
Nanobodies: From Discovery to AI-Driven Design



Simple Summary
Abstract
1. Introduction
2. Tracing the Historical Discovery of Nanobodies: Key Milestones in Nanobody Research
3. Overview of Libraries and Display Technologies for Nanobody Development
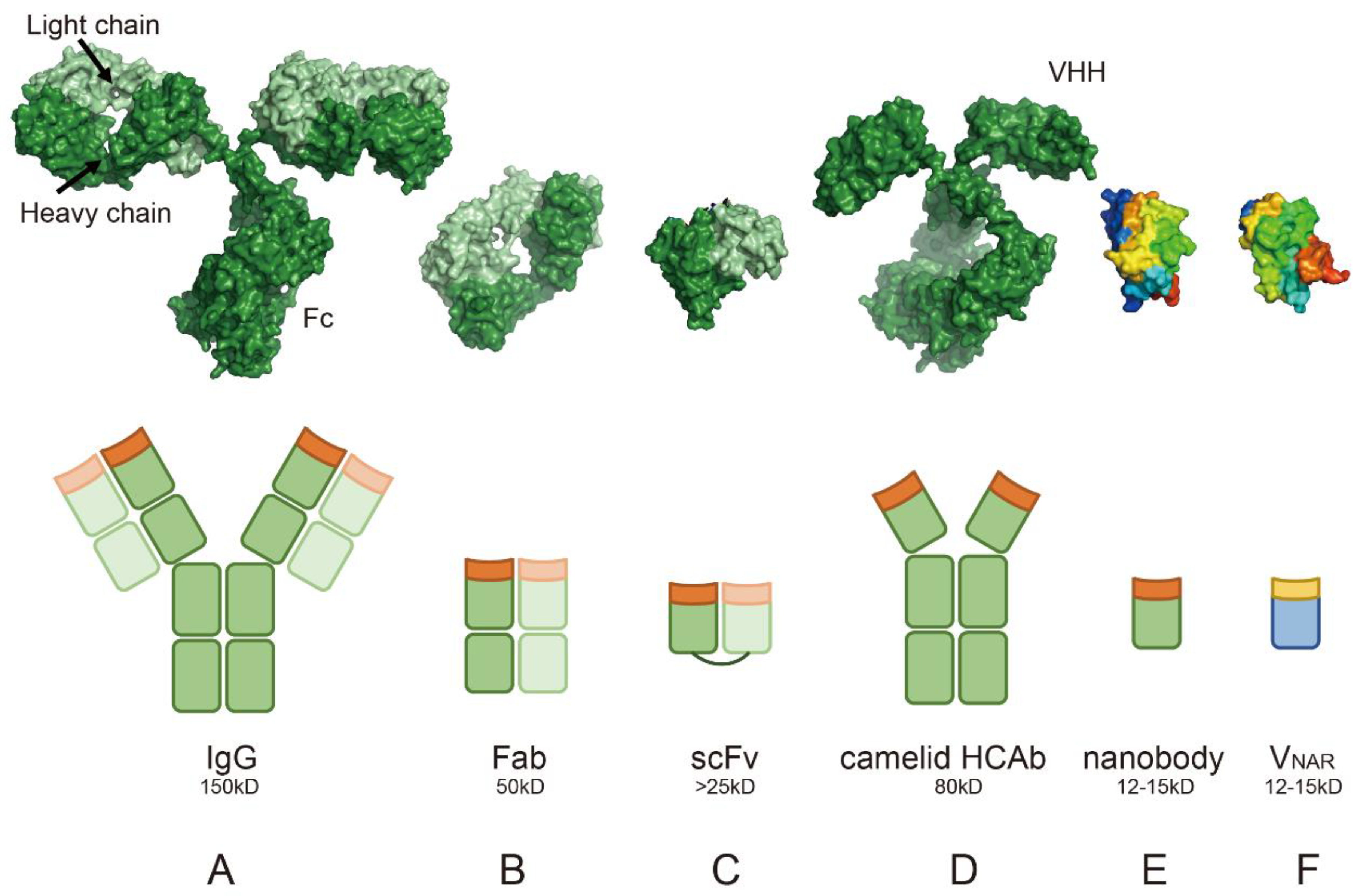
4. Clarifying the Unique Properties of Nanobodies Through Structural Biology Analysis: Advantages over Conventional Antibodies
4.1. Single-Domain Structure
4.2. Longer CDR3 Loop
4.3. Framework Region Hydrophilicity
4.4. Absence of the Light Chain
4.5. Greater Stability in Harsh Conditions
4.6. Efficient Production and Purification
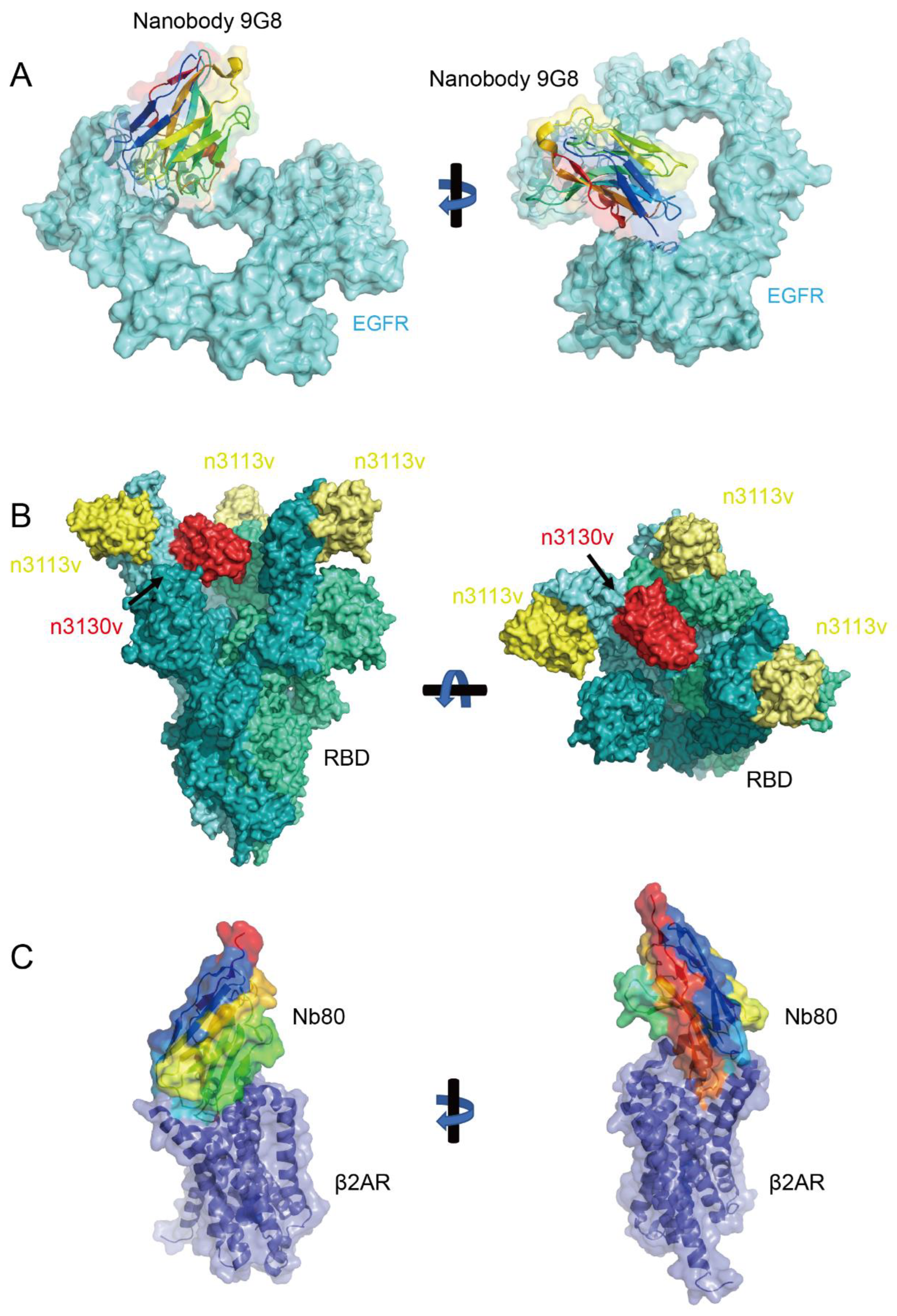
4.7. Binding to Concave Surfaces
4.8. Binding to Cryptic Epitopes
4.9. Binding to Other Hard-to-Target Regions
4.10. Comparisons with Alternative Technologies
5. Comparison of Structural Differences Among Camelid-Derived, Human-Derived, and Shark-Derived Nanobodies
6. Humanization Strategy for Camelid Nanobodies
7. Nanobodies and Their Unique Binding Mode with Specific Antigens
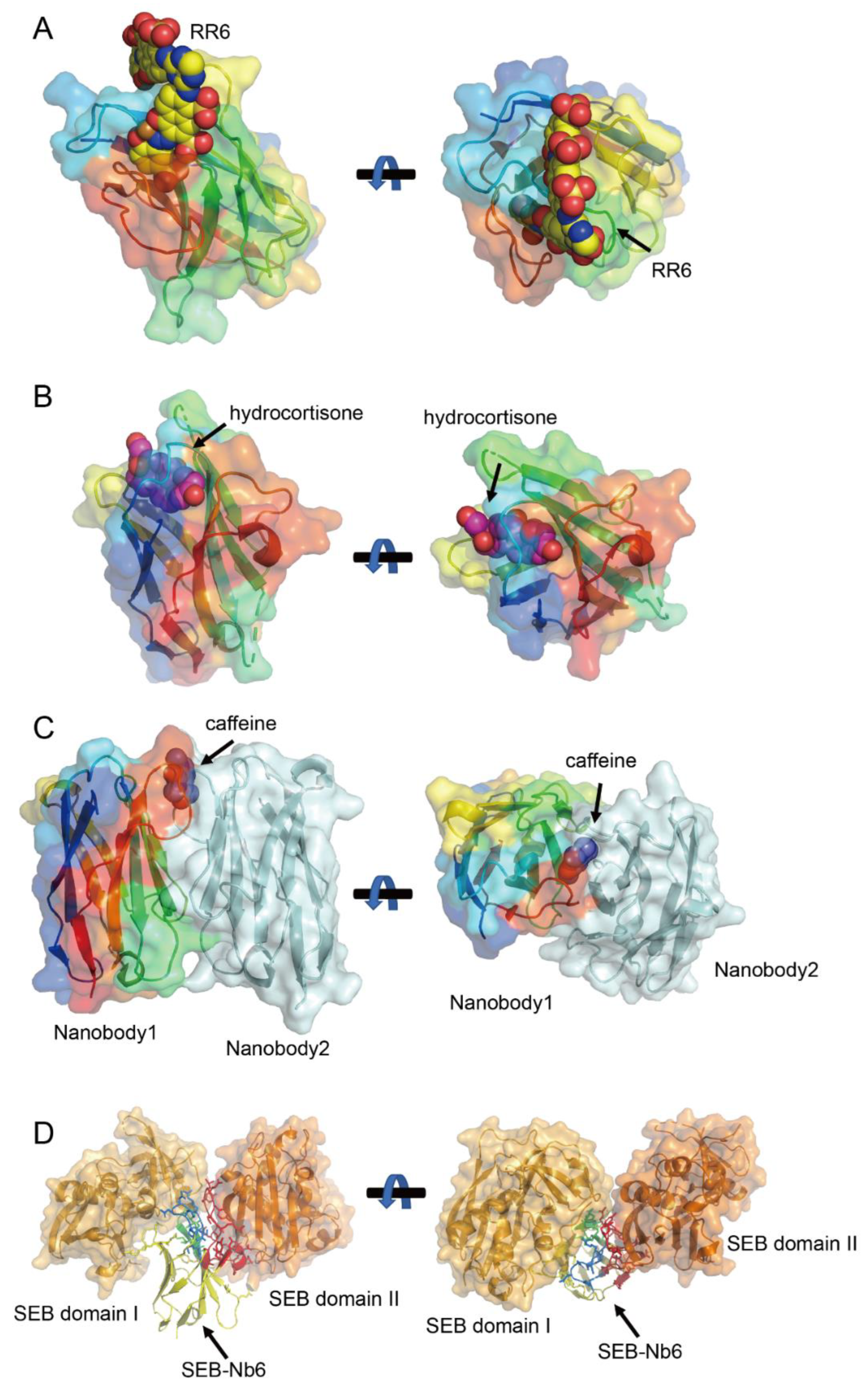
7.1. CDR1-Tunneling Modes for Small-Molecule Encapsulation
7.2. Non-Canonical Disulfide Bonds to Stabilize Compact Binding Cavities
7.3. Homodimerization-Driven Recognition for Enhanced Avidity
7.4. Intrinsic Dual-Epitope Engagement Through Spatially Segregated CDR Loops
8. Nanobodies Binding to Multiple Epitopes of the Same Antigen
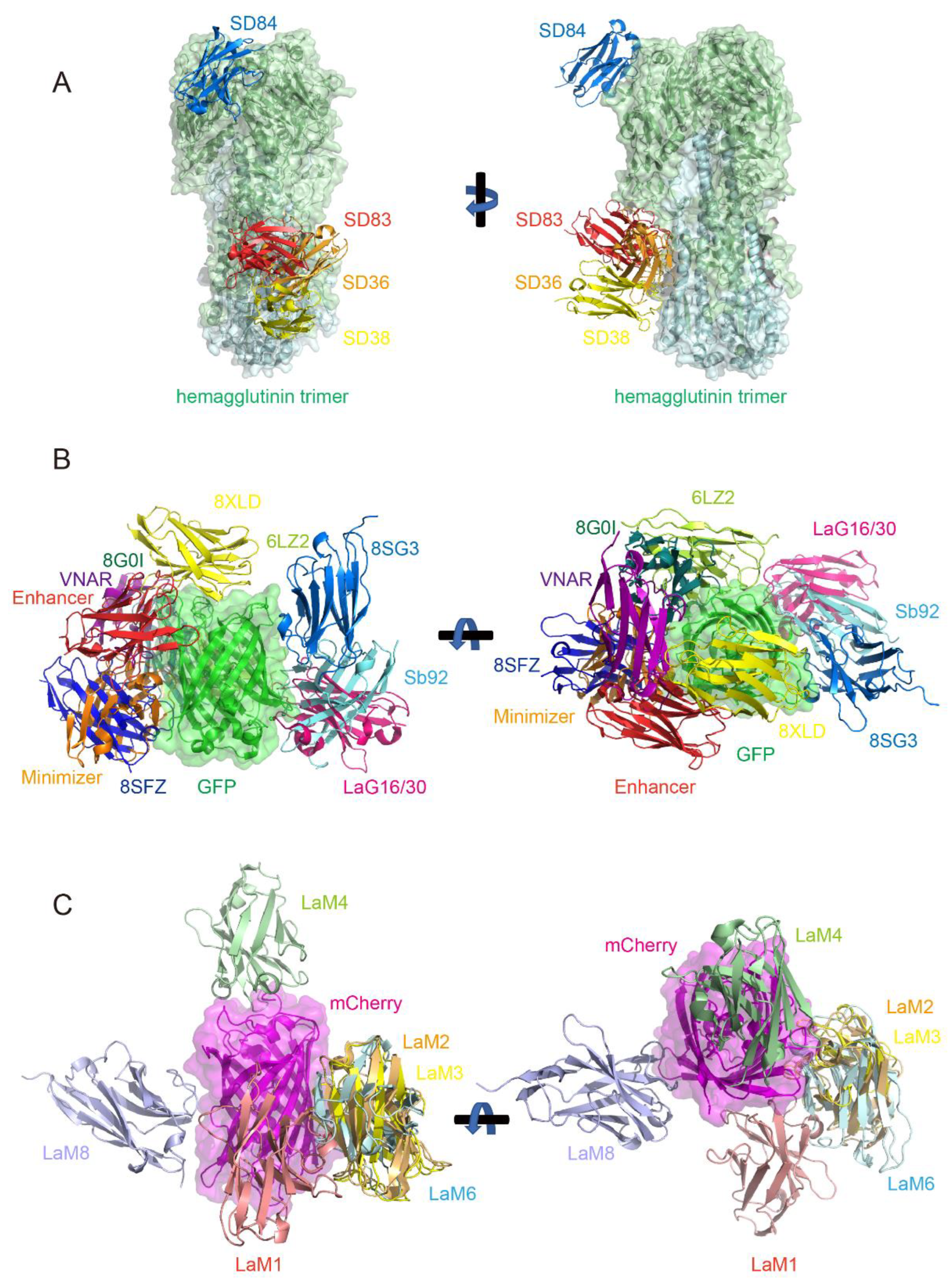
8.1. Nanobodies Targeting Different Epitopes on the COVID-19 Spike Protein
8.2. Nanobodies Binding to Different Epitopes of the Influenza Virus Hemagglutinin (HA) Protein
8.3. Nanobodies Targeting Multiple Epitopes on GFP (Green Fluorescent Protein) and RFP (Red Fluorescent Protein) mCherry
9. Leveraging Artificial Intelligence (AI) in VHH Engineering
9.1. In Silico Nanobody Structure Determination
9.2. AI Tools for Computing Individual VHH Structures
9.3. Predicting VHH Paratopes Utilizing AI
9.4. Computational Tools for Nanobody Docking and Screening
9.5. State-of-the-Art Structural Modeling Utilizing AlphaFold3
9.6. Potential Limitations in Computational Structure Determination
10. Computational Humanization Strategies Tailored for Nanobodies
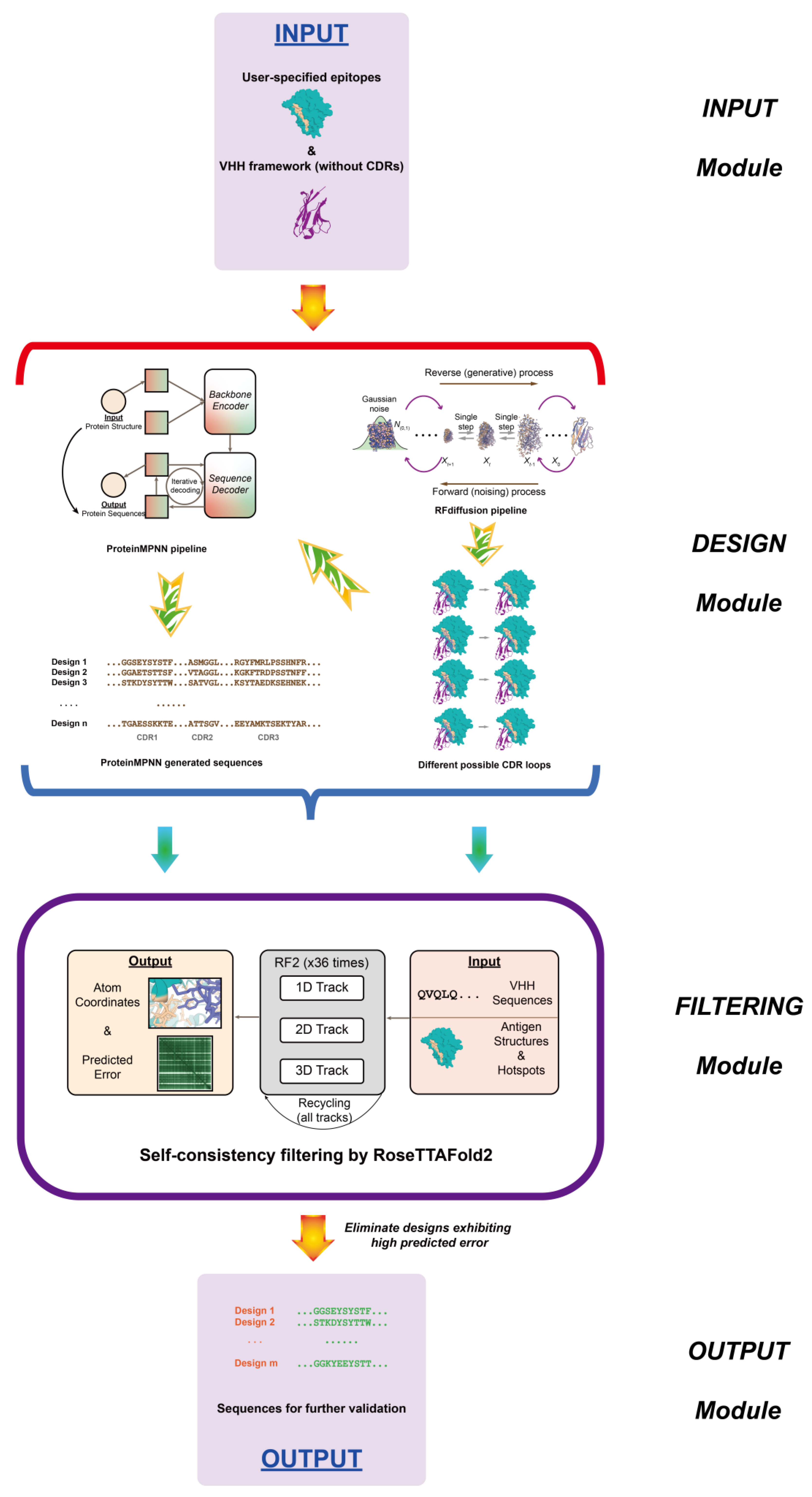
11. De Novo AI-Based Nanobody Design
12. Perspective
13. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Roux, K.H.; Greenberg, A.S.; Greene, L.; Strelets, L.; Avila, D.; McKinney, E.C.; Flajnik, M.F. Structural analysis of the nurse shark (new) antigen receptor (NAR): Molecular convergence of NAR and unusual mammalian immunoglobulins. Proc. Natl. Acad. Sci. USA 1998, 95, 11804–11809. [Google Scholar] [CrossRef] [PubMed]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.; Wohlkönig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.; Kobilka, B.K.; Steyaert, J. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Dauparas, J.; Anishchenko, I.; Bennett, N.; Bai, H.; Ragotte, R.J.; Milles, L.F.; Wicky, B.I.M.; Courbet, A.; de Haas, R.J.; Bethel, N.; et al. Robust deep learning-based protein sequence design using ProteinMPNN. Science 2022, 378, 49–56. [Google Scholar] [CrossRef]
- Desmyter, A.; Transue, T.R.; Ghahroudi, M.A.; Thi, M.H.; Poortmans, F.; Hamers, R.; Muyldermans, S.; Wyns, L. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat. Struct. Biol. 1996, 3, 803–811. [Google Scholar] [CrossRef]
- Jin, B.K.; Odongo, S.; Radwanska, M.; Magez, S. NANOBODIES®: A Review of Diagnostic and Therapeutic Applications. Int. J. Mol. Sci. 2023, 24, 5994. [Google Scholar] [CrossRef]
- Arbabi Ghahroudi, M.; Desmyter, A.; Wyns, L.; Hamers, R.; Muyldermans, S. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997, 414, 521–526. [Google Scholar] [CrossRef]
- Rothbauer, U.; Zolghadr, K.; Tillib, S.; Nowak, D.; Schermelleh, L.; Gahl, A.; Backmann, N.; Conrath, K.; Muyldermans, S.; Cardoso, M.C.; et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 2006, 3, 887–889. [Google Scholar] [CrossRef]
- Gonzalez-Sapienza, G.; Rossotti, M.A.; Tabares-da Rosa, S. Single-Domain Antibodies as Versatile Affinity Reagents for Analytical and Diagnostic Applications. Front. Immunol. 2017, 8, 977. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, T.; Muyldermans, S.; Depicker, A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 2014, 32, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.; Vincke, C.; Stijlemans, B.; Schymkowitz, J.; Decanniere, K.; Wyns, L.; Muyldermans, S.; Loris, R. Antigen binding and solubility effects upon the veneering of a camel VHH in framework-2 to mimic a VH. J. Mol. Biol. 2005, 350, 112–125. [Google Scholar] [CrossRef]
- Huang, L.; Gainkam, L.O.; Caveliers, V.; Vanhove, C.; Keyaerts, M.; De Baetselier, P.; Bossuyt, A.; Revets, H.; Lahoutte, T. SPECT imaging with 99mTc-labeled EGFR-specific nanobody for in vivo monitoring of EGFR expression. Mol. Imaging Biol. 2008, 10, 167–175. [Google Scholar] [CrossRef]
- Chakravarty, R.; Goel, S.; Cai, W. Nanobody: The “magic bullet” for molecular imaging? Theranostics 2014, 4, 386–398. [Google Scholar] [CrossRef]
- Harmand, T.J.; Islam, A.; Pishesha, N.; Ploegh, H.L. Nanobodies as in vivo, non-invasive, imaging agents. RSC Chem. Biol. 2021, 2, 685–701. [Google Scholar] [CrossRef]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.; Arnold, F.M.; Stohler, P.; Bocquet, N.; Hug, M.N.; Huber, S.; Siegrist, M.; Hetemann, L.; et al. Synthetic single domain antibodies for the conformational trapping of membrane proteins. eLife 2018, 7, e34317. [Google Scholar] [CrossRef]
- McMahon, C.; Baier, A.S.; Pascolutti, R.; Wegrecki, M.; Zheng, S.; Ong, J.X.; Erlandson, S.C.; Hilger, D.; Rasmussen, S.G.F.; Ring, A.M.; et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 2018, 25, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Fan, S.; Gai, C.; Li, B.; Wang, G. Efficacy and safety of envafolimab in the treatment of advanced dMMR/MSI-H solid tumors: A single-arm meta-analysis. Oncol. Lett. 2023, 26, 351. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kawanishi, M.; Nakanishi, M.; Yamasaki, H.; Takeuchi, T. Efficacy and safety of the anti-TNF multivalent NANOBODY® compound ozoralizumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: A 52-week result of a Phase II/III study (OHZORA trial). Mod. Rheumatol. 2023, 33, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Liu, P.; Wang, Z.; Zhu, H.; Zhong, C.; Zhong, P.; Jiang, H.; Liu, J.; Ma, Z.; Liu, X.; et al. Structural insights into the binding of nanobodies to the Staphylococcal enterotoxin B. Int. J. Biol. Macromol. 2024, 276, 133957. [Google Scholar] [CrossRef]
- Olichon, A.; de Marco, A. Preparation of a naïve library of camelid single domain antibodies. Methods Mol. Biol. 2012, 911, 65–78. [Google Scholar]
- Sabir, J.S.; Atef, A.; El-Domyati, F.M.; Edris, S.; Hajrah, N.; Alzohairy, A.M.; Bahieldin, A. Construction of naïve camelids VHH repertoire in phage display-based library. Comptes Rendus Biol. 2014, 337, 244–249. [Google Scholar] [CrossRef]
- Yan, J.; Li, G.; Hu, Y.; Ou, W.; Wan, Y. Construction of a synthetic phage-displayed Nanobody library with CDR3 regions randomized by trinucleotide cassettes for diagnostic applications. J. Transl. Med. 2014, 12, 343. [Google Scholar] [CrossRef]
- Moutel, S.; Bery, N.; Bernard, V.; Keller, L.; Lemesre, E.; de Marco, A.; Ligat, L.; Rain, J.-C.; Favre, G.; Olichon, A.; et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. eLife 2016, 5, e16228. [Google Scholar] [CrossRef]
- De Genst, E.; Saerens, D.; Muyldermans, S.; Conrath, K. Antibody repertoire development in camelids. Dev. Comp. Immunol. 2006, 30, 187–198. [Google Scholar] [CrossRef]
- Omidfar, K.; Daneshpour, M. Advances in phage display technology for drug discovery. Expert Opin. Drug Discov. 2015, 10, 651–669. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, Z.; Shao, L.; Kong, X.; Hou, X.; Tian, D.; Sun, Y.; Xiao, Y.; Yu, L. Nanobody-derived nanobiotechnology tool kits for diverse biomedical and biotechnology applications. Int. J. Nanomed. 2016, 11, 3287–3303. [Google Scholar] [CrossRef] [PubMed]
- Salema, V.; Fernández, L. Á Escherichia coli surface display for the selection of nanobodies. Microb. Biotechnol. 2017, 10, 1468–1484. [Google Scholar] [CrossRef]
- de Marco, A. Recombinant expression of nanobodies and nanobody-derived immunoreagents. Protein Expr. Purif. 2020, 172, 105645. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Applications of Nanobodies. Annu. Rev. Anim. Biosci. 2021, 9, 401–421. [Google Scholar] [CrossRef]
- Mahdavi, S.Z.B.; Oroojalian, F.; Eyvazi, S.; Hejazi, M.; Baradaran, B.; Pouladi, N.; Tohidkia, M.R.; Mokhtarzadeh, A.; Muyldermans, S. An overview on display systems (phage, bacterial, and yeast display) for production of anticancer antibodies; advantages and disadvantages. Int. J. Biol. Macromol. 2022, 208, 421–442. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Molina-Zapata, A.; Pose, A.G.; Moreno, E. Structural Insights into the Design of Synthetic Nanobody Libraries. Molecules 2022, 27, 2198. [Google Scholar] [CrossRef]
- Minatel, V.M.; Prudencio, C.R.; Barraviera, B.; Ferreira, R.S., Jr. Nanobodies: A promising approach to treatment of viral diseases. Front. Immunol. 2023, 14, 1303353. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.J.; Guntuku, G.; Palla, M.S. Advancements in nanobody generation: Integrating conventional, in silico, and machine learning approaches. Biotechnol. Bioeng. 2024, 121, 3375–3388. [Google Scholar] [CrossRef]
- Rader, C. The pComb3 Phagemid Family of Phage Display Vectors. Cold Spring Harb. Protoc. 2024, 2024, pdb.over107756. [Google Scholar] [CrossRef]
- Fridy, P.C.; Rout, M.P.; Ketaren, N.E. Nanobodies: From High-Throughput Identification to Therapeutic Development. Mol. Cell Proteom. 2024, 23, 100865. [Google Scholar] [CrossRef]
- Alexander, E.; Leong, K.W. Discovery of nanobodies: A comprehensive review of their applications and potential over the past five years. J. Nanobiotechnology 2024, 22, 661. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, I.; Egloff, P.; Hutter, C.A.J.; Kuhn, B.T.; Bräuer, P.; Newstead, S.; Dawson, R.J.P.; Geertsma, E.R.; Seeger, M.A. Generation of synthetic nanobodies against delicate proteins. Nat. Protoc. 2020, 15, 1707–1741. [Google Scholar] [CrossRef] [PubMed]
- Salema, V.; Marín, E.; Martínez-Arteaga, R.; Ruano-Gallego, D.; Fraile, S.; Margolles, Y.; Teira, X.; Gutierrez, C.; Bodelón, G.; Fernández, L. Á Selection of single domain antibodies from immune libraries displayed on the surface of E. coli cells with two β-domains of opposite topologies. PLoS ONE 2013, 8, e75126. [Google Scholar] [CrossRef]
- Salema, V.; López-Guajardo, A.; Gutierrez, C.; Mencía, M.; Fernández, L.Á. Characterization of nanobodies binding human fibrinogen selected by E. coli display. J. Biotechnol. 2016, 234, 58–65. [Google Scholar] [CrossRef]
- Ho, M.; Pastan, I. Mammalian cell display for antibody engineering. Methods Mol. Biol. 2009, 525, 337–352. [Google Scholar]
- Takahashi, K.; Sunohara, M.; Terai, T.; Kumachi, S.; Nemoto, N. Enhanced mRNA-protein fusion efficiency of a single-domain antibody by selection of mRNA display with additional random sequences in the terminal translated regions. Biophys. Physicobiol 2017, 14, 23–28. [Google Scholar] [CrossRef]
- Doshi, R.; Chen, B.R.; Vibat, C.R.T.; Huang, N.; Lee, C.-W.; Chang, G. In Vitro nanobody discovery for integral membrane protein targets. Sci. Rep. 2014, 4, 6760. [Google Scholar] [CrossRef]
- Egloff, P.; Zimmermann, I.; Arnold, F.M.; Hutter, C.A.J.; Morger, D.; Opitz, L.; Poveda, L.; Keserue, H.A.; Panse, C.; Roschitzki, B.; et al. Engineered peptide barcodes for in-depth analyses of binding protein libraries. Nat. Methods 2019, 16, 421–428. [Google Scholar] [CrossRef]
- Matsuzaki, Y.; Aoki, W.; Miyazaki, T.; Aburaya, S.; Ohtani, Y.; Kajiwara, K.; Koike, N.; Minakuchi, H.; Miura, N.; Kadonosono, T.; et al. Peptide barcoding for one-pot evaluation of sequence–function relationships of nanobodies. Sci. Rep. 2021, 11, 21516. [Google Scholar] [CrossRef]
- Harris, L.J.; Larson, S.B.; Hasel, K.W.; McPherson, A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry 1997, 36, 1581–1597. [Google Scholar] [CrossRef] [PubMed]
- Braden, B.C.; Souchon, H.; Eiselé, J.L.; Bentley, G.A.; Bhat, T.N.; Navaza, J.; Poljak, R.J. Three-dimensional structures of the free and the antigen-complexed Fab from monoclonal anti-lysozyme antibody D44.1. J. Mol. Biol. 1994, 243, 767–781. [Google Scholar] [CrossRef]
- Aÿ, J.; Keitel, T.; Küttner, G.; Wessner, H.; Scholz, C.; Hahn, M.; Höhne, W. Crystal structure of a phage library-derived single-chain Fv fragment complexed with turkey egg-white lysozyme at 2.0 A resolution. J. Mol. Biol. 2000, 301, 239–246. [Google Scholar] [CrossRef]
- Kirchhofer, A.; Helma, J.; Schmidthals, K.; Frauer, C.; Cui, S.; Karcher, A.; Pellis, M.; Muyldermans, S.; Casas-Delucchi, C.S.; Cardoso, M.C.; et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 2010, 17, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, R.L.; Dooley, H.; Flajnik, M.F.; Wilson, I.A. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science 2004, 305, 1770–1773. [Google Scholar] [CrossRef]
- Wu, X.; Chen, S.; Lin, L.; Liu, J.; Wang, Y.; Li, Y.; Li, Q.; Wang, Z. A Single Domain–Based Anti-Her2 Antibody Has Potent Antitumor Activities. Transl. Oncol. 2018, 11, 366–373. [Google Scholar] [CrossRef]
- Broos, K.; Lecocq, Q.; Xavier, C.; Bridoux, J.; Nguyen, T.T.; Corthals, J.; Schoonooghe, S.; Lion, E.; Raes, G.; Keyaerts, M.; et al. Evaluating a Single Domain Antibody Targeting Human PD-L1 as a Nuclear Imaging and Therapeutic Agent. Cancers 2019, 11, 872. [Google Scholar] [CrossRef]
- Al-Baradie, R.S. Nanobodies as versatile tools: A focus on targeted tumor therapy, tumor imaging and diagnostics. Hum. Antibodies 2020, 28, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.; Zinner, K.; Mücke, N.; Bartoschik, T.; Muyldermans, S.; Hoheisel, J.D. The structural basis of nanobody unfolding reversibility and thermoresistance. Sci. Rep. 2018, 8, 7934. [Google Scholar] [CrossRef]
- Dingus, J.G.; Tang, J.C.Y.; Amamoto, R.; Wallick, G.K.; Cepko, C.L. A general approach for stabilizing nanobodies for intracellular expression. eLife 2022, 11, e68253. [Google Scholar] [CrossRef]
- Muyldermans, S. Single domain camel antibodies: Current status. J. Biotechnol. 2001, 74, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Ackaert, C.; Smiejkowska, N.; Xavier, C.; Sterckx, Y.G.J.; Denies, S.; Stijlemans, B.; Elkrim, Y.; Devoogdt, N.; Caveliers, V.; Lahoutte, T.; et al. Immunogenicity Risk Profile of Nanobodies. Front. Immunol. 2021, 12, 632687. [Google Scholar] [CrossRef]
- Mustafa, M.I.; Mohammed, A. Revolutionizing antiviral therapy with nanobodies: Generation and prospects. Biotechnol. Rep. 2023, 39, e00803. [Google Scholar] [CrossRef]
- Suurs, F.V.; Lub-de Hooge, M.N.; de Vries, E.G.E.; de Groot, D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Stuhler, G.; Bargou, R. Bispecific and multispecific antibodies in oncology: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2024, 21, 539–560. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Ma, L.; Zhong, P.; Huang, J.; Gai, J.; Li, G.; Li, Y.; Qiao, P.; Gu, H.; Li, X.; et al. A novel inhalable nanobody targeting IL-4Rα for the treatment of asthma. J. Allergy Clin. Immunol. 2024, 154, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Peng, H.; Rader, C. From rabbit antibody repertoires to rabbit monoclonal antibodies. Exp. Mol. Med. 2017, 49, e305. [Google Scholar] [CrossRef]
- Desmyter, A.; Spinelli, S.; Roussel, A.; Cambillau, C. Camelid nanobodies: Killing two birds with one stone. Curr. Opin. Struct. Biol. 2015, 32, 1–8. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Bagchi, A.; Roovers, R.C.; van Bergen en Henegouwen, P.M.; Ferguson, K.M. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure 2013, 21, 1214–1224. [Google Scholar] [CrossRef]
- Lauwereys, M.; Arbabi Ghahroudi, M.; Desmyter, A.; Kinne, J.; Hölzer, W.; De Genst, E.; Wyns, L.; Muyldermans, S. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 1998, 17, 3512–3520. [Google Scholar] [CrossRef]
- Unger, M.; Eichhoff, A.M.; Schumacher, L.; Strysio, M.; Menzel, S.; Schwan, C.; Alzogaray, V.; Zylberman, V.; Seman, M.; Brandner, J.; et al. Selection of nanobodies that block the enzymatic and cytotoxic activities of the binary Clostridium difficile toxin CDT. Sci. Rep. 2015, 5, 7850. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhan, W.; Yang, Z.; Tu, C.; Hu, G.; Zhang, X.; Song, W.; Du, S.; Zhu, Y.; Huang, K.; et al. Broad neutralization of SARS-CoV-2 variants by an inhalable bispecific single-domain antibody. Cell 2022, 185, 1389–1401.e1318. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kobilka, B.K.; Steyaert, J. Nanobodies to Study G Protein-Coupled Receptor Structure and Function. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 19–37. [Google Scholar] [CrossRef]
- Uchański, T.; Pardon, E.; Steyaert, J. Nanobodies to study protein conformational states. Curr. Opin. Struct. Biol. 2020, 60, 117–123. [Google Scholar] [CrossRef]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef]
- Uchański, T.; Masiulis, S.; Fischer, B.; Kalichuk, V.; López-Sánchez, U.; Zarkadas, E.; Weckener, M.; Sente, A.; Ward, P.; Wohlkönig, A.; et al. Megabodies expand the nanobody toolkit for protein structure determination by single-particle cryo-EM. Nat. Methods 2021, 18, 60–68. [Google Scholar] [CrossRef]
- Wu, X.; Rapoport, T.A. Cryo-EM structure determination of small proteins by nanobody-binding scaffolds (Legobodies). Proc. Natl. Acad. Sci. USA 2021, 118, e2115001118. [Google Scholar] [CrossRef]
- Keller, L.; Bery, N.; Tardy, C.; Ligat, L.; Favre, G.; Rabbitts, T.H.; Olichon, A. Selection and Characterization of a Nanobody Biosensor of GTP-Bound RHO Activities. Antibodies 2019, 8, 8. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, S.; Zhong, P.; Wang, Z.; Liu, R.; Ding, Y. Structural insights into the binding of nanobody Rh57 to active RhoA-GTP. Biochem. Biophys. Res. Commun. 2022, 616, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A new generation of protein therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef]
- Ståhl, S.; Gräslund, T.; Eriksson Karlström, A.; Frejd, F.Y.; Nygren, P.Å.; Löfblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef]
- Dumoulin, M.; Last, A.M.; Desmyter, A.; Decanniere, K.; Canet, D.; Larsson, G.; Spencer, A.; Archer, D.B.; Sasse, J.; Muyldermans, S.; et al. A camelid antibody fragment inhibits the formation of amyloid fibrils by human lysozyme. Nature 2003, 424, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Kabat, E.A. An analysis of the sequences of the variable regions of bence jones proteins and myeloma light chains and their implications for antibody complementarity. J. Exp. Med. 1970, 132, 211–250. [Google Scholar] [CrossRef]
- Kovaleva, M.; Ferguson, L.; Steven, J.; Porter, A.; Barelle, C. Shark variable new antigen receptor biologics—A novel technology platform for therapeutic drug development. Expert Opin. Biol. Ther. 2014, 14, 1527–1539. [Google Scholar] [CrossRef]
- Streltsov, V.A.; Carmichael, J.A.; Nuttall, S.D. Structure of a shark IgNAR antibody variable domain and modeling of an early-developmental isotype. Protein Sci. 2005, 14, 2901–2909. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhu, Z.; Feng, Y.; Dimitrov, D.S. Human domain antibodies to conserved sterically restricted regions on gp120 as exceptionally potent cross-reactive HIV-1 neutralizers. Proc. Natl. Acad. Sci. USA 2008, 105, 17121–17126. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- De Pauw, T.; De Mey, L.; Debacker, J.M.; Raes, G.; Van Ginderachter, J.A.; De Groof, T.W.M.; Devoogdt, N. Current status and future expectations of nanobodies in oncology trials. Expert Opin. Investig. Drugs 2023, 32, 705–721. [Google Scholar] [CrossRef]
- Wu, Y.; Li, C.; Xia, S.; Tian, X.; Kong, Y.; Wang, Z.; Gu, C.; Zhang, R.; Tu, C.; Xie, Y.; et al. Identification of Human Single-Domain Antibodies against SARS-CoV-2. Cell Host Microbe 2020, 27, 891–898.e895. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Xiong, Y.; Hu, P.; Wu, D.; Chen, W.; Ying, T.; Zhu, Z.; Dimitrov, D.S.; Dropulic, B.; Orentas, R.J. A Unique Human Immunoglobulin Heavy Chain Variable Domain-Only CD33 CAR for the Treatment of Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 539. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Q.; Kong, Y.; Wang, Z.; Lei, C.; Li, J.; Ding, L.; Wang, C.; Cheng, Y.; Wei, Y.; et al. A highly stable human single-domain antibody-drug conjugate exhibits superior penetration and treatment of solid tumors. Mol. Ther. 2022, 30, 2785–2799. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Frenken, L.G.; Hermans, P.; Verrips, T.; Brown, K.; Tegoni, M.; Cambillau, C. Camelid heavy-chain variable domains provide efficient combining sites to haptens. Biochemistry 2000, 39, 1217–1222. [Google Scholar] [CrossRef]
- Spinelli, S.; Tegoni, M.; Frenken, L.; van Vliet, C.; Cambillau, C. Lateral recognition of a dye hapten by a llama VHH domain. J. Mol. Biol. 2001, 311, 123–129. [Google Scholar] [CrossRef]
- Fanning, S.W.; Horn, J.R. An anti-hapten camelid antibody reveals a cryptic binding site with significant energetic contributions from a nonhypervariable loop. Protein Sci. 2011, 20, 1196–1207. [Google Scholar] [CrossRef]
- Tabares-da Rosa, S.; Wogulis, L.A.; Wogulis, M.D.; González-Sapienza, G.; Wilson, D.K. Structure and specificity of several triclocarban-binding single domain camelid antibody fragments. J. Mol. Recognit. 2019, 32, e2755. [Google Scholar] [CrossRef]
- Ding, L.; Wang, Z.; Zhong, P.; Jiang, H.; Zhao, Z.; Zhang, Y.; Ren, Z.; Ding, Y. Structural insights into the mechanism of single domain VHH antibody binding to cortisol. FEBS Lett. 2019, 593, 1248–1256. [Google Scholar] [CrossRef]
- Li, J.-D.; Wu, G.-P.; Li, L.-H.; Wang, L.-T.; Liang, Y.-F.; Fang, R.-Y.; Zhang, Q.-L.; Xie, L.-L.; Shen, X.; Shen, Y.-D.; et al. Structural Insights into the Stability and Recognition Mechanism of the Antiquinalphos Nanobody for the Detection of Quinalphos in Foods. Anal. Chem. 2023, 95, 11306–11315. [Google Scholar] [CrossRef]
- Wang, W.; Gu, G.; Yin, R.; Fu, J.; Jing, M.; Shen, Z.; Lai, D.; Wang, B.; Zhou, L. A Nanobody-Based Immunoassay for Detection of Ustilaginoidins in Rice Samples. Toxins 2022, 14, 659. [Google Scholar] [CrossRef]
- Yang, H.; Vasylieva, N.; Wang, J.; Li, Z.; Duan, W.; Chen, S.; Wen, K.; Meng, H.; Yu, X.; Shen, J.; et al. Precise isolation and structural origin of an ultra-specific nanobody against chemical compound. J. Hazard. Mater. 2023, 458, 131958. [Google Scholar] [CrossRef] [PubMed]
- Ladenson, R.C.; Crimmins, D.L.; Landt, Y.; Ladenson, J.H. Isolation and characterization of a thermally stable recombinant anti-caffeine heavy-chain antibody fragment. Anal. Chem. 2006, 78, 4501–4508. [Google Scholar] [CrossRef]
- Sonneson, G.J.; Horn, J.R. Hapten-induced dimerization of a single-domain VHH camelid antibody. Biochemistry 2009, 48, 6693–6695. [Google Scholar] [CrossRef] [PubMed]
- Lesne, J.; Chang, H.J.; De Visch, A.; Paloni, M.; Barthe, P.; Guichou, J.F.; Mayonove, P.; Barducci, A.; Labesse, G.; Bonnet, J.; et al. Structural basis for chemically-induced homodimerization of a single domain antibody. Sci. Rep. 2019, 9, 1840. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Sonneson, G.J.; Hoey, R.J.; Hinerman, J.M.; Sheehy, K.; Walter, R.; Herr, A.B.; Horn, J.R. Molecular recognition requires dimerization of a VHH antibody. MAbs 2023, 15, 2215363. [Google Scholar] [CrossRef]
- Hughes, A.C.; Kirkland, M.; Du, W.; Rasooly, R.; Hernlem, B.; Tam, C.; Zhang, Y.; He, X. Development of Thermally Stable Nanobodies for Detection and Neutralization of Staphylococcal Enterotoxin B. Toxins 2023, 15, 400. [Google Scholar] [CrossRef]
- Wang, W.; Hu, Y.; Li, B.; Wang, H.; Shen, J. Applications of nanobodies in the prevention, detection, and treatment of the evolving SARS-CoV-2. Biochem. Pharmacol. 2023, 208, 115401. [Google Scholar] [CrossRef]
- Yang, Y.; Du, L. SARS-CoV-2 spike protein: A key target for eliciting persistent neutralizing antibodies. Signal Transduct. Target. Ther. 2021, 6, 95. [Google Scholar] [CrossRef]
- Hanke, L.; Vidakovics Perez, L.; Sheward, D.J.; Das, H.; Schulte, T.; Moliner-Morro, A.; Corcoran, M.; Achour, A.; Karlsson Hedestam, G.B.; Hällberg, B.M.; et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat. Commun. 2020, 11, 4420. [Google Scholar] [CrossRef]
- Schoof, M.; Faust, B.; Saunders, R.A.; Sangwan, S.; Rezelj, V.; Hoppe, N.; Boone, M.; Billesbølle, C.B.; Puchades, C.; Azumaya, C.M.; et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science 2020, 370, 1473–1479. [Google Scholar] [CrossRef]
- Schepens, B.; van Schie, L.; Nerinckx, W.; Roose, K.; Van Breedam, W.; Fijalkowska, D.; Devos, S.; Weyts, W.; De Cae, S.; Vanmarcke, S.; et al. An affinity-enhanced, broadly neutralizing heavy chain–only antibody protects against SARS-CoV-2 infection in animal models. Sci. Transl. Med. 2021, 13, eabi7826. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Liu, P.; Liu, S.; Yue, C.; Wang, X. Enhancing RBD exposure and S1 shedding by an extremely conserved SARS-CoV-2 NTD epitope. Signal Transduct. Target. Ther. 2024, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Chang, S.C. SARS-CoV-2 spike S2-specific neutralizing antibodies. Emerg. Microbes Infect. 2023, 12, 2220582. [Google Scholar] [CrossRef]
- Koenig, P.-A.; Das, H.; Liu, H.; Kümmerer, B.M.; Gohr, F.N.; Jenster, L.-M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371, eabe6230. [Google Scholar] [CrossRef]
- Laursen, N.S.; Friesen, R.H.E.; Zhu, X.; Jongeneelen, M.; Blokland, S.; Vermond, J.; van Eijgen, A.; Tang, C.; van Diepen, H.; Obmolova, G.; et al. Universal protection against influenza infection by a multidomain antibody to influenza hemagglutinin. Science 2018, 362, 598–602. [Google Scholar] [CrossRef]
- Chen, Z.-S.; Huang, H.-C.; Wang, X.; Schön, K.; Jia, Y.; Lebens, M.; Besavilla, D.F.; Murti, J.R.; Ji, Y.; Sarshad, A.A.; et al. Influenza A Virus H7 nanobody recognizes a conserved immunodominant epitope on hemagglutinin head and confers heterosubtypic protection. Nat. Commun. 2025, 16, 432. [Google Scholar] [CrossRef]
- Kubala, M.H.; Kovtun, O.; Alexandrov, K.; Collins, B.M. Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 2010, 19, 2389–2401. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Ding, Y.; Hattori, M. Structure-based engineering of anti-GFP nanobody tandems as ultra-high-affinity reagents for purification. Sci. Rep. 2020, 10, 6239. [Google Scholar] [CrossRef] [PubMed]
- Cong, A.T.Q.; Witter, T.L.; Schellenberg, M.J. High-efficiency recombinant protein purification using mCherry and YFP nanobody affinity matrices. Protein Sci. 2022, 31, e4383. [Google Scholar] [CrossRef]
- Zhong, P.; Wang, Z.; Cheng, S.; Zhang, Y.; Jiang, H.; Liu, R.; Ding, Y. Structural insights into two distinct nanobodies recognizing the same epitope of green fluorescent protein. Biochem. Biophys. Res. Commun. 2021, 565, 57–63. [Google Scholar] [CrossRef]
- Cai, H.; Yao, H.; Li, T.; Hutter, C.A.J.; Li, Y.; Tang, Y.; Seeger, M.A.; Li, D. An improved fluorescent tag and its nanobodies for membrane protein expression, stability assay, and purification. Commun. Biol. 2020, 3, 753. [Google Scholar] [CrossRef] [PubMed]
- Ketaren, N.E.; Fridy, P.C.; Malashkevich, V.; Sanyal, T.; Brillantes, M.; Thompson, M.K.; Oren, D.A.; Bonanno, J.B.; Šali, A.; Almo, S.C.; et al. Unique Binding and Stabilization Mechanisms Employed By and Engineered Into Nanobodies. bioRxiv 2023. bioRxiv:2023.10.22.563475. [Google Scholar]
- Chen, Y.L.; Xie, X.X.; Zheng, P.; Zhu, C.; Ma, H.; Khalid, Z.; Xie, Y.J.; Dang, Y.Z.; Ye, Y.; Sheng, N.; et al. Selection, identification and crystal structure of shark-derived single-domain antibodies against a green fluorescent protein. Int. J. Biol. Macromol. 2023, 247, 125852. [Google Scholar] [CrossRef] [PubMed]
- Fridy, P.C.; Li, Y.; Keegan, S.; Thompson, M.K.; Nudelman, I.; Scheid, J.F.; Oeffinger, M.; Nussenzweig, M.C.; Fenyö, D.; Chait, B.T.; et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 2014, 11, 1253–1260. [Google Scholar] [CrossRef]
- Wang, Z.; Li, L.; Hu, R.; Zhong, P.; Zhang, Y.; Cheng, S.; Jiang, H.; Liu, R.; Ding, Y. Structural insights into the binding of nanobodies LaM2 and LaM4 to the red fluorescent protein mCherry. Protein Sci. 2021, 30, 2298–2309. [Google Scholar] [CrossRef]
- Liang, H.; Ma, Z.; Wang, Z.; Zhong, P.; Li, R.; Jiang, H.; Zong, X.; Zhong, C.; Liu, X.; Liu, P.; et al. Structural Insights into the Binding of Red Fluorescent Protein mCherry-Specific Nanobodies. Int. J. Mol. Sci. 2023, 24, 6952. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, J.; Marchal, C.; Posbeyikian, A.; Harant, A.; Kamoun, S. NLR immune receptor–nanobody fusions confer plant disease resistance. Science 2023, 379, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Fauser, J.; Leschinsky, N.; Szynal, B.N.; Karginov, A.V. Engineered Allosteric Regulation of Protein Function. J. Mol. Biol. 2022, 434, 167620. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.A.; Carrasco-López, C.; Zhu, L.; Zhao, E.M.; Ravindran, P.T.; Wilson, M.Z.; Goglia, A.G.; Avalos, J.L.; Toettcher, J.E. Optogenetic control of protein binding using light-switchable nanobodies. Nat. Commun. 2020, 11, 4044. [Google Scholar] [CrossRef]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Historical Perspective and Future Outlook. Front. Immunol. 2017, 8, 1589. [Google Scholar] [CrossRef]
- Frecot, D.I.; Froehlich, T.; Rothbauer, U. 30 years of nanobodies—An ongoing success story of small binders in biological research. J. Cell Sci. 2023, 136, jcs261395. [Google Scholar] [CrossRef] [PubMed]
- Koch-Nolte, F. Nanobody-based heavy chain antibodies and chimeric antibodies. Immunol. Rev. 2024, 328, 466–472. [Google Scholar] [CrossRef]
- Kunz, S.; Durandy, M.; Seguin, L.; Feral, C.C. NANOBODY® molecule, a giga medical tool in nanodimensions. Int. J. Mol. Sci. 2023, 24, 13229. [Google Scholar] [CrossRef]
- Medina Pérez, V.M.; Baselga, M.; Schuhmacher, A.J. Single-Domain Antibodies as Antibody-Drug Conjugates: From Promise to Practice—A Systematic Review. Cancers 2024, 16, 2681. [Google Scholar] [CrossRef]
- Yu, T.; Zheng, F.; He, W.; Muyldermans, S.; Wen, Y. Single domain antibody: Development and application in biotechnology and biopharma. Immunol. Rev. 2024, 328, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Mullin, M.; McClory, J.; Haynes, W.; Grace, J.; Robertson, N.; van Heeke, G. Applications and challenges in designing VHH-based bispecific antibodies: Leveraging machine learning solutions. MAbs 2024, 16, 2341443. [Google Scholar] [CrossRef]
- Eshak, F.; Goupil-Lamy, A. AI driven approaches in nanobody epitope prediction: Are we there yet? bioRxiv 2024. [Google Scholar] [CrossRef]
- Dorn, M.; MB, E.S.; Buriol, L.S.; Lamb, L.C. Three-dimensional protein structure prediction: Methods and computational strategies. Comput. Biol. Chem. 2014, 53 Pt B, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A. Template-based protein structure modeling. Methods Mol. Biol. 2010, 673, 73–94. [Google Scholar]
- Koehler Leman, J.; Ulmschneider, M.B.; Gray, J.J. Computational modeling of membrane proteins. Proteins 2015, 83, 1–24. [Google Scholar] [CrossRef]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Fidelis, K. Protein structure prediction and model quality assessment. Drug Discov. Today 2009, 14, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Swanson, R.; Day, R.; Tsai, J. A guide to template based structure prediction. Curr. Protein Pept. Sci. 2009, 10, 270–285. [Google Scholar] [CrossRef] [PubMed]
- Ruffolo, J.A.; Sulam, J.; Gray, J.J. Antibody structure prediction using interpretable deep learning. Patterns 2022, 3, 100406. [Google Scholar] [CrossRef]
- Abanades, B.; Georges, G.; Bujotzek, A.; Deane, C.M. ABlooper: Fast accurate antibody CDR loop structure prediction with accuracy estimation. Bioinformatics 2022, 38, 1877–1880. [Google Scholar] [CrossRef]
- Gowthaman, R.; Pierce, B.G. TCRmodel: High resolution modeling of T cell receptors from sequence. Nucleic Acids Res. 2018, 46, W396–W401. [Google Scholar] [CrossRef]
- Lapidoth, G.; Parker, J.; Prilusky, J.; Fleishman, S.J. AbPredict 2: A server for accurate and unstrained structure prediction of antibody variable domains. Bioinformatics 2019, 35, 1591–1593. [Google Scholar] [CrossRef]
- Weitzner, B.D.; Kuroda, D.; Marze, N.; Xu, J.; Gray, J.J. Blind prediction performance of RosettaAntibody 3.0: Grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins 2014, 82, 1611–1623. [Google Scholar] [CrossRef]
- Cohen, T.; Halfon, M.; Schneidman-Duhovny, D. NanoNet: Rapid and accurate end-to-end nanobody modeling by deep learning. Front. Immunol. 2022, 13, 958584. [Google Scholar] [CrossRef]
- Chowdhury, R.; Bouatta, N.; Biswas, S.; Floristean, C.; Kharkar, A.; Roy, K.; Rochereau, C.; Ahdritz, G.; Zhang, J.; Church, G.M.; et al. Single-sequence protein structure prediction using a language model and deep learning. Nat. Biotechnol. 2022, 40, 1617–1623. [Google Scholar] [CrossRef]
- Erckert, K.; Rost, B. Assessing the role of evolutionary information for enhancing protein language model embeddings. Sci. Rep. 2024, 14, 20692. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, T.K. Chasing long-range evolutionary couplings in the AlphaFold era. Biopolymers 2023, 114, e23530. [Google Scholar] [CrossRef] [PubMed]
- Rahimzadeh, F.; Mohammad Khanli, L.; Salehpoor, P.; Golabi, F.; PourBahrami, S. Unveiling the evolution of policies for enhancing protein structure predictions: A comprehensive analysis. Comput. Biol. Med. 2024, 179, 108815. [Google Scholar] [CrossRef]
- Ruffolo, J.A.; Gray, J.J.; Sulam, J. Deciphering antibody affinity maturation with language models and weakly supervised learning. arXiv 2021, arXiv:2112.07782. [Google Scholar]
- Ruffolo, J.A.; Chu, L.-S.; Mahajan, S.P.; Gray, J.J. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. Nat. Commun. 2023, 14, 2389. [Google Scholar] [CrossRef]
- Abanades, B.; Wong, W.K.; Boyles, F.; Georges, G.; Bujotzek, A.; Deane, C.M. ImmuneBuilder: Deep-learning models for predicting the structures of immune proteins. Commun. Biol. 2023, 6, 575. [Google Scholar] [CrossRef]
- Liberis, E.; Veličković, P.; Sormanni, P.; Vendruscolo, M.; Liò, P. Parapred: Antibody paratope prediction using convolutional and recurrent neural networks. Bioinformatics 2018, 34, 2944–2950. [Google Scholar] [CrossRef]
- Kalemati, M.; Noroozi, A.; Shahbakhsh, A.; Koohi, S. ParaAntiProt provides paratope prediction using antibody and protein language models. Sci. Rep. 2024, 14, 29141. [Google Scholar] [CrossRef]
- Li, J.; Kang, G.; Wang, J.; Yuan, H.; Wu, Y.; Meng, S.; Wang, P.; Zhang, M.; Wang, Y.; Feng, Y.; et al. Affinity maturation of antibody fragments: A review encompassing the development from random approaches to computational rational optimization. Int. J. Biol. Macromol. 2023, 247, 125733. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Yamamori, Y.; Tomii, K. Protein-protein interaction prediction methods: From docking-based to AI-based approaches. Biophys. Rev. 2022, 14, 1341–1348. [Google Scholar] [CrossRef]
- van Noort, C.W.; Honorato, R.V.; Bonvin, A. Information-driven modeling of biomolecular complexes. Curr. Opin. Struct. Biol. 2021, 70, 70–77. [Google Scholar] [CrossRef]
- Verkhivker, G. Structural and Computational Studies of the SARS-CoV-2 Spike Protein Binding Mechanisms with Nanobodies: From Structure and Dynamics to Avidity-Driven Nanobody Engineering. Int. J. Mol. Sci. 2022, 23, 2928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wu, T.; Wang, W.; Zhang, L.; Gong, X. Review and Comparative Analysis of Methods and Advancements in Predicting Protein Complex Structure. Interdiscip. Sci. 2024, 16, 261–288. [Google Scholar] [CrossRef]
- Tam, C.; Kumar, A.; Zhang, K.Y.J. NbX: Machine learning-Guided Re-Ranking of Nanobody–Antigen Binding Poses. Pharmaceuticals 2021, 14, 968. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Buchanan, A.; Taddese, B.; Deane, C.M. DLAB: Deep learning methods for structure-based virtual screening of antibodies. Bioinformatics 2022, 38, 377–383. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Jiménez-Gutiérrez, D.E.; Moreno, E. Structural Modeling of Nanobodies: A Benchmark of State-of-the-Art Artificial Intelligence Programs. Molecules 2023, 28, 3991. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Wang, W.; Peng, Z.; Yang, J. Single-sequence protein structure prediction using supervised transformer protein language models. Nat. Comput. Sci. 2022, 2, 804–814. [Google Scholar] [CrossRef]
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; et al. High-resolution de novo structure prediction from primary sequence. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hitawala, F.N.; Gray, J.J. What does AlphaFold3 learn about antigen and nanobody docking, and what remains unsolved? bioRxiv 2024. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. bioRxiv:2021.2010.2004.463034. [Google Scholar]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112–7127. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef]
- Norden, A.; Moon, J.Y.; Javadi, S.S.; Munawar, L.; Maul, J.T.; Wu, J.J. Anti-drug antibodies of IL-23 inhibitors for psoriasis: A systematic review. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1171–1177. [Google Scholar] [CrossRef]
- Pizano-Martinez, O.; Mendieta-Condado, E.; Vázquez-Del Mercado, M.; Martínez-García, E.A.; Chavarria-Avila, E.; Ortuño-Sahagún, D.; Márquez-Aguirre, A.L. Anti-Drug Antibodies in the Biological Therapy of Autoimmune Rheumatic Diseases. J. Clin. Med. 2023, 12, 3271. [Google Scholar] [CrossRef]
- Sun, X.; Cui, Z.; Wang, Q.; Liu, L.; Ding, X.; Wang, J.; Cai, X.; Li, B.; Li, X. Formation and clinical effects of anti-drug antibodies against biologics in psoriasis treatment: An analysis of current evidence. Autoimmun. Rev. 2024, 23, 103530. [Google Scholar] [CrossRef] [PubMed]
- Velikova, T.; Sekulovski, M.; Peshevska-Sekulovska, M. Immunogenicity and Loss of Effectiveness of Biologic Therapy for Inflammatory Bowel Disease Patients Due to Anti-Drug Antibody Development. Antibodies 2024, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Vultaggio, A.; Perlato, M.; Nencini, F.; Vivarelli, E.; Maggi, E.; Matucci, A. How to Prevent and Mitigate Hypersensitivity Reactions to Biologicals Induced by Anti-Drug Antibodies? Front. Immunol. 2021, 12, 765747. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.L.; Raybould, M.I.J.; Wong, A.; Deane, C.M. Prospects for the computational humanization of antibodies and nanobodies. Front. Immunol. 2024, 15, 1399438. [Google Scholar] [CrossRef]
- Carroll, M.; Rosenbaum, E.; Viswanathan, R. Computational Methods to Predict Conformational B-Cell Epitopes. Biomolecules 2024, 14, 983. [Google Scholar] [CrossRef]
- Delgado, M.; Garcia-Sanz, J.A. Therapeutic Monoclonal Antibodies against Cancer: Present and Future. Cells 2023, 12, 2837. [Google Scholar] [CrossRef]
- Hanning, K.R.; Minot, M.; Warrender, A.K.; Kelton, W.; Reddy, S.T. Deep mutational scanning for therapeutic antibody engineering. Trends Pharmacol. Sci. 2022, 43, 123–135. [Google Scholar] [CrossRef]
- Martín-Galiano, A.J.; McConnell, M.J. Using Omics Technologies and Systems Biology to Identify Epitope Targets for the Development of Monoclonal Antibodies Against Antibiotic-Resistant Bacteria. Front. Immunol. 2019, 10, 2841. [Google Scholar] [CrossRef]
- Ng, C.L.; Lim, T.S.; Choong, Y.S. Application of Computational Techniques in Antibody Fc-Fused Molecule Design for Therapeutics. Mol. Biotechnol. 2024, 66, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Marks, C.; Hummer, A.M.; Chin, M.; Deane, C.M. Humanization of antibodies using a machine learning approach on large-scale repertoire data. Bioinformatics 2021, 37, 4041–4047. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Wernery, U.; Muyldermans, S.; Nguyen, V.K. Emergence and evolution of functional heavy-chain antibodies in Camelidae. Dev. Comp. Immunol. 2003, 27, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.K.; Desmyter, A.; Muyldermans, S. Functional heavy-chain antibodies in Camelidae. Adv. Immunol. 2001, 79, 261–296. [Google Scholar]
- Nguyen, V.K.; Hamers, R.; Wyns, L.; Muyldermans, S. Camel heavy-chain antibodies: Diverse germline VHH and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 2000, 19, 921–930. [Google Scholar] [CrossRef]
- Soler, M.A.; Medagli, B.; Wang, J.; Oloketuyi, S.; Bajc, G.; Huang, H.; Fortuna, S.; de Marco, A. Effect of Humanizing Mutations on the Stability of the Llama Single-Domain Variable Region. Biomolecules 2021, 11, 163. [Google Scholar] [CrossRef]
- Gordon, G.L.; Capel, H.L.; Guloglu, B.; Richardson, E.; Stafford, R.L.; Deane, C.M. A comparison of the binding sites of antibodies and single-domain antibodies. Front. Immunol. 2023, 14, 1231623. [Google Scholar] [CrossRef]
- Sang, Z.; Xiang, Y.; Bahar, I.; Shi, Y. Llamanade: An open-source computational pipeline for robust nanobody humanization. Structure 2022, 30, 418–429.e413. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.H.; Huang, K.; Tu, H.; Adler, A.S. Monoclonal antibody humanness score and its applications. BMC Biotechnol. 2013, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Ramon, A.; Ali, M.; Atkinson, M.; Saturnino, A.; Didi, K.; Visentin, C.; Ricagno, S.; Xu, X.; Greenig, M.; Sormanni, P. Assessing antibody and nanobody nativeness for hit selection and humanization with AbNatiV. Nat. Mach. Intell. 2024, 6, 74–91. [Google Scholar] [CrossRef]
- Prihoda, D.; Maamary, J.; Waight, A.; Juan, V.; Fayadat-Dilman, L.; Svozil, D.; Bitton, D.A. BioPhi: A platform for antibody design, humanization, and humanness evaluation based on natural antibody repertoires and deep learning. MAbs 2022, 14, 2020203. [Google Scholar] [CrossRef] [PubMed]
- Wollacott, A.M.; Xue, C.; Qin, Q.; Hua, J.; Bohnuud, T.; Viswanathan, K.; Kolachalama, V.B. Quantifying the nativeness of antibody sequences using long short-term memory networks. Protein Eng. Des. Sel. 2019, 32, 347–354. [Google Scholar] [CrossRef]
- Ma, J.; Wu, F.; Xu, T.; Xu, S.; Liu, W.; Yan, D.; Bai, Q.; Yao, J. An adaptive autoregressive diffusion approach to design active humanized antibody and nanobody. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kovaltsuk, A.; Leem, J.; Kelm, S.; Snowden, J.; Deane, C.M.; Krawczyk, K. Observed Antibody Space: A Resource for Data Mining Next-Generation Sequencing of Antibody Repertoires. J. Immunol. 2018, 201, 2502–2509. [Google Scholar] [CrossRef]
- Olsen, T.H.; Boyles, F.; Deane, C.M. Observed Antibody Space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci. 2022, 31, 141–146. [Google Scholar] [CrossRef]
- Raybould, M.I.J.; Kovaltsuk, A.; Marks, C.; Deane, C.M. CoV-AbDab: The coronavirus antibody database. Bioinformatics 2021, 37, 734–735. [Google Scholar] [CrossRef]
- Raybould, M.I.J.; Marks, C.; Lewis, A.P.; Shi, J.; Bujotzek, A.; Taddese, B.; Deane, C.M. Thera-SAbDab: The Therapeutic Structural Antibody Database. Nucleic Acids Res. 2020, 48, D383–D388. [Google Scholar] [CrossRef]
- Kortemme, T. De novo protein design-From new structures to programmable functions. Cell 2024, 187, 526–544. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Jin, X.; Yang, C.; Wang, Z.; Min, X.; Ge, S. De novo protein design in the age of artificial intelligence. Sheng Wu Gong Cheng Xue Bao 2024, 40, 3912–3929. [Google Scholar] [PubMed]
- Meinen, B.A.; Bahl, C.D. Breakthroughs in computational design methods open up new frontiers for de novo protein engineering. Protein Eng. Des. Sel. 2021, 34, gzab007. [Google Scholar] [CrossRef]
- Pan, X.; Kortemme, T. Recent advances in de novo protein design: Principles, methods, and applications. J. Biol. Chem. 2021, 296, 100558. [Google Scholar] [CrossRef]
- Winnifrith, A.; Outeiral, C.; Hie, B.L. Generative artificial intelligence for de novo protein design. Curr. Opin. Struct. Biol. 2024, 86, 102794. [Google Scholar] [CrossRef] [PubMed]
- Woolfson, D.N. A brief history of de novo protein design: Minimal, rational, and computational. J. Mol. Biol. 2021, 433, 167160. [Google Scholar] [CrossRef]
- Huang, P.-S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Shin, J.-E.; Riesselman, A.J.; Kollasch, A.W.; McMahon, C.; Simon, E.; Sander, C.; Manglik, A.; Kruse, A.C.; Marks, D.S. Protein design and variant prediction using autoregressive generative models. Nat. Commun. 2021, 12, 2403. [Google Scholar] [CrossRef]
- Watson, J.L.; Juergens, D.; Bennett, N.R.; Trippe, B.L.; Yim, J.; Eisenach, H.E.; Ahern, W.; Borst, A.J.; Ragotte, R.J.; Milles, L.F.; et al. De novo design of protein structure and function with RFdiffusion. Nature 2023, 620, 1089–1100. [Google Scholar] [CrossRef]
- Bennett, N.R.; Watson, J.L.; Ragotte, R.J.; Borst, A.J.; See, D.L.; Weidle, C.; Biswas, R.; Yu, Y.; Shrock, E.L.; Ault, R.; et al. Atomically accurate de novo design of antibodies with RFdiffusion. bioRxiv 2025. [Google Scholar] [CrossRef]
- Baek, M.; Anishchenko, I.; Humphreys, I.R.; Cong, Q.; Baker, D.; DiMaio, F. Efficient and accurate prediction of protein structure using RoseTTAFold2. bioRxiv 2023. [Google Scholar] [CrossRef]
- Wang, R.; Wu, F.; Gao, X.; Wu, J.; Zhao, P.; Yao, J. IgGM: A generative model for functional antibody and nanobody design. bioRxiv 2024. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Milestone | Description | References |
|---|---|---|---|
| 1993 | Discovery of heavy-chain-only antibodies (HCAbs) | HCAbs were first discovered in camelids (camels, llamas, and alpacas) by C Hamers-Casterman and colleagues, marking the discovery of single-domain antibodies (VHH). | [1] |
| 1996 | First nanobody crystal structure | The first crystal structures of nanobodies were solved, providing insights into their binding mechanisms and how the CDR3 loop plays a critical role in their high specificity and affinity. | [7,8] |
| 1997 | First large-scale nanobody production and screening platform | The feasibility of producing and screening nanobodies from camelid heavy-chain antibodies was demonstrated, enabling their large-scale application, providing a robust platform for developing nanobodies with high specificity and stability. | [9] |
| 2000s | Early applications in diagnostics | Nanobodies started being applied for diagnostic purposes, including biosensors and immunoassays. Their small size and stability made them suitable for rapid diagnostics, including the detection of bacteria, viruses, and toxins. | [10,11,12] |
| 2001 | The term “nanobody” was introduced | The term “nanobody” introduced by Ablynx in 2001 describes single-domain antibody fragments derived from camelid heavy-chain-only antibodies and is a registered trademark of Ablynx NV. | [8] |
| 2005–2009 | Humanization of nanobodies | Efforts to humanize nanobodies began, making them suitable for use in human therapies. This reduced their potential immunogenicity while preserving their functional activity. | [4,13] |
| 2008 | Nanobody for in vivo imaging | Nanobodies were successfully used for in vivo imaging due to their small size and ability to target tumor biomarkers, offering a more efficient alternative to traditional antibodies for molecular imaging. | [14,15,16] |
| 2010s | Nanobody engineering advances | New techniques such as ribosome display and yeast display were employed to create large nanobody libraries, improving screening and selection processes for targeted nanobody therapies and diagnostics. | [17,18] |
| 2018 | First nanobody-based therapeutic approved | Caplacizumab (Cablivi), developed by Ablynx, became the first nanobody-based therapeutic approved in the EU for treating thrombotic thrombocytopenic purpura (TTP). | [19] |
| 2021 | First pan-tumor PD-L1 antibody and subcutaneous immunotherapy approved | Envafolimab (KN035, Envorria), co-developed by CanSino Biologics and 3D Medicines, became the first approved subcutaneous PD-L1 antibody and the first pan-tumor oncology therapy for unresectable or metastatic microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) solid tumors in China. | [20] |
| 2022 | First nanobody-based CAR-T therapy approved | Carvykti (ciltacabtagene autoleucel), developed by Legend Biotech and Janssen, became the first CAR-T therapy using nanobody technology approved in the U.S. for relapsed/refractory multiple myeloma (RRMM). Its design incorporates a dual-BCMA-targeting nanobody. | [21] |
| 2022 | First nanobody for autoimmune disease approved | Ozoralizumab (Nanozora), developed by Ablynx, became the first nanobody-based therapy for autoimmune disease approved in Japan for treating rheumatoid arthritis (RA). Its trivalent bispecific VHH design combines two anti-TNFα domains (targeting inflammation) and one anti-HSA domain (prolonging half-life). | [22] |
| 2020s | AI-driven nanobody design | The integration of AI in nanobody engineering began, with AI tools such as AlphaFold2 and ProteinMPNN enhancing the prediction of nanobody structures and enabling the rational design of multiepitope nanobodies for targeted therapeutic and diagnostic applications. | [6,23] |
| Display Technology | Description | Key Features | Advantages | Disadvantages |
|---|---|---|---|---|
| Phage Display [42] | Involves the expression of peptides or antibodies on the surface of bacteriophages, where the genetic information is linked to the displayed protein. | High-diversity libraries, easy cloning, rapid screening | High-throughput screening, easy to scale up, well established | Limited to larger targets, less efficient for membrane-bound proteins, phage amplification may be required |
| Ribosome Display [17,43] | An in vitro method where mRNA is linked to its translated protein, forming a stable mRNA–protein complex that can be used for screening. | No need for host cell transformation, no bacterial growth | No transformation required, high-diversity libraries (up to 1013), high sensitivity | Labor-intensive, slower screening process compared to phage display |
| Yeast Display [18] | Displays peptides or antibodies on the surface of yeast cells. The displayed proteins are directly linked to the yeast genome. | Yeast cells serve as both expression and selection systems | Single-cell resolution, real-time binding analysis, simpler than mammalian systems | Lower throughput compared to phage display, limited to eukaryotic targets, needs yeast transformation |
| Bacterial Display (Escherichia coli) [44,45] | Proteins are displayed on the surface of bacteria, which can then be selected based on binding to a target. | Fast expression, uses bacterial systems. | High expression levels, simple and cost-effective | Limited to smaller targets, lower display efficiency compared to yeast, less versatile for complex proteins |
| Mammalian Display [46] | A type of display where proteins are expressed on the surface of mammalian cells. This allows for the display of complex proteins. | Suitable for complex and membrane proteins, similar to human systems. | Better mimic of natural systems, good for membrane proteins and intracellular interactions | Expensive, requires specialized equipment, lower throughput than yeast or phage display |
| mRNA Display [47,48] | In vitro method where mRNA is linked to its corresponding protein, creating a stable mRNA–protein complex for screening. | Does not require cell-based transformation, used for in vitro selections | Huge diversity of libraries, high sensitivity and speed, can work without transformation | Requires specialized equipment, labor-intensive for large libraries |
| Property | Camelid-Derived (VHH) | Human-Derived | Shark-Derived (VNAR) |
|---|---|---|---|
| CDR3 Loop | Long and flexible | Shorter and less flexible | Elongated, highly flexible |
| Stability | High, due to hydrophilic framework mutations | Moderate, requires engineering | Extremely high, naturally stable |
| Size | ~12–15 kDa | ~15 kDa | ~12 kDa (smallest fragment) |
| Antigen Access | Cryptic and concave epitopes | Protein antigens, less cryptic sites | Narrow grooves, cryptic epitopes |
| Natural Solubility | High | Moderate, engineered for solubility | Very high |
| Primary Application | Protein antigens (therapeutics, diagnostics) | Humanized therapies, low immunogenicity | Extreme environments, non-protein antigens |
| Model Name | VHH Applicability | Model Type | Main Function | Code Availability (Most Recent) | Web Server or Colab Availability (Most Recent) | Reference |
|---|---|---|---|---|---|---|
| AlphaFold2 | Yes | Transformer | Protein monomer structure computation | https://github.com/google-deepmind/alphafold, accessed on 4 April 2025 | https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb, accessed on 4 April 2025 | [23] |
| DeepAb | Yes | RNN(biLSTM + LSTM) ResNet | Antibody Fv and VHH structure prediction | https://github.com/RosettaCommons/DeepAb, accessed on 4 April 2025 | https://colab.research.google.com/github/RosettaCommons/DeepAb/blob/main/DeepAb.ipynb, accessed on 4 April 2025 | [144] |
| ABlooper | No | GNN(E(n)-EGNN) | Antibody CDR loop prediction | https://github.com/brennanaba/ABlooper, accessed on 4 April 2025 | Not found | [145] |
| NanoNet | Yes | ResNet | High-throughput VHH structure determination | https://github.com/dina-lab3D/NanoNet, accessed on 4 April 2025 | https://bio3d.cs.huji.ac.il/nanonet/, accessed on 4 April 2025 | [149] |
| IgFold | Yes | Transformer(AntiBERTy [154] based on BERT) | Antibody and VHH structure prediction | https://github.com/Graylab/IgFold, accessed on 4 April 2025 | https://colab.research.google.com/github/Graylab/IgFold/blob/main/IgFold.ipynb, accessed on 4 April 2025 | [155] |
| ImmuneBuilder | Yes | Transformer(Based on AlphaFold-Multimer [171]) | Antibody, VHH and T-cell receptor structure prediction | https://github.com/brennanaba/ImmuneBuilder, accessed on 4 April 2025 | https://colab.research.google.com/github/brennanaba/ImuneBuilder/blob/main/notebook/ImmuneBuilder.ipynb, accessed on 4 April 2025 | [156] |
| Parapred | No | RNN(LSTM) CNN | Antibody paratope prediction | https://github.com/eliberis/parapred, accessed on 4 April 2025 | Not found | [157] |
| ParaAntiProt | Yes | Transformer(Based on ProtTrans [172]) CNN | Antibody and VHH paratope prediction | https://github.com/Alirzeanoroozi/ParaAntiProt, accessed on 4 April 2025 | Not found | [158] |
| NbX | Yes | Decision Tree | Nanobody binding pose prediction | https://github.com/johnnytam100/NbX, accessed on 4 April 2025 | Not found | [164] |
| DLAB | No | CNN | Antibody virtual screening | https://github.com/con-schneider/dlab-public, accessed on 4 April 2025 | Not found | [165] |
| AlphaFold3 | Yes | Transformer(Adapted from AF2 [23]) | Structural modeling of monomers and multimers for biomacromolecules | https://github.com/google-deepmind/alphafold3, accessed on 4 April 2025 | https://alphafoldserver.com/, accessed on 4 April 2025 | [5] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Ding, Y. Nanobodies: From Discovery to AI-Driven Design. Biology 2025, 14, 547. https://doi.org/10.3390/biology14050547
Zhu H, Ding Y. Nanobodies: From Discovery to AI-Driven Design. Biology. 2025; 14(5):547. https://doi.org/10.3390/biology14050547
Chicago/Turabian StyleZhu, Haoran, and Yu Ding. 2025. "Nanobodies: From Discovery to AI-Driven Design" Biology 14, no. 5: 547. https://doi.org/10.3390/biology14050547
APA StyleZhu, H., & Ding, Y. (2025). Nanobodies: From Discovery to AI-Driven Design. Biology, 14(5), 547. https://doi.org/10.3390/biology14050547