
Identification of a Novel hsa_circ_0058058/miR-324-5p Axis and Prognostic/Predictive Molecules for Acute Myeloid Leukemia Outcome by Bioinformatics-Based Analysis

 , , , , and
, , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Identification of circRNAs in LAML
2.2. Identification of circRNAs-Mediated miRNA in LAML
2.3. Prediction of miRNA Targets
2.4. Verification of Gene Expression in LAML Cohort I
2.5. Verification of Gene Expression in LAML Cohort II
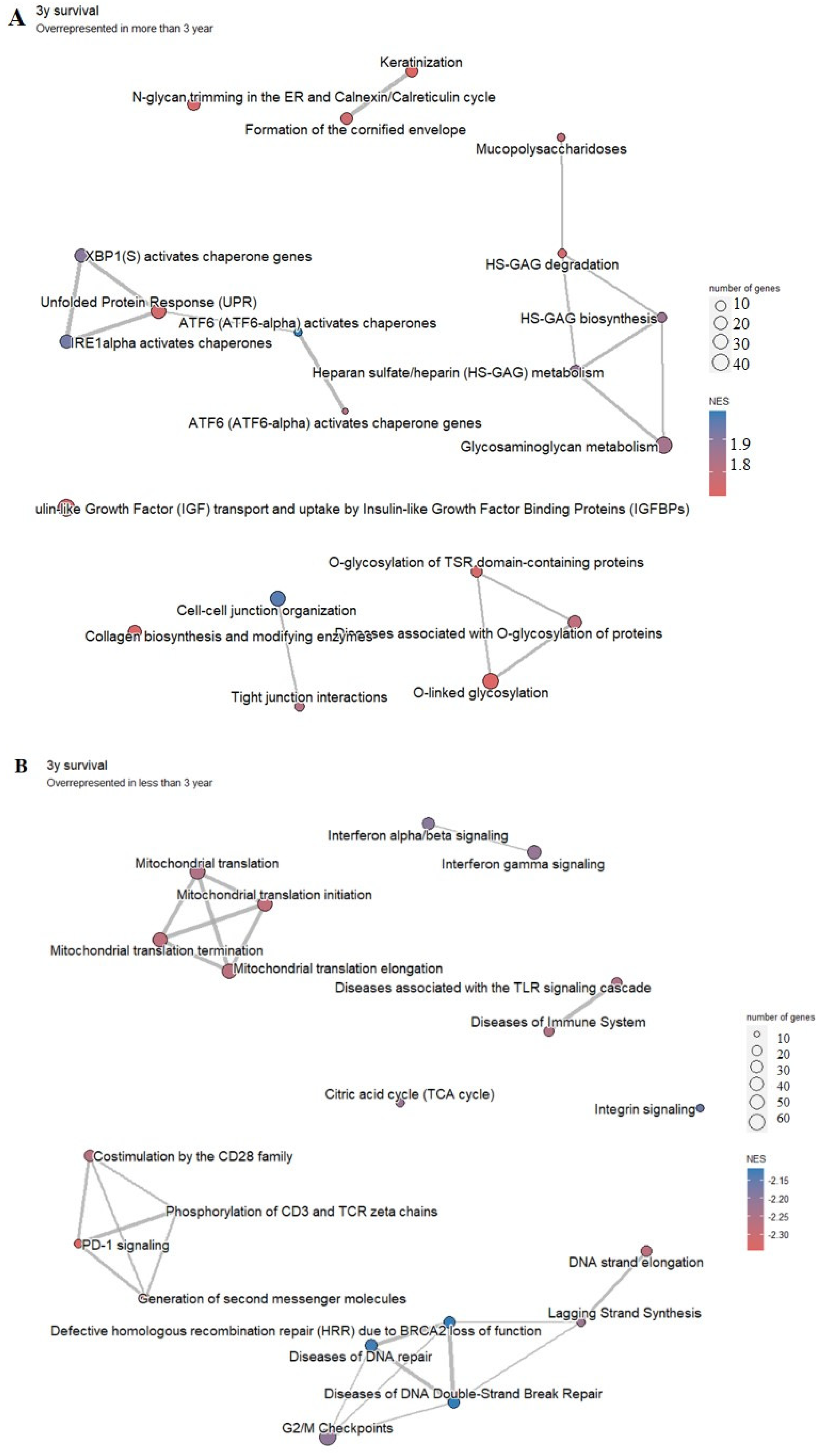
2.6. Survival Analysis of AP1G1 and SP1
2.7. Construction of Protein–Protein Interaction (PPI) Network
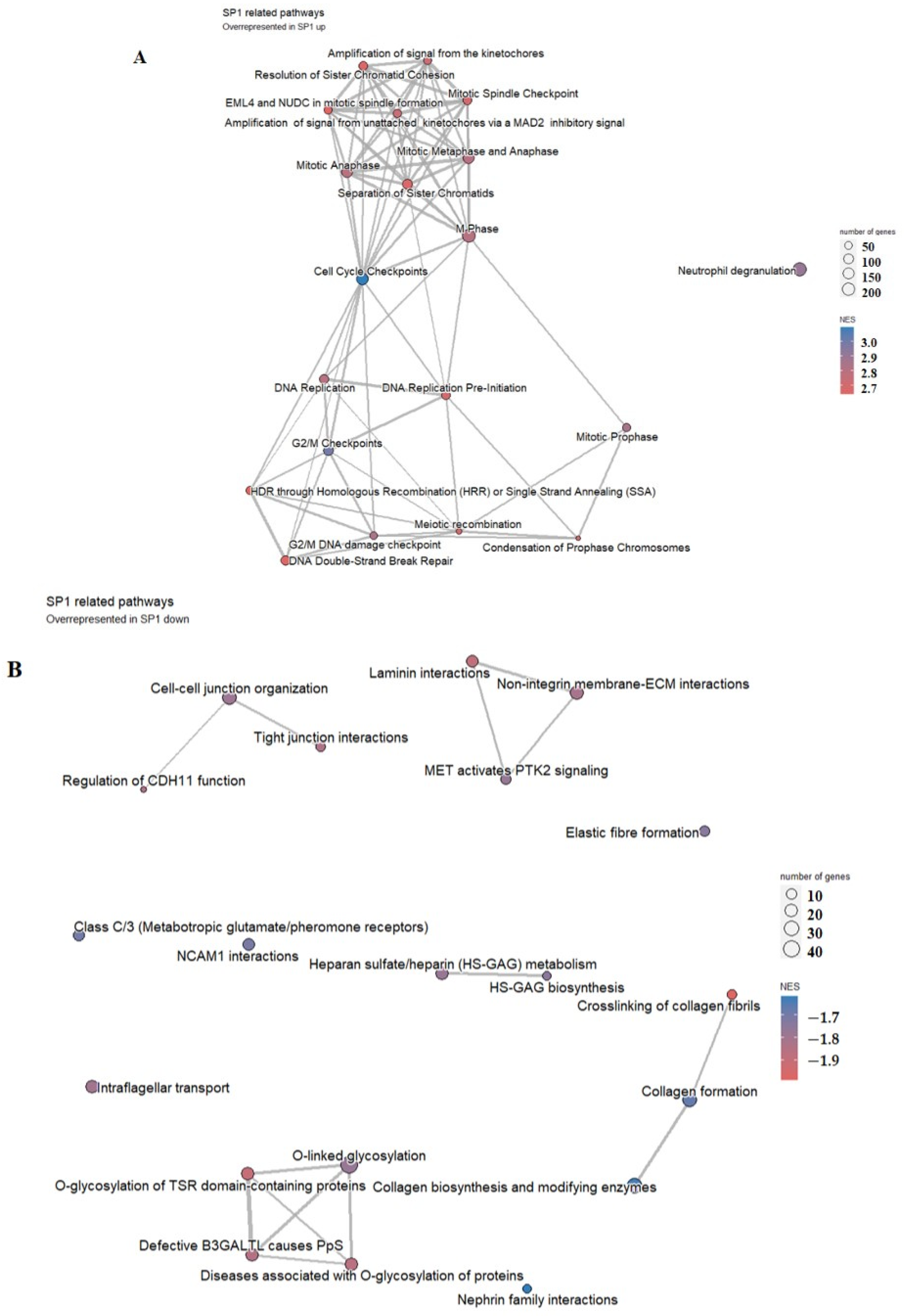
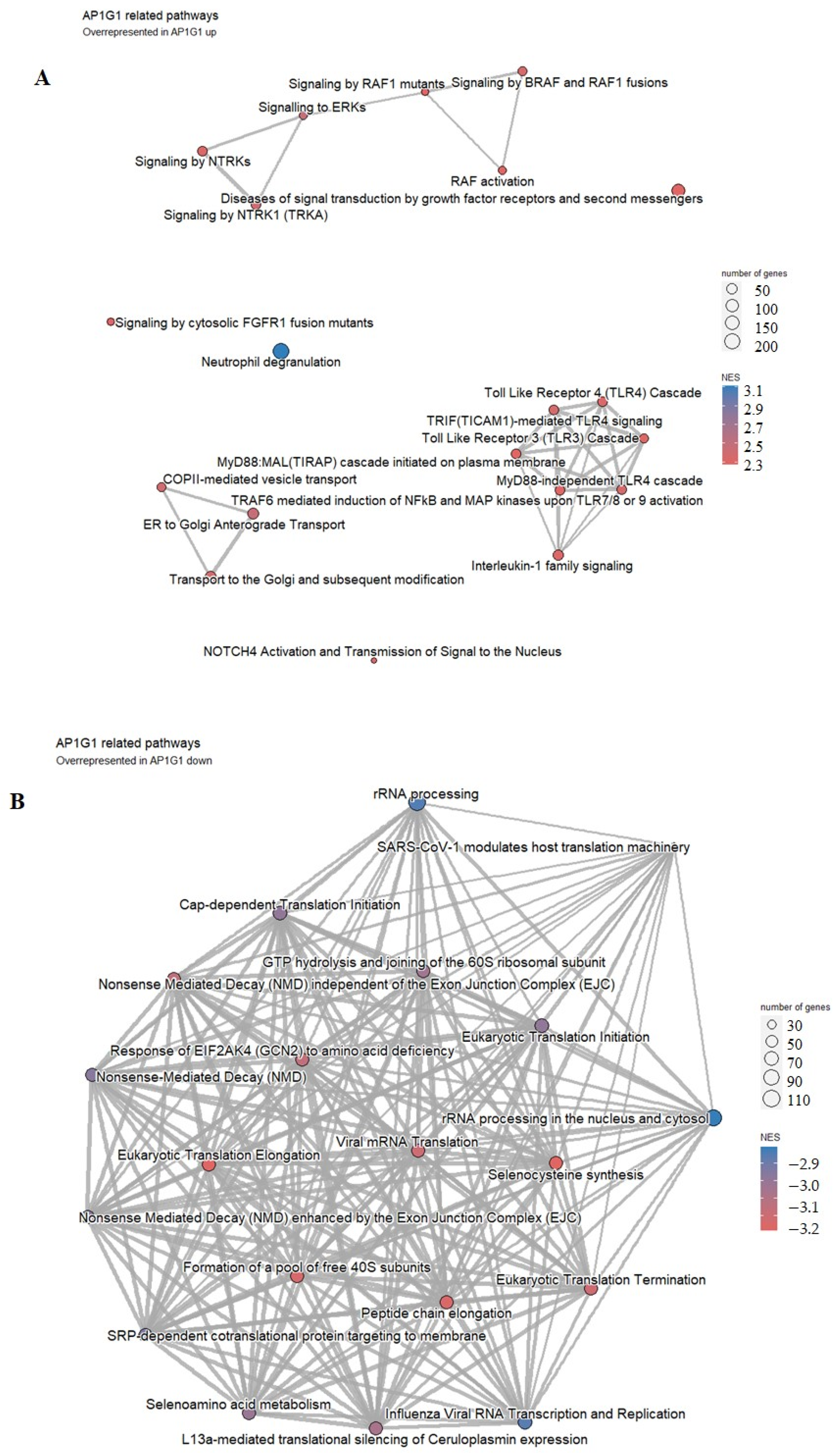
2.8. Functions of AP1G1 and SP1
3. Results
3.1. Identification of CircRNAs in LAML
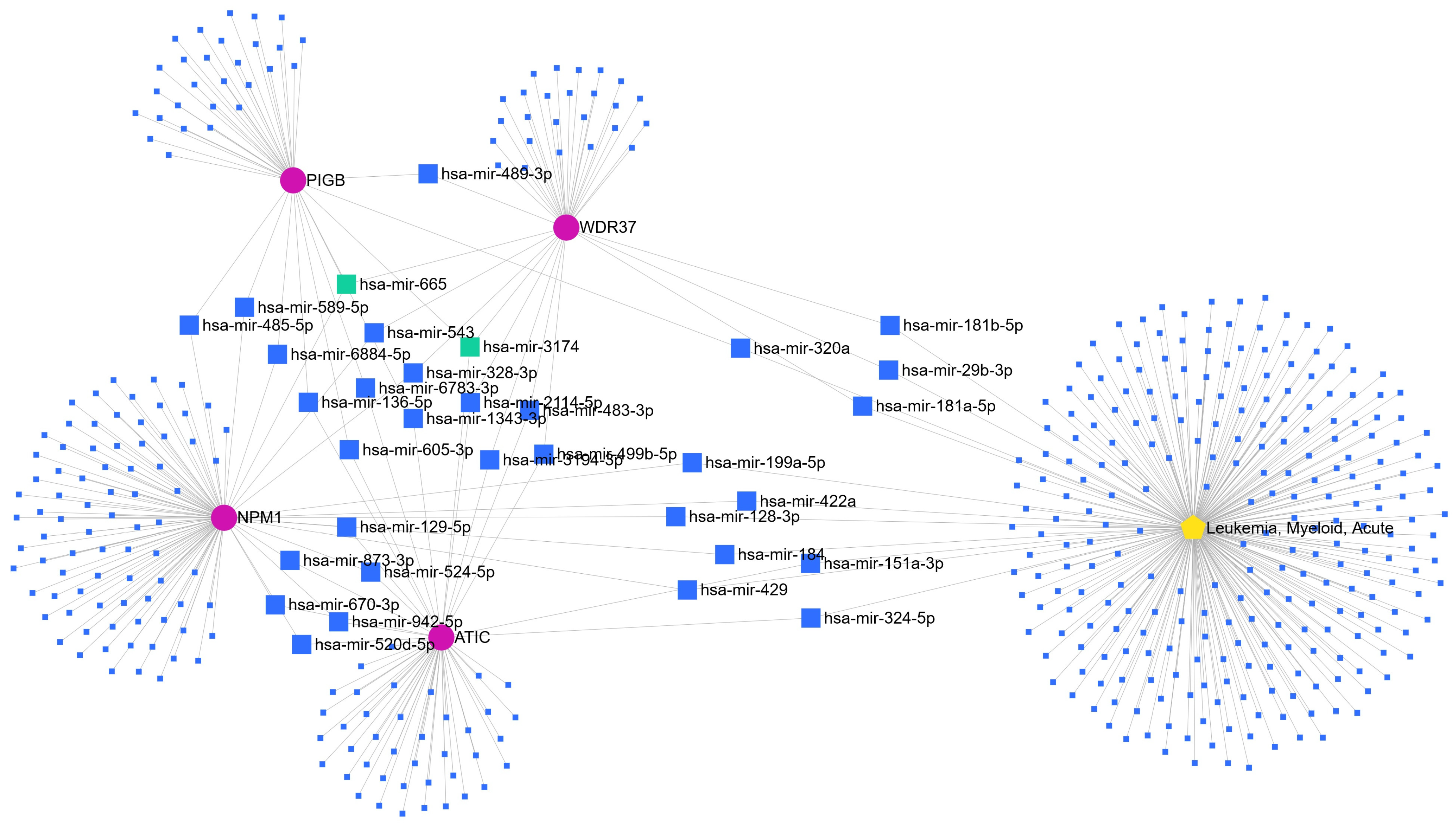
3.2. Identification of circRNA-Mediated miRNA in LAML
3.3. Prediction of miRNA Targets
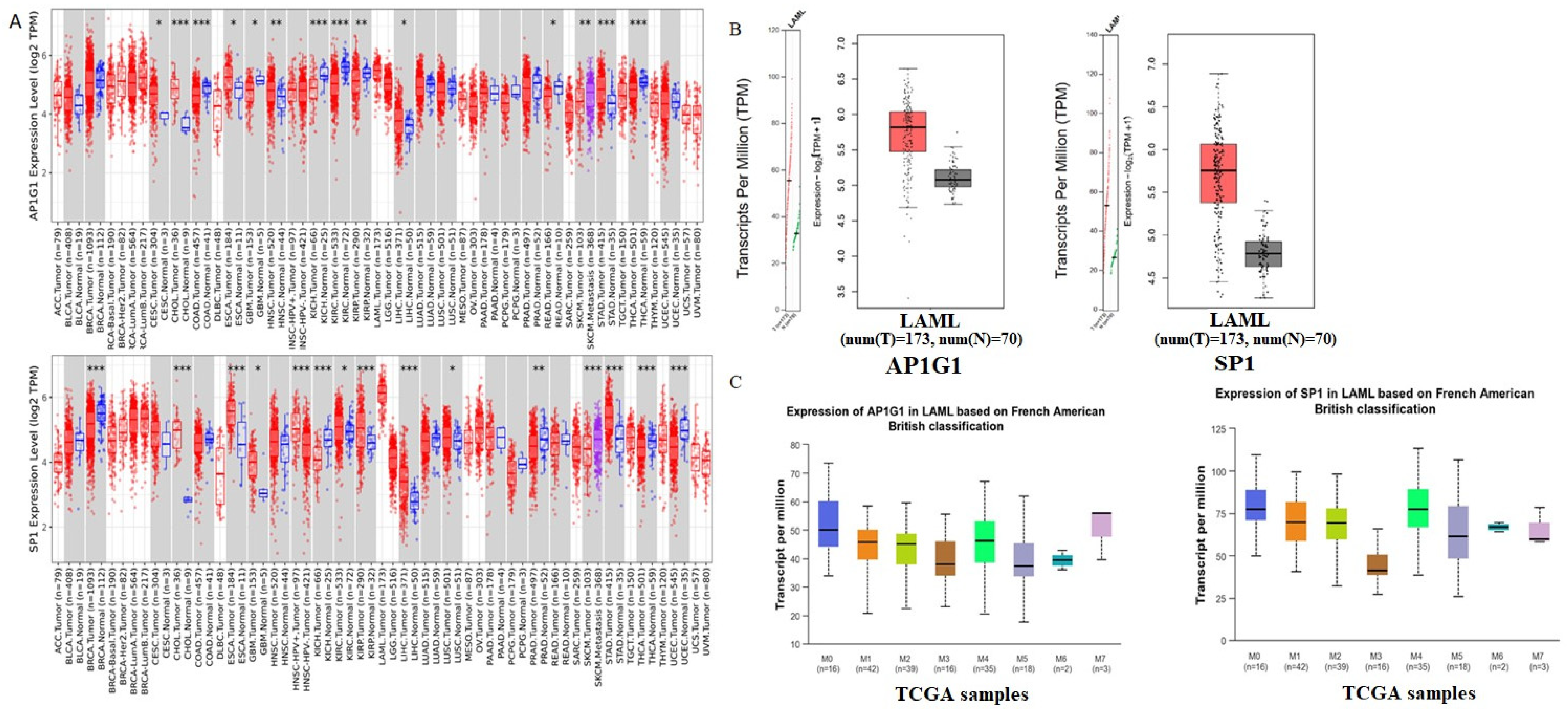
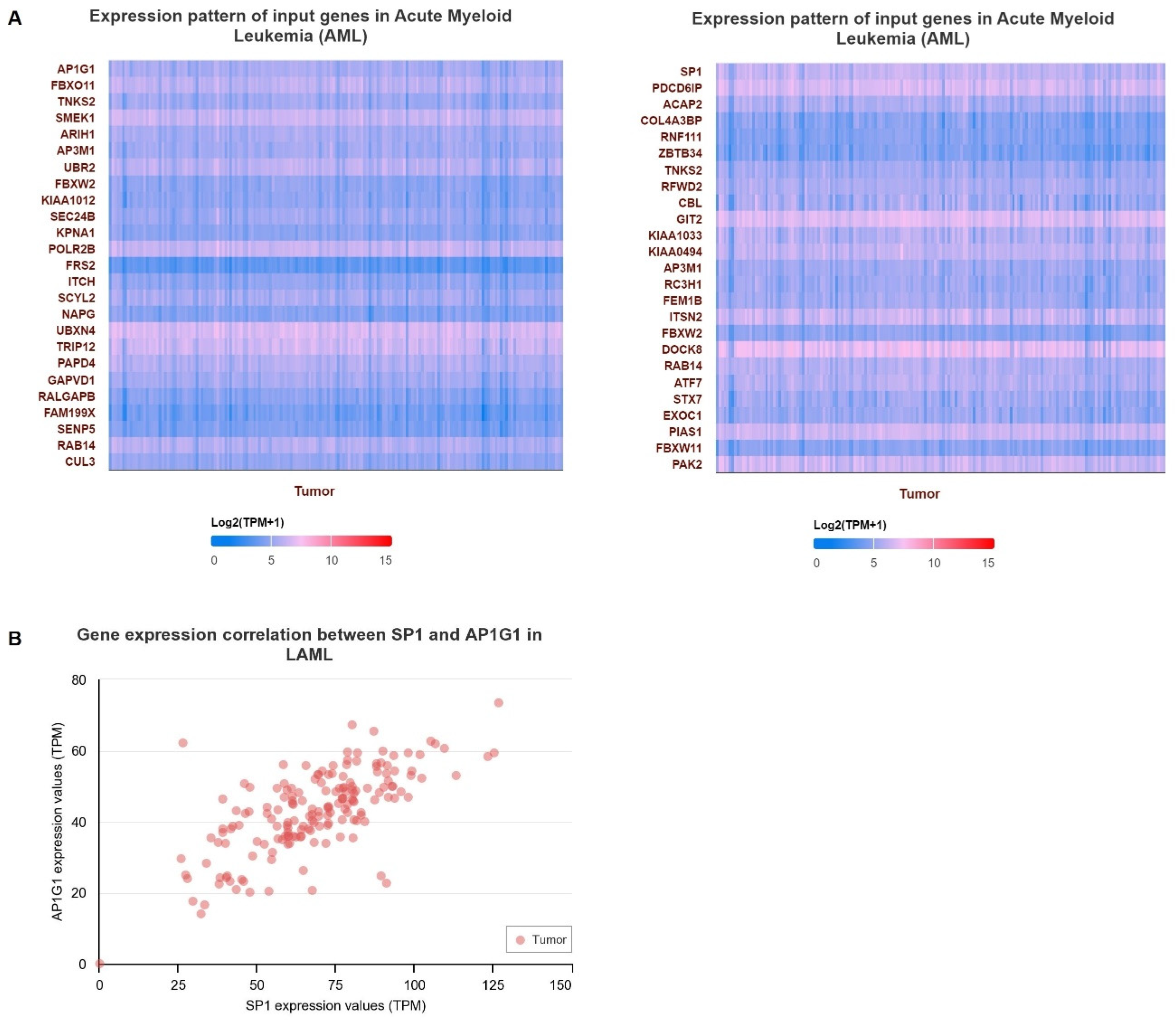
3.4. Verification of Gene Expression in LAML
3.5. Survival Analysis of AP1G1 and SP1
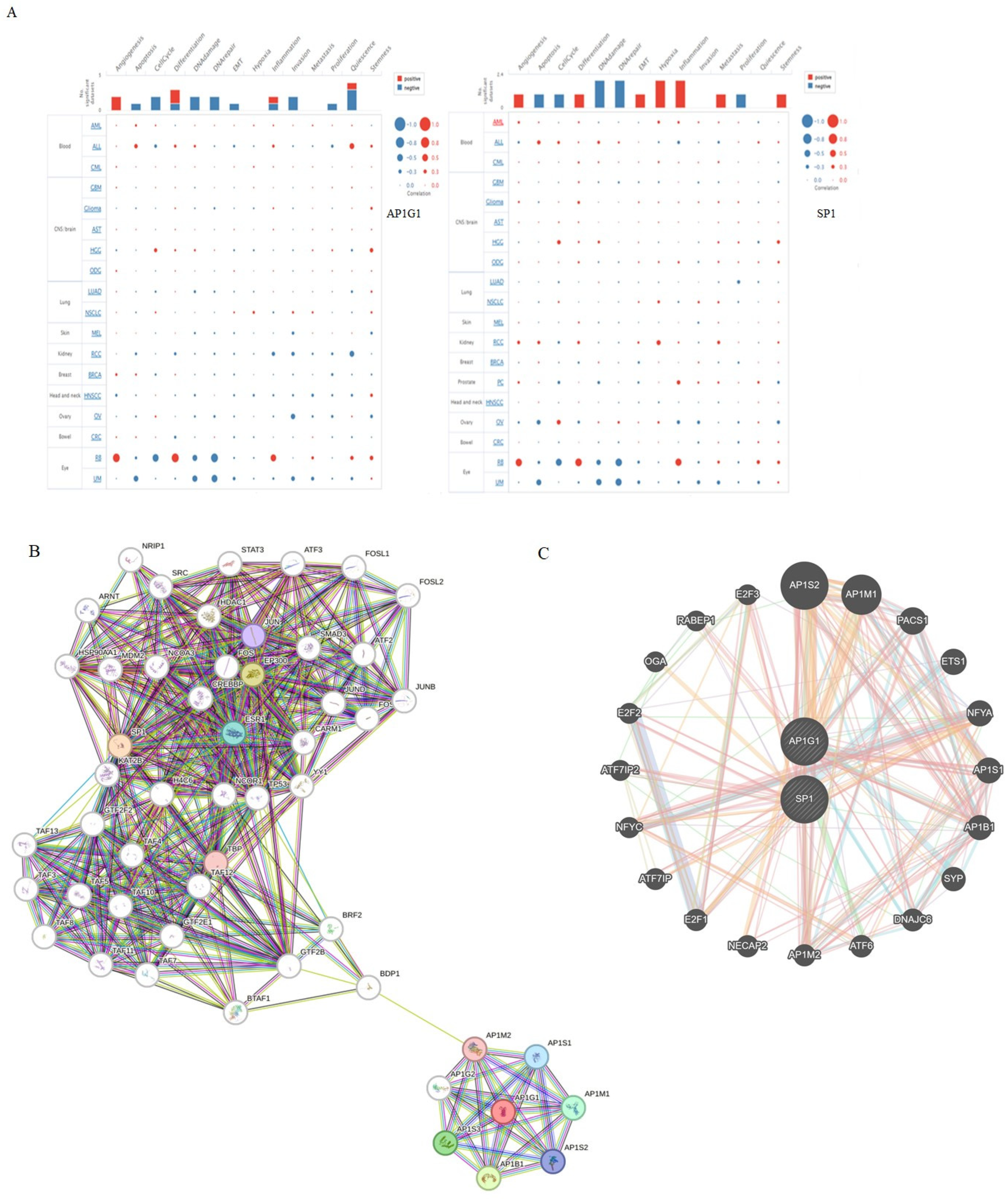
3.6. Construction of Protein–Protein Interaction (PPI) Network
3.7. Functions of AP1G1 and SP1
3.8. DGE-LAML Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, W.; Cheng, F. Circular RNA circCRKL inhibits the proliferation of acute myeloid leukemia cells via the miR-196a-5p/miR-196b-5p/p27 axis. Bioengineered 2021, 12, 7704–7713. [Google Scholar] [CrossRef] [PubMed]
- Moussa Agha, D.; Rouas, R.; Najar, M.; Bouhtit, F.; Naamane, N.; Fayyad-Kazan, H.; Bron, D.; Meuleman, N.; Lewalle, P.; Merimi, M. Identification of Acute Myeloid Leukemia Bone Marrow Circulating MicroRNAs. Int. J. Mol. Sci. 2020, 21, 7065. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ning, X.; Yan, X.; Song, L. Circ_0104700 contributes to acute myeloid leukemia progression by enhancing MCM2 expression through targeting miR-665. Hematology 2023, 28, 2227489. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, X.; Liu, J.; Jin, Y.; Wang, W. Circular RNA circ_0004277 Inhibits Acute Myeloid Leukemia Progression through MicroRNA-134-5p / Single stranded DNA binding protein 2. Bioengineered 2022, 13, 9662–9673. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Zheng, Y.L.; Su, X.; Wang, X.Q. Crosstalk Between MicroRNAs and Circular RNAs in Human Diseases: A Bibliographic Study. Front. Cell. Dev. Biol. 2021, 9, 754880. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Yang, L. Regulation of circRNA biogenesis. RNA Biol. 2015, 12, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Zheng, H.; Wu, Z.; Chen, M.; Huang, Y. Circular RNA-protein interactions: Functions, mechanisms, and identification. Theranostics 2020, 10, 3503–3517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xin, Y. Circular RNAs: A new frontier for cancer diagnosis and therapy. J. Hematol. Oncol. 2018, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Okcanoğlu, T.B.; Gündüz, C. Circular RNAs in leukemia. Biomed. Rep. 2019, 10, 87–91. [Google Scholar]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome. Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Karadag, A. Comparison of Prognostic miRNA Signature in Patients with Acute and Chronic Myeloid Leukemia by Bioinformatic Analysis. Med. Rec. 2022, 4, 447–453. [Google Scholar] [CrossRef]
- Khan, S.; Ayub, H.; Khan, T.; Wahid, F. MicroRNA biogenesis, gene silencing mechanisms and role in breast, ovarian and prostate cancer. Biochimie 2019, 167, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2021, 28, 127–138. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, W.; Zhang, P.; Chen, J.; Qian, H.; Zhang, X.; Wenrong, X. Circular RNAs: Emerging cancer biomarkers and targets. J. Exp. Clin. Cancer Res. 2017, 36, 152. [Google Scholar] [CrossRef]
- Tian, Y.; Xing, Y.; Zhang, Z.; Peng, R.; Zhang, L.; Sun, Y. Bioinformatics Analysis of Key Genes and circRNA-miRNA-mRNA Regulatory Network in Gastric Cancer. Biomed. Res. Int. 2020, 2020, 2862701. [Google Scholar] [CrossRef]
- Lei, X.; Fang, Z.; Guo, L. Predicting circRNA–disease associations based on improved collaboration filtering recommendation system with multiple data. Front. Genet. 2019, 10, 897. [Google Scholar] [CrossRef]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef]
- Dudekula, D.B.; Panda, A.C.; Grammatikakis, I.; De, S.; Abdelmohsen, K.; Gorospe, M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016, 13, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, 127–131. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A web server for comprehensive analysis of tumor- infiltrating immune cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From reads to genes to pathways: Differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Research 2016, 5, 1438. [Google Scholar] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, 561–658. [Google Scholar] [CrossRef] [PubMed]
- Warde-farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yan, M.; Zhang, G.; Liu, W.; Deng, C.; Liao, G.; Xu, L.; Luo, T.; Yan, H.; Long, Z.; et al. CancerSEA: A cancer single-cell state atlas. Nucleic Acids Res. 2019, 47, D900–D908. [Google Scholar] [CrossRef] [PubMed]
- Guangchuang, Y.; Qing-Yu, H. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar]
- Liang, N.; Mi, L.; Li, J.; Li, T.; Chen, J.; Dionigi, G.; Guan, H.; Sun, H. Pan-Cancer Analysis of the Oncogenic and Prognostic Role of PKM2: A Potential Target for Survival and Immunotherapy. Biomed. Res. Int. 2023, 2023, 3375109. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Zheng, G.; Ning, Q.; Zheng, J.; Dong, D. Translation and functional roles of circular RNAs in human cancer. Mol. Cancer 2020, 19, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhan, L.; Huang, K.; Wang, X. The functions and clinical significance of circRNAs in hematological malignancies. J. Hematol. Oncol. 2020, 13, 138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xia, P.; Liu, J.; Chen, Z.; Ma, W.; Yuan, Y. Atic inhibits autophagy in hepatocellular cancer through the akt/foxo3 pathway and serves as a prognostic signature for modeling patient survival. Int. J. Biol. Sci. 2021, 17, 4442–4458. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, G.; Dang, Y.; He, R.; Liu, A.; Ma, J.; Wang, C. Upregulation of ATIC in multiple myeloma tissues based on tissue microarray and gene microarrays. Int. J. Lab. Hematol. 2021, 43, 409–417. [Google Scholar] [CrossRef]
- Niu, N.; Zeng, J.; Ke, X.; Zheng, W.; Fu, C.; Lv, S.; Fu, J.; Yu, Y. ATIC facilitates cell growth and migration by upregulating Myc expression in lung adenocarcinoma. Oncol. Lett. 2022, 23, 131. [Google Scholar] [CrossRef]
- Zhou, Q.; Lei, C.; Cui, F.; Chen, H.; Cao, X. Circ-ATIC regulates esophageal squamous cell carcinoma growth and metastasis through miR-1294/PBX3 pathway. Heliyon 2023, 9, e12916. [Google Scholar] [CrossRef]
- Li, W.; Zhong, C.; Jiao, J.; Li, P.; Cui, B.; Ji, C.; Ma, D. Characterization of hsa_circ_0004277 as a new biomarker for acute myeloid leukemia via circular RNA profile and bioinformatics analysis. Int. J. Mol. Sci. 2017, 18, 597. [Google Scholar] [CrossRef]
- Kadkhoda, S.; Hussen, B.M.; Eslami, S.; Ghafouri-Fard, S. A review on the role of miRNA-324 in various diseases. Front. Genet. 2022, 13, 950162. [Google Scholar] [CrossRef]
- Bhise, N.S.; Chauhan, L.; Shin, M.; Cao, X.; Pounds, S.; Lamba, V.; Lamba, J.K. MicroRNA-mRNA pairs associated with outcome in AML: From in vitro cell-based studies to AML patients. Front. Pharmacol. 2016, 6, 324. [Google Scholar] [CrossRef]
- Wu, B.; Wang, F.; Wang, Y.; Deng, X.; Wu, W. CircATIC Contributes to Multiple Myeloma Progression via miR-324-5p-Dependent Regulation of HGF. Biochem. Genet. 2022, 60, 2515–2532. [Google Scholar] [CrossRef]
- Leoncini, P.P.; Vitullo, P.; Reddel, S.; Tocco, V.; Paganelli, V.; Stocchi, F.; Mariggiò, E.; Massa, M.; Nigita, G.; Veneziano, D.; et al. MicroRNA profiling of paediatric AML with FLT-ITD or MLL-rearrangements: Expression signatures and in vitro modulation of miR-221-3p and miR-222-3p with BRD4/HATs inhibitors. Oncol. Rep. 2022, 48, 221. [Google Scholar] [CrossRef] [PubMed]
- Takatsu, H.; Sakurai, M.; Shin, H.W.; Murakami, K.; Nakayama, K. Identification and characterization of novel clathrin adaptor-related proteins. J. Biol. Chem. 1998, 273, 24693–24700. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Lu, Y.; Qiu, S.; Wang, Y.; Qin, J.; Fan, Z. AP1G1 is involved in cetuximab-mediated downregulation of ASCT2-EGFR complex and sensitization of human head and neck squamous cell carcinoma cells to ROS-induced apoptosis. Cancer Lett. 2017, 408, 33–42. [Google Scholar] [CrossRef]
- Johnson, K.R.; Gagnon, L.H.; Chang, B. A hypomorphic mutation of the gamma-1 adaptin gene (Ap1g1) causes inner ear, retina, thyroid, and testes abnormalities in mice. Mamm. Genome 2016, 27, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, H.; Jiang, P. MicroRNA-382 inhibits cell growth and migration in colorectal cancer by targeting SP1. Biol. Res. 2018, 51, 51. [Google Scholar] [CrossRef]
- Liu, B.; Ma, H.; Liu, Q.; Xiao, Y.; Pan, S.; Zhou, H.; Jia, L. MiR-29b/Sp1/FUT4 axis modulates the malignancy of leukemia stem cells by regulating fucosylation via Wnt/β-catenin pathway in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2023, 42, 208. [Google Scholar] [CrossRef]
- Seznec, J.; Silkenstedt, B.; Naumann, U. Therapeutic effects of the Sp1 inhibitor mithramycin A in glioblastoma. J. Neurooncol. 2011, 101, 365–377. [Google Scholar] [CrossRef]
- Lin, R.K.; Wu, C.Y.; Chang, J.W.; Juan, L.J.; Hsu, H.S.; Chen, C.Y.; Lu, Y.Y.; Tang, Y.A.; Yang, Y.C.; Yang, P.C.; et al. Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res. 2010, 70, 5807–5817. [Google Scholar] [CrossRef]
- Monteleone, E.; Orecchia, V.; Corrieri, P.; Schiavone, D.; Avalle, L.; Moiso, E.; Savino, A.; Molineris, I.; Provero, P.; Poli, V. SP1 and STAT3 functionally synergize to induce the RhoU small GTPase and a subclass of non-canonical WNT responsive genes correlating with poor prognosis in breast cancer. Cancers 2019, 11, 101. [Google Scholar] [CrossRef]
- Xu, X.; Wang, X.; Chen, Q.; Zheng, A.; Li, D.; Meng, Z.; Li, X.; Cai, H.; Li, W.; Huang, S.; et al. Sp1 promotes tumour progression by remodelling the mitochondrial network in cervical cancer. J. Transl. Med. 2023, 21, 307. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, L.; Xiao, M.; Zhang, Z.; Shen, J.; Anuchapreeda, S.; Tima, S.; Chiampanichayakul, S.; Xiao, Z. PD-L1 regulates cell proliferation and apoptosis in acute myeloid leukemia by activating PI3K-AKT signaling pathway. Sci. Rep. 2022, 12, 11444. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wu, T.; Zhou, X.; Xie, S.; Sun, H.; Sun, Y.; Li, Y. Progress of research on PD-1/PD-L1 in leukemia. Front. Immunol. 2023, 14, 1265299. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CircRNA ID | Location | Gene | Expression Levels |
|---|---|---|---|
| hsa_circ_0075001, circNPM1 | chr5:170817054-170827214 | NPM1 | Up |
| hsa_circ_0004277 | chr10:1125950-1126416 | WDR37 | Down |
| hsa_circ_0035381 | chr15:55621921-55634000 | PIGB | Up |
| hsa_circ_0004136 | chr6:73713630-73751785 | KCNQ5 | Up |
| hsa_circ_0058058 | chr2:216177220-216190861 | ATIC | Up |
| hsa_circ_0017446 | chr10:1125950-1132297 | WDR37 | Down |
| AP1G1 | RBL1 | ERLIN2 | DNAJC11 | SMARCA4 |
| VDAC1 | PIGM | RAN | CUEDC2 | PAFAH1B1 |
| ELAVL1 | UBE2I | SP1 | SLC16A1 | SETD5 |
| THAP11 | STRN | CHTOP | ARF3 | HTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misir, S.; Ozer Yaman, S.; Petrović, N.; Šami, A.; Akidan, O.; Hepokur, C.; Aliyazicioglu, Y. Identification of a Novel hsa_circ_0058058/miR-324-5p Axis and Prognostic/Predictive Molecules for Acute Myeloid Leukemia Outcome by Bioinformatics-Based Analysis. Biology 2024, 13, 487. https://doi.org/10.3390/biology13070487
Misir S, Ozer Yaman S, Petrović N, Šami A, Akidan O, Hepokur C, Aliyazicioglu Y. Identification of a Novel hsa_circ_0058058/miR-324-5p Axis and Prognostic/Predictive Molecules for Acute Myeloid Leukemia Outcome by Bioinformatics-Based Analysis. Biology. 2024; 13(7):487. https://doi.org/10.3390/biology13070487
Chicago/Turabian StyleMisir, Sema, Serap Ozer Yaman, Nina Petrović, Ahmad Šami, Osman Akidan, Ceylan Hepokur, and Yuksel Aliyazicioglu. 2024. "Identification of a Novel hsa_circ_0058058/miR-324-5p Axis and Prognostic/Predictive Molecules for Acute Myeloid Leukemia Outcome by Bioinformatics-Based Analysis" Biology 13, no. 7: 487. https://doi.org/10.3390/biology13070487
APA StyleMisir, S., Ozer Yaman, S., Petrović, N., Šami, A., Akidan, O., Hepokur, C., & Aliyazicioglu, Y. (2024). Identification of a Novel hsa_circ_0058058/miR-324-5p Axis and Prognostic/Predictive Molecules for Acute Myeloid Leukemia Outcome by Bioinformatics-Based Analysis. Biology, 13(7), 487. https://doi.org/10.3390/biology13070487