


Identification of Key Nucleotide Metabolism Genes in Diabetic Retinopathy Based on Bioinformatics Analysis and Experimental Verification
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Processing
2.2. Identification of DEGs
2.3. GO and KEGG Enrichment Analysis
2.4. Construction of Protein–Protein Interaction (PPI) Network and Selection of Core Genes
2.5. Identification and Validation of Biomarkers
2.6. Chromosome and Subcellular Localization
2.7. Correlation Analysis and Gene–Gene Interaction (GGI) Network
2.8. Gene Set Enrichment Analysis (GSEA)
2.9. Gene Set Variation Analysis (GSVA)
2.10. Immuno-Infiltration Analysis
2.11. miRNA and Transcription Factor (TF) Prediction
2.12. Drug Prediction and Molecular Docking
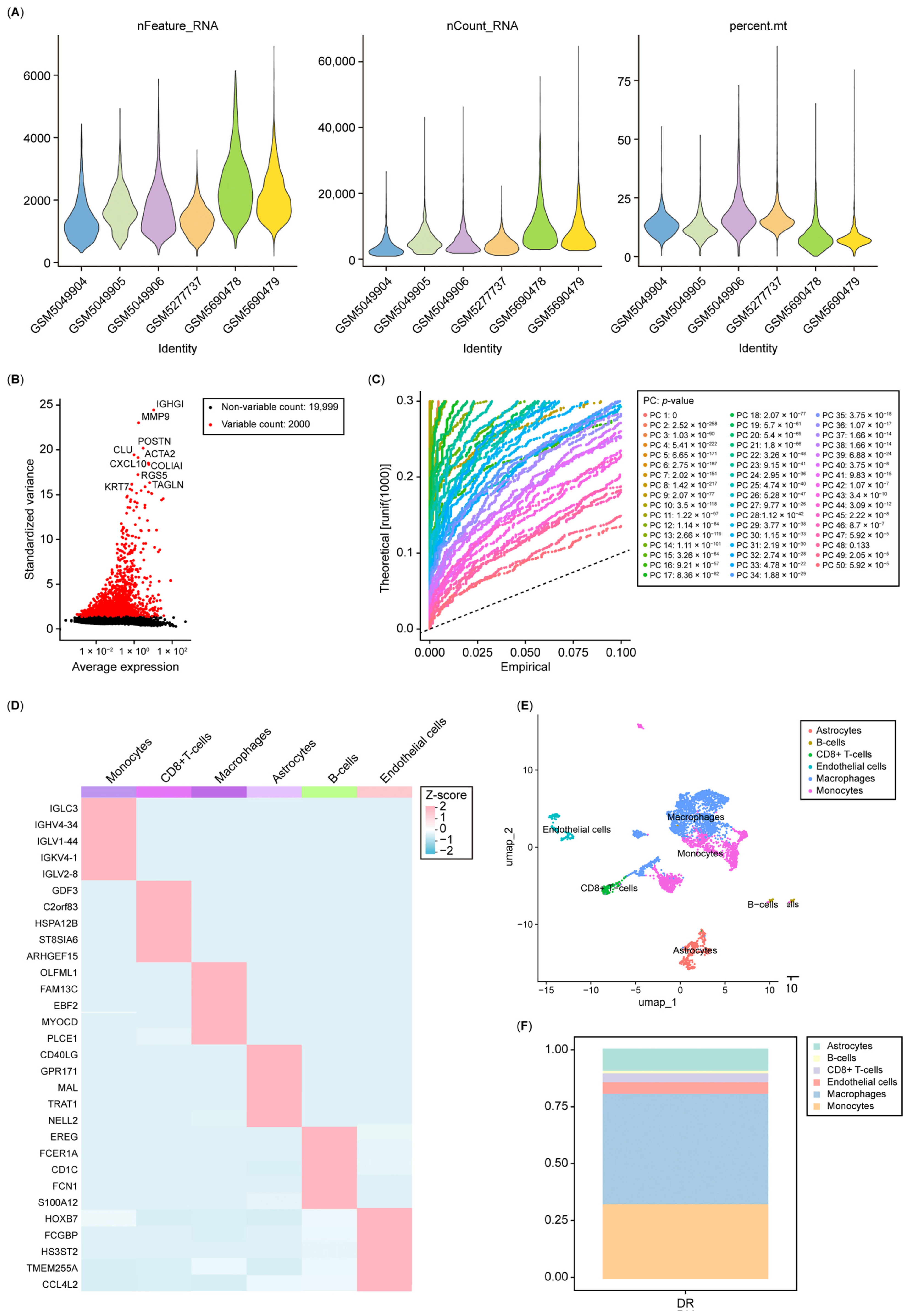
2.13. scRNA-Seq Analysis
2.14. RT-PCR
2.15. Statistical Analysis
3. Results
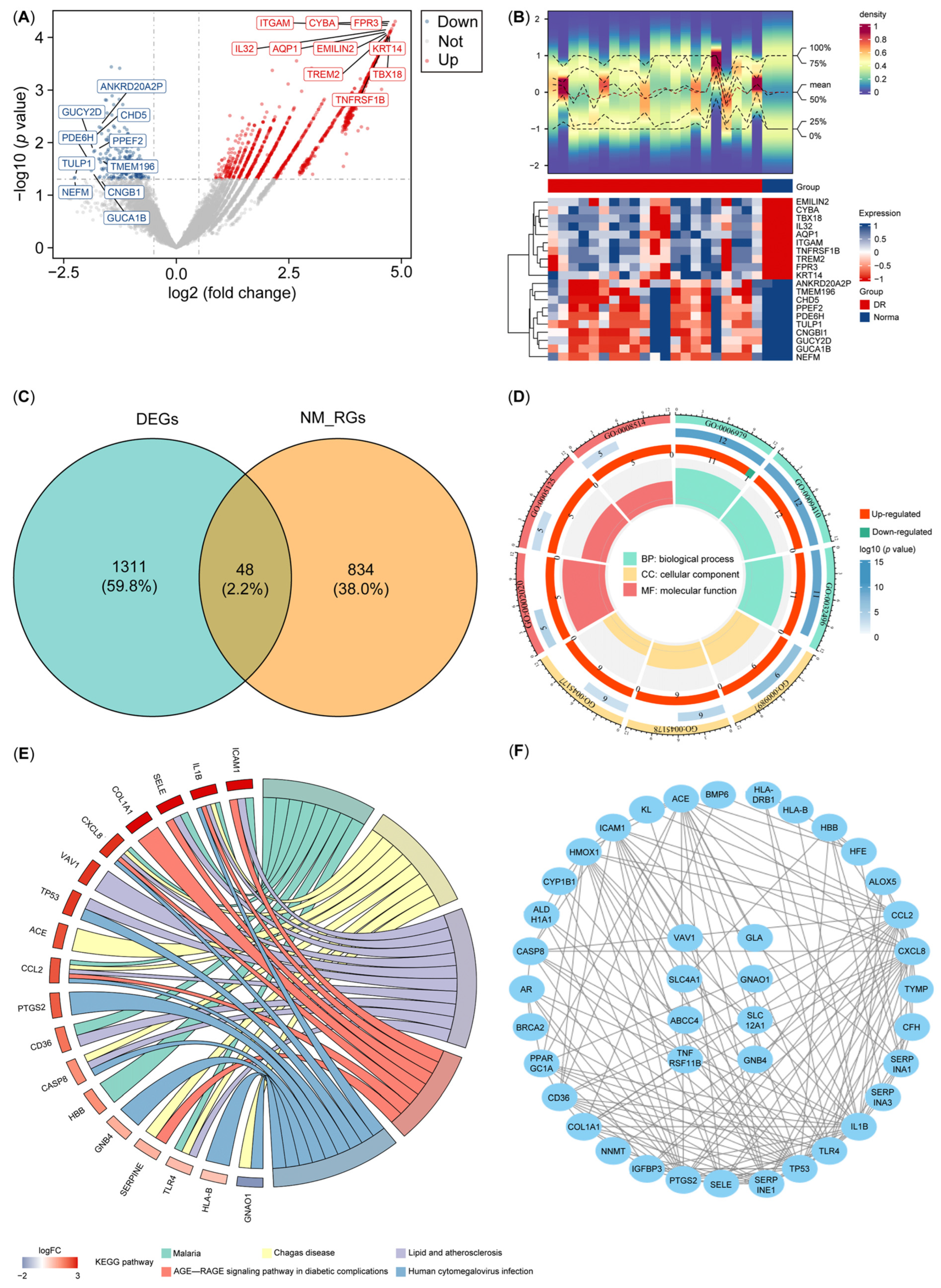
3.1. Screening and Functional Enrichment Analysis of DE-NMRGs
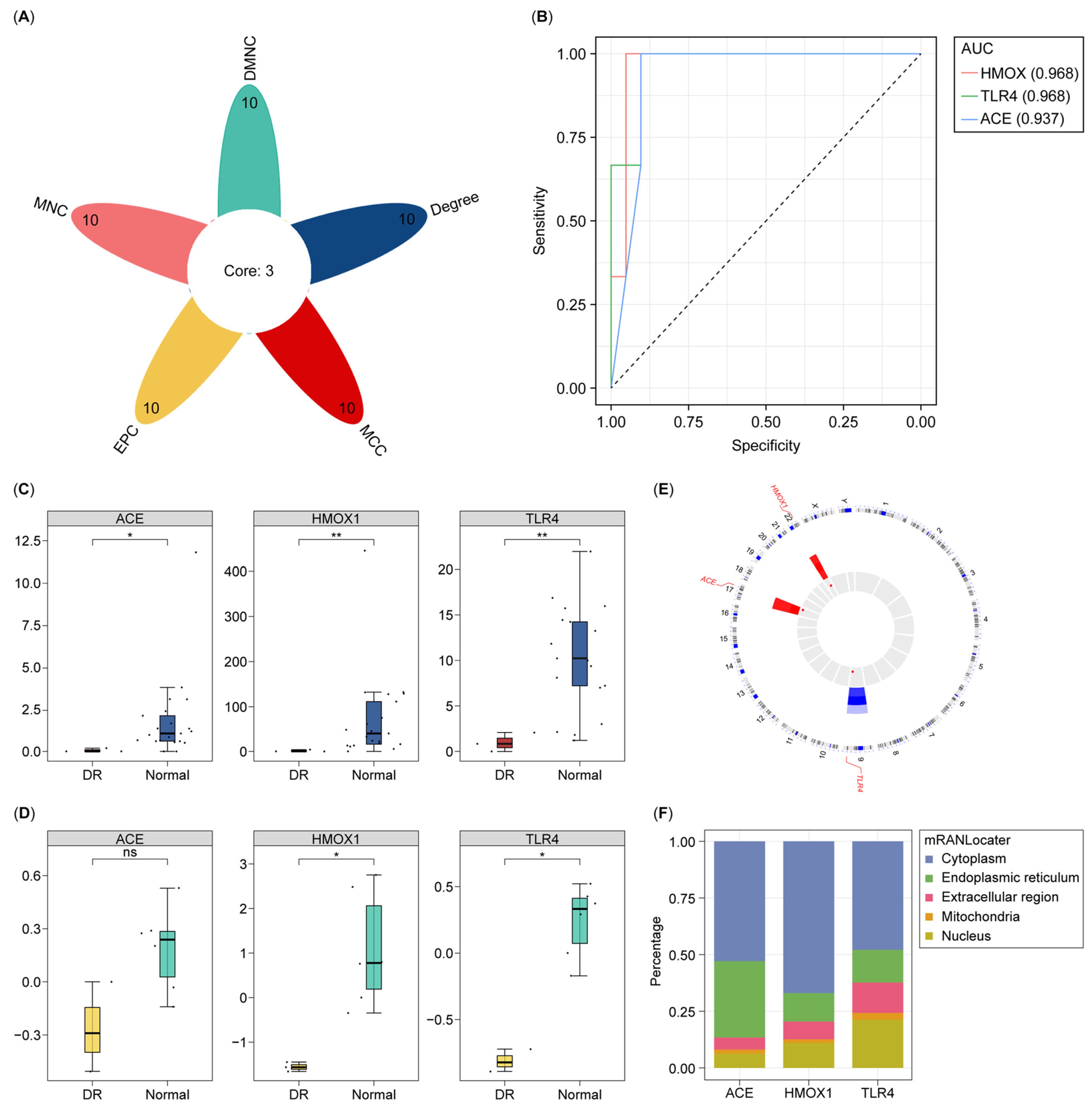
3.2. Identification and Assessment of All Biomarkers for DR
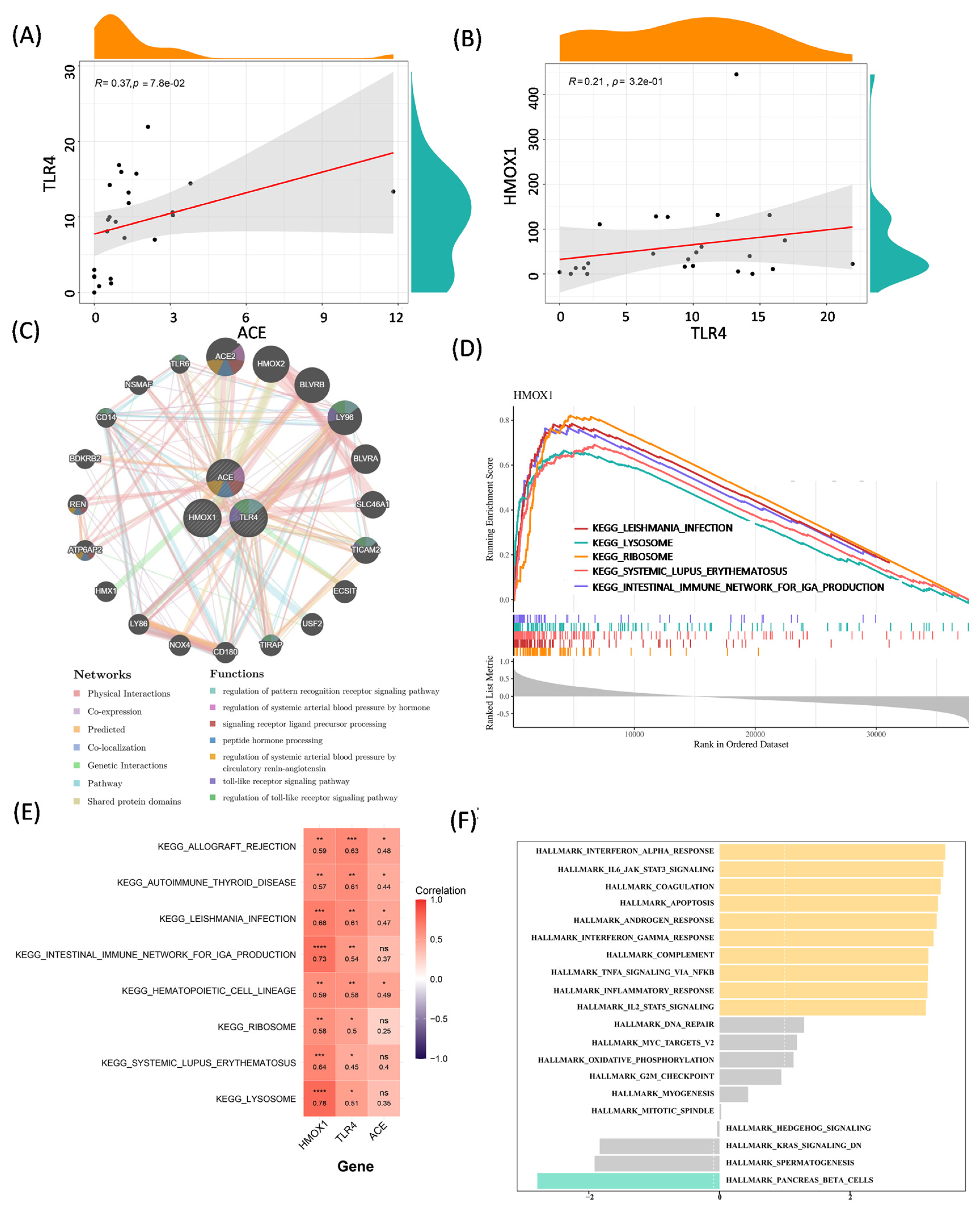
3.3. Correlation Analysis and Function Exploration of Biomarkers
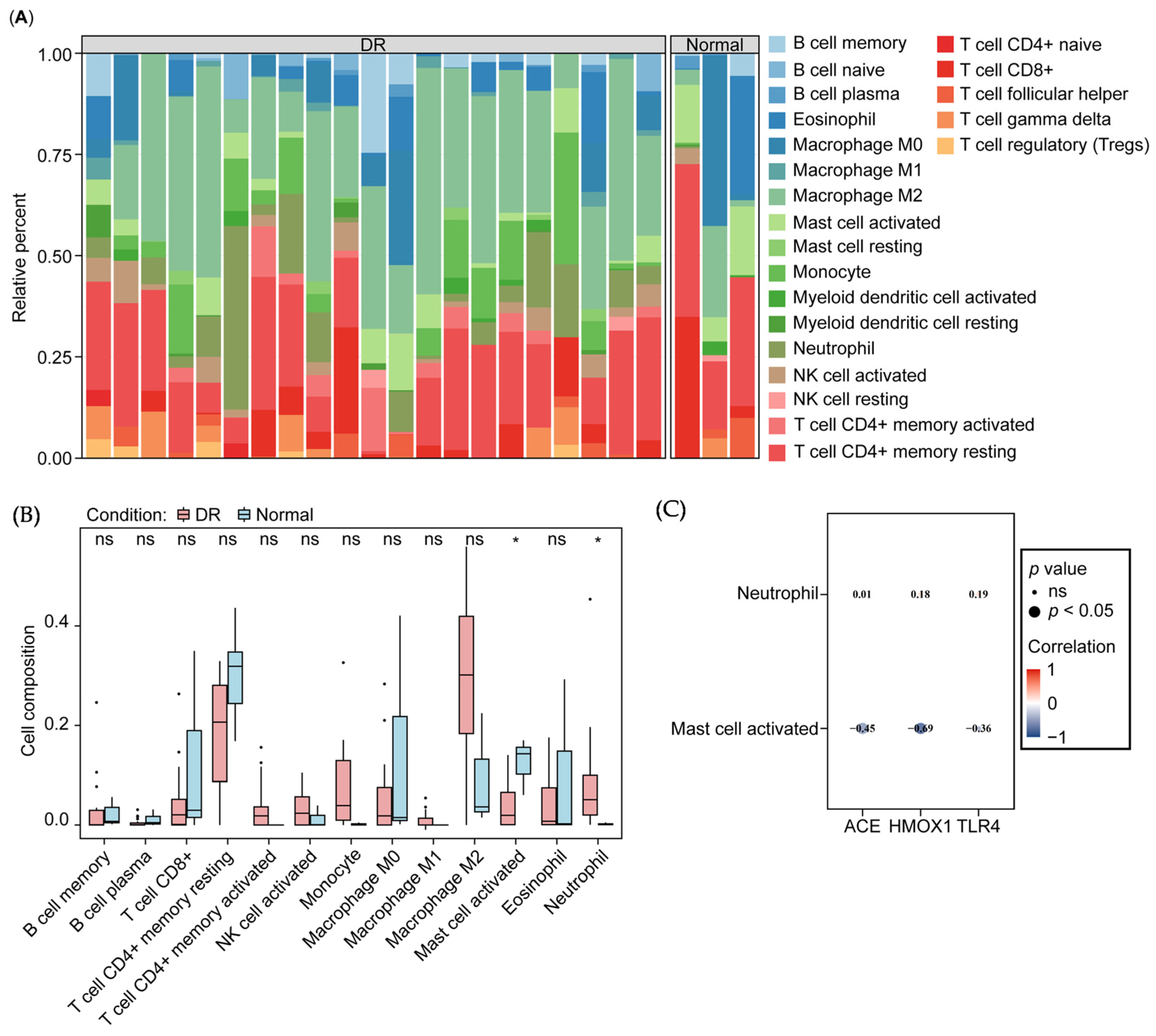
3.4. Correlation of Biomarkers and Immune Cells
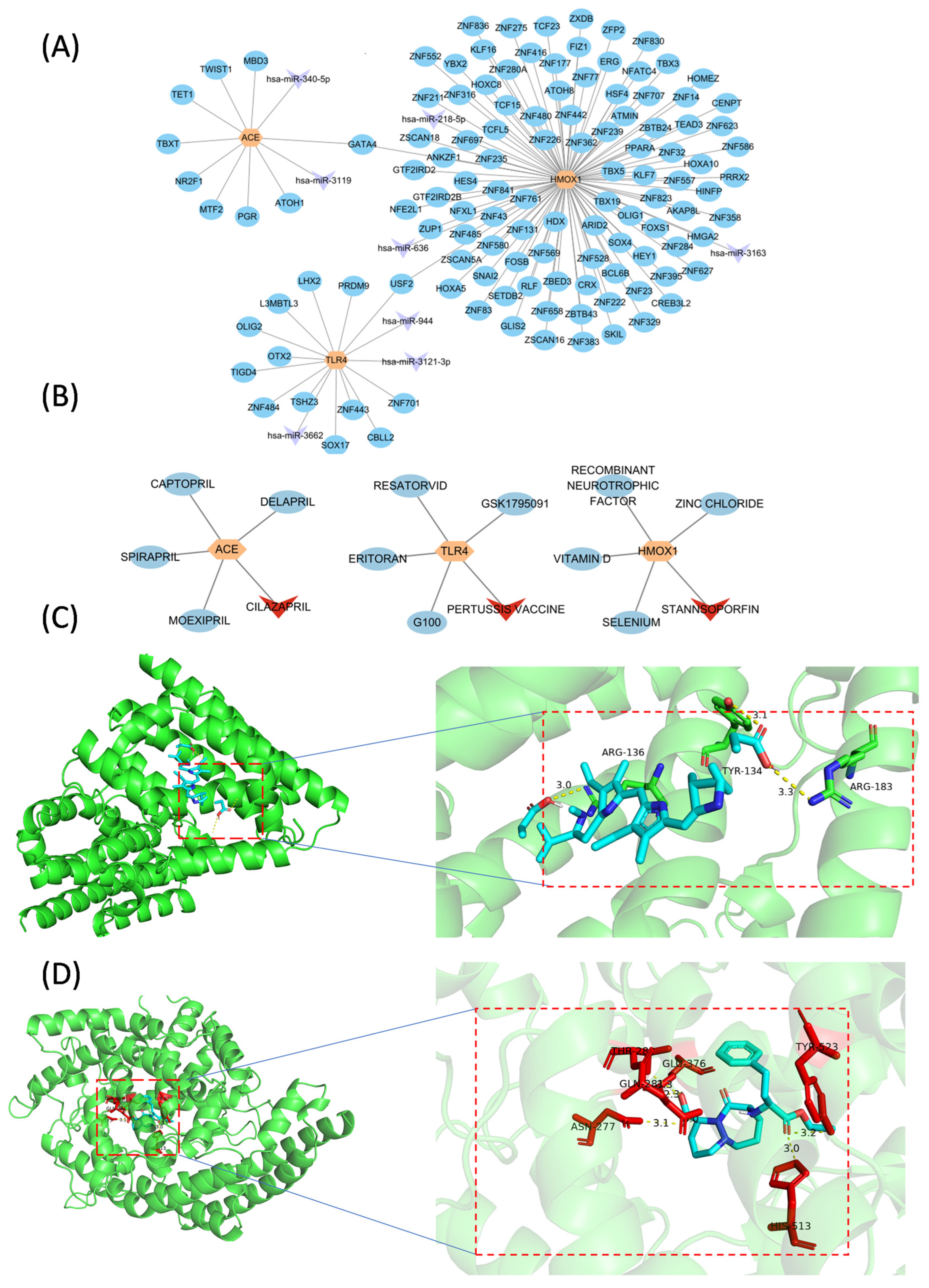
3.5. Molecular Regulatory and Drug–Gene Networks
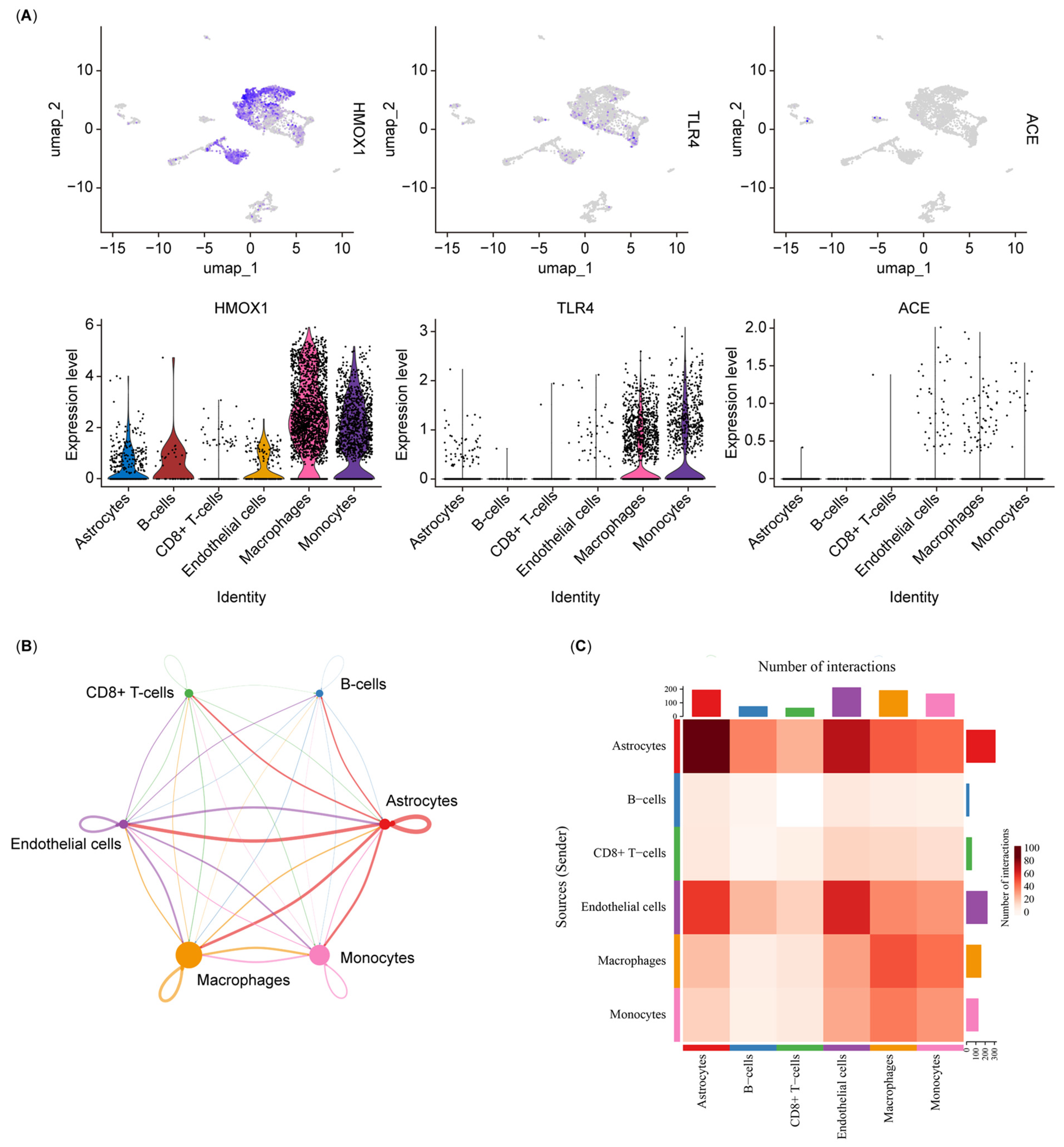
3.6. Single-Cell Analysis Re-Indicated That Nucleotide Metabolism Interacts with the Immune System in Diabetic Retinopathy
3.7. Validation of Biomarker Expression via RT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, N.; Wei, L.; Liu, D.; Zhang, Q.; Xia, X.; Ding, L.; Xiong, S. Identification and Validation of Autophagy-Related Genes in Diabetic Retinopathy. Front. Endocrinol. 2022, 13, 867600. [Google Scholar] [CrossRef] [PubMed]
- Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Rossino, M.G.; Dal Monte, M.; Casini, G. Relationships Between Neurodegeneration and Vascular Damage in Diabetic Retinopathy. Front. Neurosci. 2019, 13, 1172. [Google Scholar] [CrossRef]
- Santos, J.M.; Mohammad, G.; Zhong, Q.; Kowluru, R.A. Diabetic retinopathy, superoxide damage and antioxidants. Curr. Pharm. Biotechnol. 2011, 12, 352–361. [Google Scholar] [CrossRef]
- Meng, Z.; Chen, Y.; Wu, W.; Yan, B.; Meng, Y.; Liang, Y.; Yao, X.; Luo, J. Exploring the Immune Infiltration Landscape and M2 Macrophage-Related Biomarkers of Proliferative Diabetic Retinopathy. Front. Endocrinol. 2022, 13, 841813. [Google Scholar] [CrossRef]
- Li, C.; Cai, Q. Two ferroptosis-specific expressed genes NOX4 and PARP14 are considered as potential biomarkers for the diagnosis and treatment of diabetic retinopathy and atherosclerosis. Diabetol. Metab. Syndr. 2024, 16, 61. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, Q. Gene Biomarkers Related to Th17 Cells in Macular Edema of Diabetic Retinopathy: Cutting-Edge Comprehensive Bioinformatics Analysis and In Vivo Validation. Front. Immunol. 2022, 13, 858972. [Google Scholar] [CrossRef]
- Chatziralli, I.; Dimitriou, E.; Theodossiadis, G.; Bourouki, E.; Bagli, E.; Kitsos, G.; Theodossiadis, P. Intravitreal ranibizumab versus vitrectomy for recurrent vitreous haemorrhage after pars plana vitrectomy for proliferative diabetic retinopathy: A prospective study. Int. Ophthalmol. 2020, 40, 841–847. [Google Scholar] [CrossRef]
- Lim, J.W.; Lee, S.J.; Sung, J.Y.; Kim, J.S.; Nam, K.Y. Effect of prophylactic anti-VEGF injections on the prevention of recurrent vitreous hemorrhage in PDR patients after PRP. Sci. Rep. 2022, 12, 14484. [Google Scholar] [CrossRef]
- Berk Ergun, S.; Toklu, Y.; Cakmak, H.B.; Raza, S.; Simsek, S. The effect of intravitreal bevacizumab as a pretreatment of vitrectomy for diabetic vitreous hemorrhage on recurrent hemorrhage. Semin. Ophthalmol. 2015, 30, 177–180. [Google Scholar] [CrossRef]
- Yan, K.; Gao, L.N.; Cui, Y.L.; Zhang, Y.; Zhou, X. The cyclic AMP signaling pathway: Exploring targets for successful drug discovery (Review). Mol. Med. Rep. 2016, 13, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Zeller, M.; Guan, K.; Wunder, F.; Wagner, M.; El-Armouche, A. PDE2 at the crossway between cAMP and cGMP signalling in the heart. Cell. Signal. 2017, 38, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Shu, T.; Zhou, Y.; Yan, C. The perspective of cAMP/cGMP signaling and cyclic nucleotide phosphodiesterases in aortic aneurysm and dissection. Vascul. Pharmacol. 2024, 154, 107278. [Google Scholar] [CrossRef] [PubMed]
- Delhaye, S.; Bardoni, B. Role of phosphodiesterases in the pathophysiology of neurodevelopmental disorders. Mol. Psychiatry 2021, 26, 4570–4582. [Google Scholar] [CrossRef]
- Pålsson-McDermott, E.M.; O’Neill, L.A.J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020, 30, 300–314. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Gu, Q.; Wei, H.F. PLAG1 Promotes High Glucose-Induced Angiogenesis and Migration of Retinal Endothelial Cells by Regulating the Wnt/β-Catenin Signalling Pathway. Folia Biol. 2022, 68, 25–32. [Google Scholar] [CrossRef]
- Wang, N.; Ding, L.; Liu, D.; Zhang, Q.; Zheng, G.; Xia, X.; Xiong, S. Molecular investigation of candidate genes for pyroptosis-induced inflammation in diabetic retinopathy. Front. Endocrinol. 2022, 13, 918605. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, P.; Niu, S.; Xie, J.; Liu, Y.; Liu, Y.; Zhao, N.; Cheng, C.; Lu, P. Targeting nucleotide metabolic pathways in colorectal cancer by integrating scRNA-seq, spatial transcriptome, and bulk RNA-seq data. Funct. Integr. Genom. 2024, 24, 72. [Google Scholar] [CrossRef]
- Gustavsson, E.K.; Zhang, D.; Reynolds, R.H.; Garcia-Ruiz, S.; Ryten, M. ggtranscript: An R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 2022, 38, 3844–3846. [Google Scholar] [CrossRef]
- Zuguang, G.; Roland, E.; Matthias, S. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar]
- Mao, W.; Ding, J.; Li, Y.; Huang, R.; Wang, B. Inhibition of cell survival and invasion by Tanshinone IIA via FTH1: A key therapeutic target and biomarker in head and neck squamous cell carcinoma. Exp. Ther. Med. 2022, 24, 521. [Google Scholar] [CrossRef] [PubMed]
- Tianzhi, W.; Erqiang, H.; Shuangbin, X.; Meijun, C.; Pingfan, G.; Zehan, D.; Tingze, F.; Lang, Z.; Wenli, T.; Li, Z.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Frédérique, L.; Natalia, T.; Alexandre, H.; Natacha, T.; Xavier, R.; Jean-Charles, S.; Markus, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef]
- Tang, Q.; Nie, F.; Kang, J.; Chen, W. mRNALocater: Enhance the prediction accuracy of eukaryotic mRNA subcellular localization by using model fusion strategy. Mol. Ther. 2021, 29, 2617–2623. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The multiMiR R package and database: Integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Mao, X.; Chen, M.; Wu, X.; Zhu, T.; Liu, Y.; Zhang, Z.; Fan, W.; Xie, P.; Yuan, S.; et al. Single-Cell Transcriptomics Reveals Novel Role of Microglia in Fibrovascular Membrane of Proliferative Diabetic Retinopathy. Diabetes 2022, 71, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.J.; Joglekar, M.V.; Hardikar, A.A.; Keech, A.C.; O’Neal, D.N.; Januszewski, A.S. Biomarkers in Diabetic Retinopathy. Rev. Diabet. Stud. 2015, 12, 159–195. [Google Scholar] [CrossRef]
- Lin, K.Y.; Hsih, W.H.; Lin, Y.B.; Wen, C.Y.; Chang, T.J. Update in the epidemiology, risk factors, screening, and treatment of diabetic retinopathy. J. Diabetes Investig. 2021, 12, 1322–1325. [Google Scholar] [CrossRef]
- Adki, K.M.; Kulkarni, Y.A. Potential Biomarkers in Diabetic Retinopathy. Curr. Diabetes Rev. 2020, 16, 971–983. [Google Scholar]
- Cosgrove, M. Perinatal and infant nutrition. Nucleotides. Nutrition 1998, 14, 748–751. [Google Scholar] [CrossRef]
- Yang, K.; Liu, J.; Zhang, X.; Ren, Z.; Gao, L.; Wang, Y.; Lin, W.; Ma, X.; Hao, M.; Kuang, H. H3 Relaxin Alleviates Migration, Apoptosis and Pyroptosis Through P2X7R-Mediated Nucleotide Binding Oligomerization Domain-Like Receptor Protein 3 Inflammasome Activation in Retinopathy Induced by Hyperglycemia. Front. Pharmacol. 2020, 11, 603689. [Google Scholar] [CrossRef]
- Sumarriva, K.; Uppal, K.; Ma, C.; Herren, D.J.; Wang, Y.; Chocron, I.M.; Warden, C.; Mitchell, S.L.; Burgess, L.G.; Goodale, M.P.; et al. Arginine and Carnitine Metabolites Are Altered in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3119–3126. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Cheng, Y.; Liu, K.; Zou, H.; You, Z. Identification of potential ferroptosis-related biomarkers and a pharmacological compound in diabetic retinopathy based on machine learning and molecular docking. Front. Endocrinol. 2022, 13, 988506. [Google Scholar] [CrossRef]
- Huang, Y.; Peng, J.; Liang, Q. Identification of key ferroptosis genes in diabetic retinopathy based on bioinformatics analysis. PLoS ONE 2023, 18, e0280548. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, G.; Aminuddin, F.; Akhabir, L.; He, J.Q.; Shumansky, K.; Connett, J.E.; Anthonisen, N.R.; Abboud, R.T.; Paré, P.D.; Sandford, A.J. Effect of heme oxygenase-1 polymorphisms on lung function and gene expression. BMC Med. Genet. 2011, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Chau, L.Y. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 2002, 8, 240–246. [Google Scholar] [CrossRef]
- Meng, Z.; Liang, H.; Zhao, J.; Gao, J.; Liu, C.; Ma, X.; Liu, J.; Liang, B.; Jiao, X.; Cao, J.; et al. HMOX1 upregulation promotes ferroptosis in diabetic atherosclerosis. Life Sci. 2021, 284, 119935. [Google Scholar] [CrossRef]
- Bayan, N.; Yazdanpanah, N.; Rezaei, N. Role of toll-like receptor 4 in diabetic retinopathy. Pharmacol. Res. 2022, 175, 105960. [Google Scholar] [CrossRef]
- Hotinger, J.A.; Gallagher, A.H.; May, A.E. Phage-Related Ribosomal Proteases (Prps): Discovery, Bioinformatics, and Structural Analysis. Antibiotics 2022, 11, 1109. [Google Scholar] [CrossRef]
- Xiao, Z.; Yang, M.; Fang, L.; Lv, Q.; He, Q.; Deng, M.; Liu, X.; Chen, X.; Chen, M.; Xie, X.; et al. Extracellular nucleotide inhibits cell proliferation and negatively regulates Toll-like receptor 4 signalling in human progenitor endothelial cells. Cell Biol. Int. 2012, 36, 625–633. [Google Scholar] [CrossRef]
- Molugu, T.R.; Oita, R.C.; Chawla, U.; Camp, S.M.; Brown, M.F.; Garcia, J.G.N. Nicotinamide phosphoribosyltransferase purification using SUMO expression system. Anal Biochem. 2020, 598, 113597. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Garcia, V.; Joseph, G.; Shkolnik, B.; Ding, Y.; Zhang, F.F.; Gotlinger, K.; Falck, J.R.; Dakarapu, R.; Capdevila, J.H.; Bernstein, K.E.; et al. Angiotensin II receptor blockade or deletion of vascular endothelial ACE does not prevent vascular dysfunction and remodeling in 20-HETE-dependent hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R71–R78. [Google Scholar] [CrossRef]
- Abbas, S.; Raza, S.T.; Ahmed, F.; Ahmad, A.; Rizvi, S.; Mahdi, F. Association of genetic polymorphism of PPARγ-2, ACE, MTHFR, FABP-2 and FTO genes in risk prediction of type 2 diabetes mellitus. J. Biomed. Sci. 2013, 20, 80. [Google Scholar] [CrossRef] [PubMed]
- Cattelan, F.; Hysa, E.; Gotelli, E.; Pizzorni, C.; Bica, P.F.; Grosso, M.; Barisione, E.; Paolino, S.; Carmisciano, L.; Sulli, A.; et al. Microvascular capillaroscopic abnormalities and occurrence of antinuclear autoantibodies in patients with sarcoidosis. Rheumatol. Int. 2022, 42, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Vasudeva, K.; Balyan, R.; Munshi, A. ACE-Triggered Hypertension Incites Stroke: Genetic, Molecular, and Therapeutic Aspects. Neuromol. Med. 2020, 22, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Kushida, T.; Quan, S.; Yang, L.; Ikehara, S.; Kappas, A.; Abraham, N.G. A significant role for the heme oxygenase-1 gene in endothelial cell cycle progression. Biochem. Biophys. Res. Commun. 2002, 291, 68–75. [Google Scholar] [CrossRef]
- Banus, S.; Stenger, R.M.; Gremmer, E.R.; Dormans, J.A.; Mooi, F.R.; Kimman, T.G.; Vandebriel, R.J. The role of Toll-like receptor-4 in pertussis vaccine-induced immunity. BMC Immunol. 2008, 9, 21. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Gene | Sequence (5′ to 3′) |
|---|---|---|
| HMOX1 F | HMOX1 | AAGACTGCGTTCCTGCTCAAC |
| HMOX1 R | AAAGCCCTACAGCAACTGTCG | |
| TRL4 F | TRL4 | AGACCTGTCCCTGAACCCTAT |
| TRL4 R | CGATGGACTTCTAAACCAGCCA | |
| ACE F | ACE | CCACGTCCCGGAAATATGAAG |
| ACE R | AGTCCCCTGCATCTACATAGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Gong, J. Identification of Key Nucleotide Metabolism Genes in Diabetic Retinopathy Based on Bioinformatics Analysis and Experimental Verification. Biology 2025, 14, 409. https://doi.org/10.3390/biology14040409
Wang W, Gong J. Identification of Key Nucleotide Metabolism Genes in Diabetic Retinopathy Based on Bioinformatics Analysis and Experimental Verification. Biology. 2025; 14(4):409. https://doi.org/10.3390/biology14040409
Chicago/Turabian StyleWang, Wei, and Jianyang Gong. 2025. "Identification of Key Nucleotide Metabolism Genes in Diabetic Retinopathy Based on Bioinformatics Analysis and Experimental Verification" Biology 14, no. 4: 409. https://doi.org/10.3390/biology14040409
APA StyleWang, W., & Gong, J. (2025). Identification of Key Nucleotide Metabolism Genes in Diabetic Retinopathy Based on Bioinformatics Analysis and Experimental Verification. Biology, 14(4), 409. https://doi.org/10.3390/biology14040409