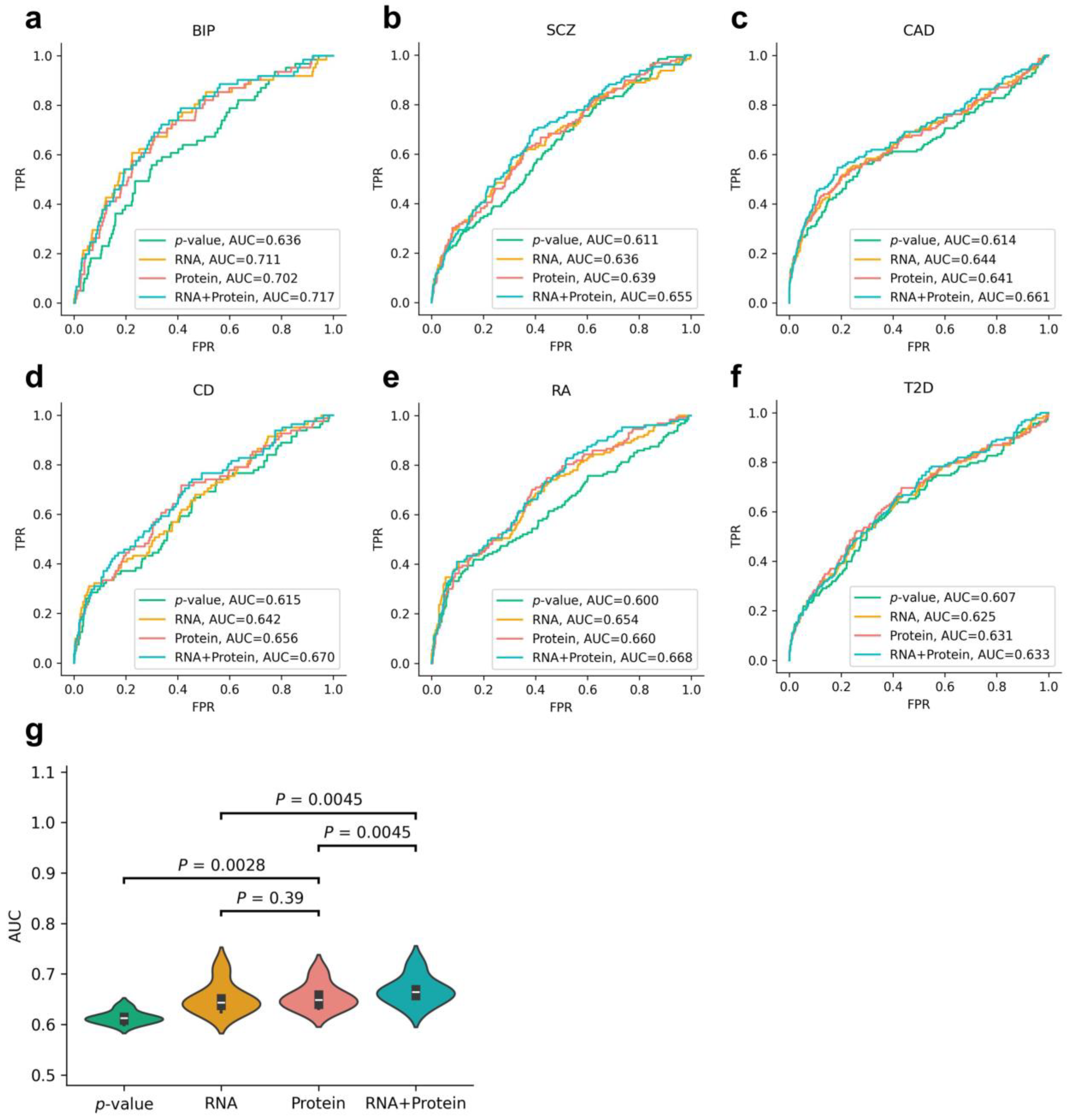
3.3. Evaluation of Disease-Associated Genes
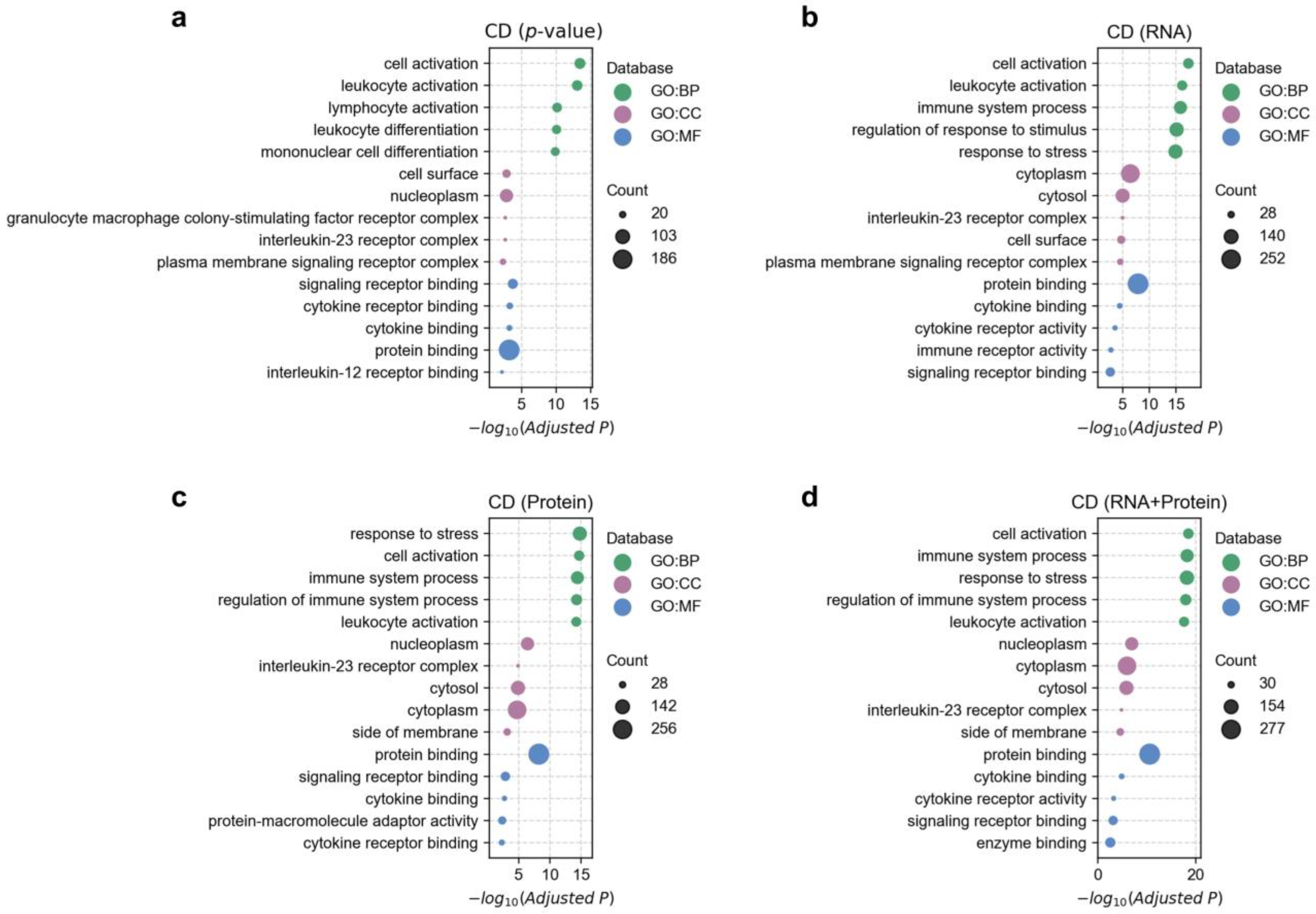
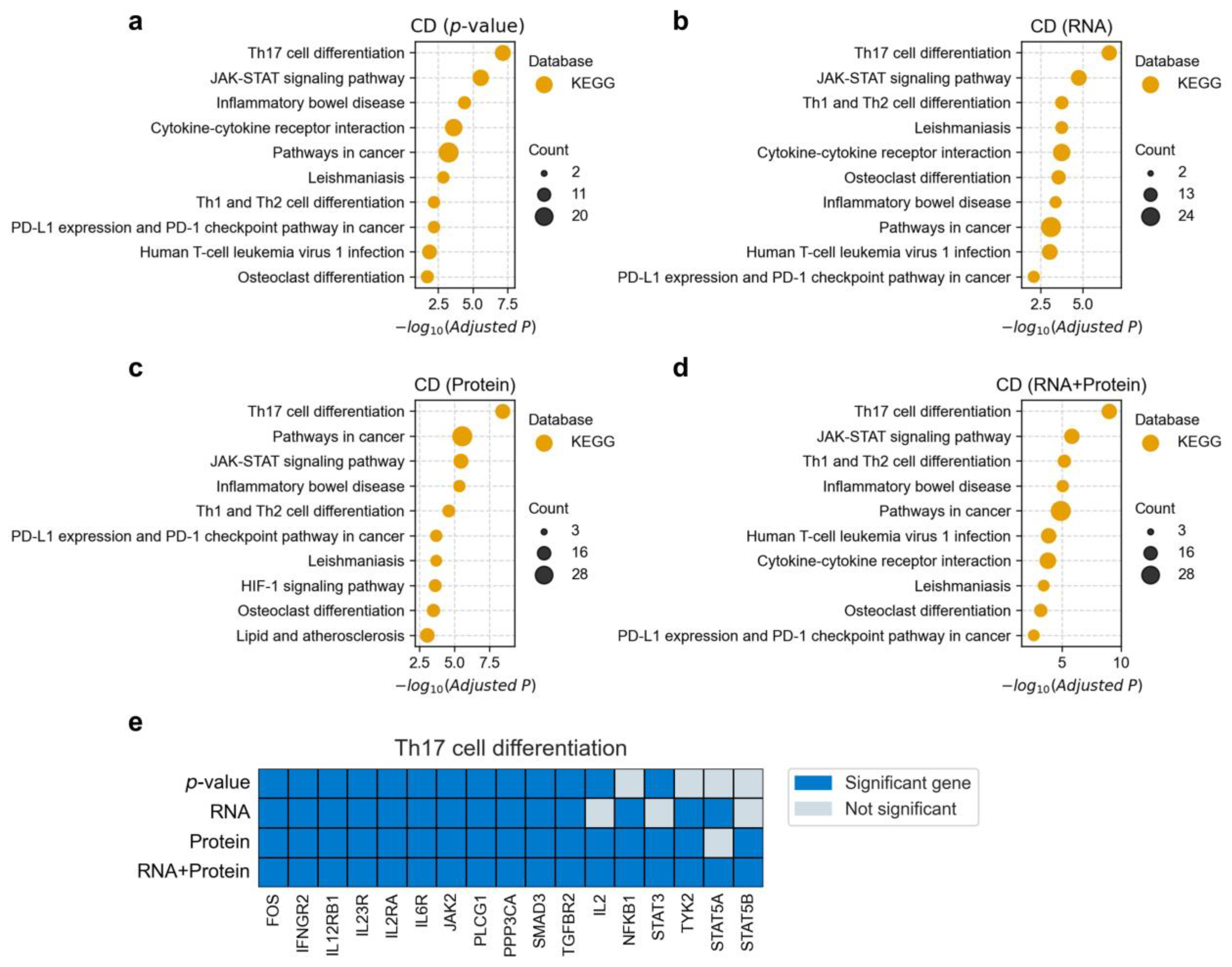
3.4. Functional Enrichment Analysis of Disease-Associated Genes
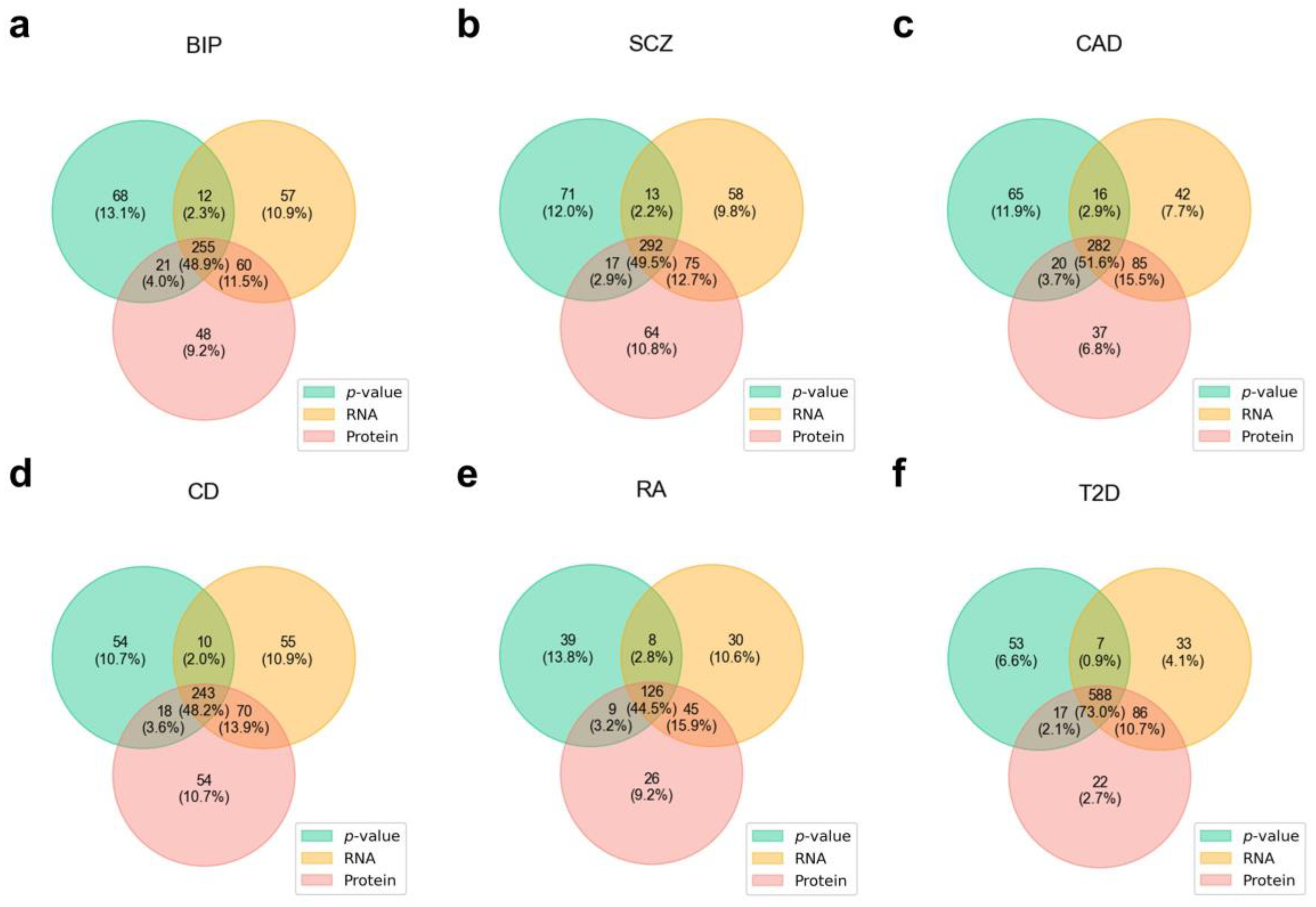
3.5. Unique Disease-Gene Associations Identified by Protein-Based Fine-Mapping
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GWAS | genome-wide association study |
| RNA | ribonucleic acid |
| GTEx | genotype-tissue expression |
| CI | confidence interval |
| ROC | receiver operating characteristic |
| AUC | area under the curve |
| HGNC | HUGO Gene Nomenclature Committee |
| LD | linkage disequilibrium |
| MHC | major histocompatibility complex |
| BIP | bipolar disorder |
| SCZ | schizophrenia |
| CAD | coronary artery disease |
| CD | Crohn’s disease |
| RA | rheumatoid arthritis |
| T2D | type 2 diabetes |
| FDR | false discovery rate |
| API | application programming interface |
| GO | Gene Ontology |
| PGC | Psychiatric Genomics Consortium |
References
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xue, C.; Dai, S.; Chen, S.; Chen, P.; Sham, P.C.; Wang, H.; Li, M. DESE: Estimating driver tissues by selective expression of genes associated with complex diseases or traits. Genome Biol. 2019, 20, 233. [Google Scholar] [CrossRef] [PubMed]
- Calderon, D.; Bhaskar, A.; Knowles, D.A.; Golan, D.; Raj, T.; Fu, A.Q.; Pritchard, J.K. Inferring Relevant Cell Types for Complex Traits by Using Single-Cell Gene Expression. Am. J. Hum. Genet. 2017, 101, 686–699. [Google Scholar] [CrossRef]
- Cano-Gamez, E.; Trynka, G. From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases. Front. Genet. 2020, 11, 424. [Google Scholar] [CrossRef]
- Finucane, H.K.; Reshef, Y.A.; Anttila, V.; Slowikowski, K.; Gusev, A.; Byrnes, A.; Gazal, S.; Loh, P.R.; Lareau, C.; Shoresh, N.; et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 2018, 50, 621–629. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; Snyder, M.P. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallstrom, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.H.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Mullins, N.; Forstner, A.J.; O’Connell, K.S.; Coombes, B.; Coleman, J.; Qiao, Z.; Als, T.D.; Bigdeli, T.B.; Borte, S.; Bryois, J.; et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 2021, 53, 817–829. [Google Scholar] [CrossRef]
- Trubetskoy, V.; Pardinas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef] [PubMed]
- van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef] [PubMed]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Verma, A.; Huffman, J.E.; Rodriguez, A.; Conery, M.; Liu, M.; Ho, Y.L.; Kim, Y.; Heise, D.A.; Guare, L.; Panickan, V.A.; et al. Diversity and scale: Genetic architecture of 2068 traits in the VA Million Veteran Program. Science 2024, 385, eadj1182. [Google Scholar] [CrossRef]
- Li, M.; Jiang, L.; Mak, T.; Kwan, J.; Xue, C.; Chen, P.; Leung, H.C.; Cui, L.; Li, T.; Sham, P.C. A powerful conditional gene-based association approach implicated functionally important genes for schizophrenia. Bioinformatics 2019, 35, 628–635. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; Hill, D.P.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids. Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids. Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]
- Andreasen, N.C.; Pierson, R. The role of the cerebellum in schizophrenia. Biol. Psychiatry 2008, 64, 81–88. [Google Scholar] [CrossRef]
- Faris, P.; Pischedda, D.; Palesi, F.; D’Angelo, E. New clues for the role of cerebellum in schizophrenia and the associated cognitive impairment. Front. Cell. Neurosci. 2024, 18, 1386583. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Weiss, Y.; Navon, Y.; Milo, I.; Azulay, N.; Keren, L.; Fuchs, S.; Ben-Zvi, D.; Noor, E.; Milo, R. The total mass, number, and distribution of immune cells in the human body. Proc. Natl. Acad. Sci. USA 2023, 120, e1986456176. [Google Scholar] [CrossRef] [PubMed]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Kadura, S.; Raghu, G. Rheumatoid arthritis-interstitial lung disease: Manifestations and current concepts in pathogenesis and management. Eur. Respir. Rev. 2021, 30, 210011. [Google Scholar] [CrossRef]
- Wilsher, M.; Voight, L.; Milne, D.; Teh, M.; Good, N.; Kolbe, J.; Williams, M.; Pui, K.; Merriman, T.; Sidhu, K.; et al. Prevalence of airway and parenchymal abnormalities in newly diagnosed rheumatoid arthritis. Respir. Med. 2012, 106, 1441–1446. [Google Scholar] [CrossRef]
- Norton, S.; Koduri, G.; Nikiphorou, E.; Dixey, J.; Williams, P.; Young, A. A study of baseline prevalence and cumulative incidence of comorbidity and extra-articular manifestations in RA and their impact on outcome. Rheumatology 2013, 52, 99–110. [Google Scholar] [CrossRef]
- Demoruelle, M.K.; Solomon, J.J.; Fischer, A.; Deane, K.D. The lung may play a role in the pathogenesis of rheumatoid arthritis. Int. J. Clin. Rheumtol. 2014, 9, 295–309. [Google Scholar] [CrossRef]
- Cavagna, L.; Monti, S.; Grosso, V.; Boffini, N.; Scorletti, E.; Crepaldi, G.; Caporali, R. The multifaceted aspects of interstitial lung disease in rheumatoid arthritis. BioMed Res. Int. 2013, 2013, 759760. [Google Scholar] [CrossRef]
- Merz, K.E.; Thurmond, D.C. Role of Skeletal Muscle in Insulin Resistance and Glucose Uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Krycer, J.R.; Kearney, A.L.; Hocking, S.L.; James, D.E. Muscle and adipose tissue insulin resistance: Malady without mechanism? J. Lipid. Res. 2019, 60, 1720–1732. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Jiang, L.; Zhou, M.; Long, Q.; Chen, Y.; Li, X.; Peng, W.; Yang, Q.; Li, M. PCGA: A comprehensive web server for phenotype-cell-gene association analysis. Nucleic Acids. Res. 2022, 50, W568–W576. [Google Scholar] [CrossRef]
- Jia, P.; Dai, Y.; Hu, R.; Pei, G.; Manuel, A.M.; Zhao, Z. TSEA-DB: A trait-tissue association map for human complex traits and diseases. Nucleic Acids. Res. 2020, 48, D1022–D1030. [Google Scholar] [CrossRef]
- Soppert, J.; Lehrke, M.; Marx, N.; Jankowski, J.; Noels, H. Lipoproteins and lipids in cardiovascular disease: From mechanistic insights to therapeutic targeting. Adv. Drug Deliv. Rev. 2020, 159, 4–33. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, P.; Ray, K.K.; Cannon, C.P. High-density lipoprotein and coronary heart disease: Current and future therapies. J. Am. Coll. Cardiol. 2010, 55, 1283–1299. [Google Scholar] [CrossRef]
- Maddur, M.S.; Miossec, P.; Kaveri, S.V.; Bayry, J. Th17 cells: Biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am. J. Pathol. 2012, 181, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lu, Q.; Liu, Y.; Shi, Z.; Hu, L.; Zeng, Z.; Tu, Y.; Xiao, Z.; Xu, Q. Th17 Cells in Inflammatory Bowel Disease: Cytokines, Plasticity, and Therapies. J. Immunol. Res. 2021, 2021, 8816041. [Google Scholar] [CrossRef]
- Jiang, W.; Su, J.; Zhang, X.; Cheng, X.; Zhou, J.; Shi, R.; Zhang, H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm. Res. 2014, 63, 943–950. [Google Scholar] [CrossRef]
- Chen, L.; Ruan, G.; Cheng, Y.; Yi, A.; Chen, D.; Wei, Y. The role of Th17 cells in inflammatory bowel disease and the research progress. Front. Immunol. 2022, 13, 1055914. [Google Scholar] [CrossRef]
- Kosmaczewska, A.; Ciszak, L.; Swierkot, J.; Szteblich, A.; Kosciow, K.; Frydecka, I. Exogenous IL-2 controls the balance in Th1, Th17, and Treg cell distribution in patients with progressive rheumatoid arthritis treated with TNF-alpha inhibitors. Inflammation 2015, 38, 765–774. [Google Scholar] [CrossRef]
- Wei, L.; Laurence, A.; O’Shea, J.J. New insights into the roles of Stat5a/b and Stat3 in T cell development and differentiation. Semin. Cell Dev. Biol. 2008, 19, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, L.; Xue, C.; Li, M.J.; Li, M. A conditional gene-based association framework integrating isoform-level eQTL data reveals new susceptibility genes for schizophrenia. eLife 2022, 11, e70779. [Google Scholar] [CrossRef] [PubMed]
- Solmi, M.; Radua, J.; Olivola, M.; Croce, E.; Soardo, L.; Salazar, D.P.G.; Il, S.J.; Kirkbride, J.B.; Jones, P.; Kim, J.H.; et al. Age at onset of mental disorders worldwide: Large-scale meta-analysis of 192 epidemiological studies. Mol. Psychiatry 2022, 27, 281–295. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Disease Name | PMID | Source | Sample Size |
|---|---|---|---|---|
| BIP | Bipolar disorder | 34002096 | PGC | 413,466 |
| SCZ | Schizophrenia | 35396580 | PGC | 320,404 |
| CAD | Coronary artery disease | 29212778 | GWAS Catalog | 296,525 |
| CD | Crohn’s disease | 28067908 | GWAS Catalog | 40,266 |
| RA | Rheumatoid arthritis | 24390342 | GWAS Catalog | 57,284 |
| T2D | Type 2 diabetes | 39024449 | GWAS Catalog | 432,648 |
| Disease | Gene | P (Protein) | P (RNA) | PubMed Count | Associated Tissue | Rank (Protein) | Rank (RNA) |
|---|---|---|---|---|---|---|---|
| BIP | CREB1 | 7.93 × 10−5 | 0.054 | 15 | BrainCerebellum | 0.864 | 0.424 |
| BIP | NME2 | 8.56 × 10−6 | 1.000 | 0 | BrainCerebellum | 0.780 | 0.019 |
| SCZ | HSPD1 | 1.05 × 10−14 | 0.115 | 11 | BrainCerebellum | 0.797 | 0.354 |
| SCZ | CENPA | 1.20 × 10−6 | 0.268 | 1 | BrainCortex | 0.876 | 0.185 |
| CAD | SMARCA4 | 3.29 × 10−23 | 0.021 | 11 | ArteryCoronary | 0.812 | 0.052 |
| CAD | TNRC6B | 4.84 × 10−5 | 0.073 | 0 | ArteryCoronary | 0.943 | 0.013 |
| CD | STAT3 | 9.13 × 10−5 | 0.012 | 151 | Spleen | 0.799 | 0.593 |
| CD | RAD50 | 4.95 × 10−13 | 0.011 | 1 | Spleen | 0.809 | 0.010 |
| RA | ARCN1 | 4.14 × 10−5 | 1.000 | 53 | Spleen | 0.720 | 0.161 |
| RA | SMARCC2 | 4.71 × 10−5 | 1.000 | 0 | Spleen | 0.867 | 0.215 |
| T2D | CYP17A1 | 4.08 × 10−9 | 1.000 | 7 | EsophagusMuscle | 0.818 | 0.296 |
| T2D | GPN1 | 8.61 × 10−6 | 1.000 | 0 | EsophagusMuscle | 0.818 | 0.194 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, C.; Zhou, M. Integrating Proteomics and GWAS to Identify Key Tissues and Genes Underlying Human Complex Diseases. Biology 2025, 14, 554. https://doi.org/10.3390/biology14050554
Xue C, Zhou M. Integrating Proteomics and GWAS to Identify Key Tissues and Genes Underlying Human Complex Diseases. Biology. 2025; 14(5):554. https://doi.org/10.3390/biology14050554
Chicago/Turabian StyleXue, Chao, and Miao Zhou. 2025. "Integrating Proteomics and GWAS to Identify Key Tissues and Genes Underlying Human Complex Diseases" Biology 14, no. 5: 554. https://doi.org/10.3390/biology14050554
APA StyleXue, C., & Zhou, M. (2025). Integrating Proteomics and GWAS to Identify Key Tissues and Genes Underlying Human Complex Diseases. Biology, 14(5), 554. https://doi.org/10.3390/biology14050554