
Genomic Diversity and Recombination Analysis of the Spike Protein Gene from Selected Human Coronaviruses
 , , and
, , and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieval of Viral Genome Sequences
2.2. Genome Analyses of HCoVs
2.3. Recombination Pattern Analyses among HCoVs
3. Results
3.1. Genome Analyses of HCoVs
3.2. Phylogenetic Analyses
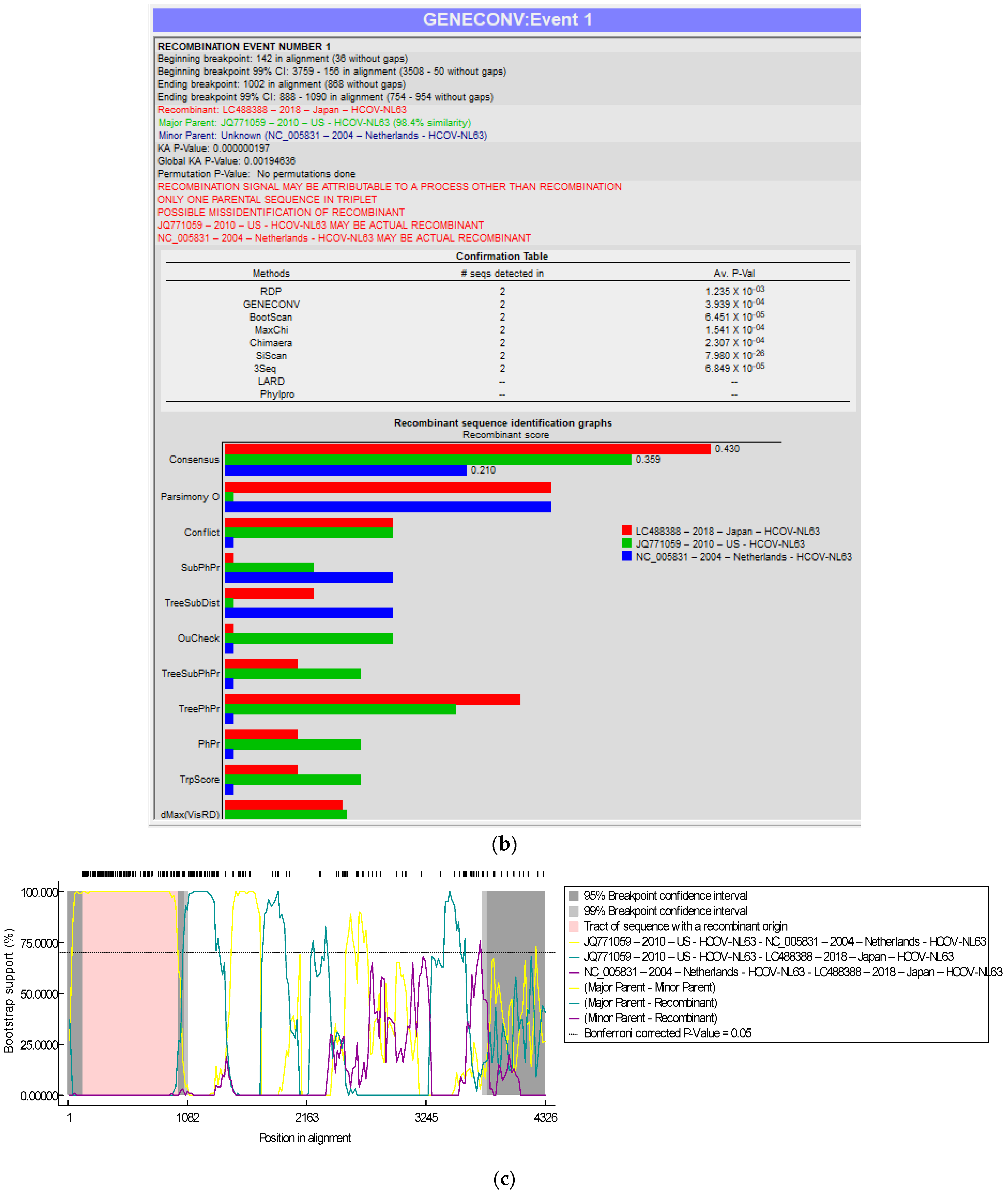
3.3. Recombination Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect. Genet. Evol. 2020, 85, 104502. [Google Scholar] [CrossRef] [PubMed]
- Cheng, V.C.; Lau, S.K.; Woo, P.C.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007, 20, 660–694. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Bolles, M.; Donaldson, E.; Baric, R. SARS-CoV and emergent coronaviruses: Viral determinants of interspecies transmission. Curr. Opin. Virol. 2011, 1, 624–634. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Yip, C.C.; Tse, H.; Tsoi, H.W.; Cheng, V.C.; Lee, P.; Tang, B.S.; Cheung, C.H.; Lee, R.A.; et al. Coronavirus HKU1 and other coronavirus infections in Hong Kong. J. Clin. Microbiol. 2006, 44, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Rasool, S.; Fielding, B.C. Understanding Human Coronavirus HCoV-NL63. Open Virol. J. 2010, 4, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human Coronaviruses: A Review of Virus-Host Interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Che, X.Y.; Di, B.; Zhao, G.P.; Wang, Y.D.; Qiu, L.W.; Hao, W.; Wang, M.; Qin, P.Z.; Liu, Y.F.; Chan, K.H.; et al. A patient with asymptomatic severe acute respiratory syndrome (SARS) and antigenemia from the 2003–2004 community outbreak of SARS in Guangzhou, China. Clin. Infect. Dis. 2006, 43, e1–e5. [Google Scholar] [CrossRef] [PubMed]
- Tsang, K.W.; Ho, P.L.; Ooi, G.C.; Yee, W.K.; Wang, T.; Chan-Yeung, M.; Lam, W.K.; Seto, W.H.; Yam, L.Y.; Cheung, T.M.; et al. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Peiris, M.; Perlman, S. Unresolved questions in the zoonotic transmission of MERS. Curr. Opin. Virol. 2022, 52, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Azhar, E.I.; Hashem, A.M.; El-Kafrawy, S.A.; Sohrab, S.S.; Aburizaiza, A.S.; Farraj, S.A.; Hassan, A.M.; Al-Saeed, M.S.; Jamjoom, G.A.; Madani, T.A. Detection of the Middle East respiratory syndrome coronavirus genome in an air sample originating from a camel barn owned by an infected patient. mBio 2014, 5, e01450-14. [Google Scholar] [CrossRef] [PubMed]
- Azhar, E.I.; El-Kafrawy, S.A.; Farraj, S.A.; Hassan, A.M.; Al-Saeed, M.S.; Hashem, A.M.; Madani, T.A. Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 2014, 370, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Meyer, B.; Muller, M.A.; Corman, V.M.; Al-Masri, M.; Hossain, R.; Madani, H.; Sieberg, A.; Bosch, B.J.; Lattwein, E.; et al. Transmission of MERS-coronavirus in household contacts. N. Engl. J. Med. 2014, 371, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.S.; Azhar, E.I.; Kim, Y.J.; Memish, Z.A.; Oh, M.D.; Zumla, A. Middle East respiratory syndrome coronavirus: Risk factors and determinants of primary, household, and nosocomial transmission. Lancet Infect. Dis. 2018, 18, e217–e227. [Google Scholar] [CrossRef] [PubMed]
- Reusken, C.B.; Messadi, L.; Feyisa, A.; Ularamu, H.; Godeke, G.J.; Danmarwa, A.; Dawo, F.; Jemli, M.; Melaku, S.; Shamaki, D.; et al. Geographic distribution of MERS coronavirus among dromedary camels, Africa. Emerg. Infect. Dis. 2014, 20, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.D.; Park, W.B.; Park, S.W.; Choe, P.G.; Bang, J.H.; Song, K.H.; Kim, E.S.; Kim, H.B.; Kim, N.J. Middle East respiratory syndrome: What we learned from the 2015 outbreak in the Republic of Korea. Korean J. Intern. Med. 2018, 33, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Sohrab, S.S.; Azhar, E.I. Genetic diversity of MERS-CoV spike protein gene in Saudi Arabia. J. Infect. Public. Health 2020, 13, 709–717. [Google Scholar] [CrossRef]
- Zhou, Z.; Hui, K.P.Y.; So, R.T.Y.; Lv, H.; Perera, R.; Chu, D.K.W.; Gelaye, E.; Oyas, H.; Njagi, O.; Abayneh, T.; et al. Phenotypic and genetic characterization of MERS coronaviruses from Africa to understand their zoonotic potential. Proc. Natl. Acad. Sci. USA 2021, 118, e2103984118. [Google Scholar] [CrossRef] [PubMed]
- Azhar, E.I.; Velavan, T.P.; Rungsung, I.; Traore, T.; Hui, D.S.; McCloskey, B.; El-Kafrawy, S.A.; Zumla, A. Middle East respiratory syndrome coronavirus-a 10-year (2012–2022) global analysis of human and camel infections, genomic sequences, lineages, and geographical origins. Int. J. Infect. Dis. 2023, 131, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Parkhe, P.; Verma, S. Evolution, Interspecies Transmission, and Zoonotic Significance of Animal Coronaviruses. Front. Vet. Sci. 2021, 8, 719834. [Google Scholar] [CrossRef] [PubMed]
- Solomon, M.; Liang, C. Human coronaviruses: The emergence of SARS-CoV-2 and management of COVID-19. Virus Res. 2022, 319, 198882. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Giorgi, E.E.; Marichann, M.H.; Foley, B.; Xiao, C.; Kong, X.P.; Chen, Y.; Korber, B.; Gao, F.; Gnanakaran, S. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Grunewald, M.; Perlman, S. Coronaviruses: An Updated Overview of Their Replication and Pathogenesis. In Coronaviruses; Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2203, pp. 1–29. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Park, Y.J.; Walls, A.C.; Wang, Z.; Sauer, M.M.; Li, W.; Tortorici, M.A.; Bosch, B.J.; DiMaio, F.; Veesler, D. Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat. Struct. Mol. Biol. 2019, 26, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Parra-Lucares, A.; Segura, P.; Rojas, V.; Pumarino, C.; Saint-Pierre, G.; Toro, L. Emergence of SARS-CoV-2 Variants in the World: How Could This Happen? Life 2022, 12, 194. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Mou, H.H.; Smits, S.L.; Dekkers, D.H.W.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further Evidence for Bats as the Evolutionary Source of Middle East Respiratory Syndrome Coronavirus. mBio 2017, 8, e00373-17. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.W.; Hui, K.P.Y.; Perera, R.; Miguel, E.; Niemeyer, D.; Zhao, J.; Channappanavar, R.; Dudas, G.; Oladipo, J.O.; Traore, A.; et al. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proc. Natl. Acad. Sci. USA 2018, 115, 3144–3149. [Google Scholar] [CrossRef] [PubMed]
- El-Kafrawy, S.A.; Corman, V.M.; Tolah, A.M.; Al Masaudi, S.B.; Hassan, A.M.; Muller, M.A.; Bleicker, T.; Harakeh, S.M.; Alzahrani, A.A.; Alsaaidi, G.A.; et al. Enzootic patterns of Middle East respiratory syndrome coronavirus in imported African and local Arabian dromedary camels: A prospective genomic study. Lancet Planet Health 2019, 3, e521–e528. [Google Scholar] [CrossRef] [PubMed]
- Ngere, I.; Hunsperger, E.A.; Tong, S.; Oyugi, J.; Jaoko, W.; Harcourt, J.L.; Thornburg, N.J.; Oyas, H.; Muturi, M.; Osoro, E.M.; et al. Outbreak of Middle East Respiratory Syndrome Coronavirus in Camels and Probable Spillover Infection to Humans in Kenya. Viruses 2022, 14, 1743. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ali, A.; Walelign, E.; Demissie, G.F.; El Masry, I.; Abayneh, T.; Getachew, B.; Krishnan, P.; Ng, D.Y.M.; Gardner, E.; et al. Genetic diversity and molecular epidemiology of Middle East Respiratory Syndrome Coronavirus in dromedaries in Ethiopia, 2017–2020. Emerg. Microbes Infect. 2023, 12, e2164218. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Meng, K.; Meng, G. Genomic recombination events may reveal the evolution of coronavirus and the origin of SARS-CoV-2. Sci. Rep. 2020, 10, 21617. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Yi, S.V. On the origin and evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.L.; Wu, C.C.; Li, X.; Song, Y.H.; Yao, X.M.; Wu, X.K.; Duan, Y.G.; Zhang, H.; Wang, Y.R.; Qian, Z.H.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Scovino, A.M.; Dahab, E.C.; Vieira, G.F.; Freire-de-Lima, L.; Freire-de-Lima, C.G.; Morrot, A. SARS-CoV-2’s Variants of Concern: A Brief Characterization. Front. Immunol. 2022, 13, 834098. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses 2022, 14, 1239. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sharma, K.; Shaw, D.; Bhargava, A.; Negi, S.S. Mosaic Recombination Inflicted Various SARS-CoV-2 Lineages to Emerge into Novel Virus Variants: A Review Update. Indian J. Clin. Biochem 2022, 38, 418–425. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Koopmans, M. Tracking SARS-CoV-2 variants and resources. Nat. Methods 2023, 20, 489–490. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef] [PubMed]
- Nikonova, A.A.; Faizuloev, E.B.; Gracheva, A.V.; Isakov, I.Y.; Zverev, V.V. Genetic Diversity and Evolution of the Biological Features of the Pandemic SARS-CoV-2. Acta Naturae 2021, 13, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Liu, Z.; Chen, D. Human coronaviruses: Origin, host and receptor. J. Clin. Virol. 2022, 155, 105246. [Google Scholar] [CrossRef] [PubMed]
- Chathappady House, N.N.; Palissery, S.; Sebastian, H. Corona Viruses: A Review on SARS, MERS and COVID-19. Microbiol. Insights 2021, 14, 11786361211002481. [Google Scholar] [CrossRef] [PubMed]
- Jo, W.K.; de Oliveira-Filho, E.F.; Rasche, A.; Greenwood, A.D.; Osterrieder, K.; Drexler, J.F. Potential zoonotic sources of SARS-CoV-2 infections. Transbound. Emerg. Dis. 2021, 68, 1824–1834. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Kan, B.; Wang, M.; Jing, H.; Xu, H.; Jiang, X.; Yan, M.; Liang, W.; Zheng, H.; Wan, K.; Liu, Q.; et al. Molecular evolution analysis and geographic investigation of severe acute respiratory syndrome coronavirus-like virus in palm civets at an animal market and on farms. J. Virol. 2005, 79, 11892–11900. [Google Scholar] [CrossRef] [PubMed]
- Gussow, A.B.; Auslander, N.; Faure, G.; Wolf, Y.I.; Zhang, F.; Koonin, E.V. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 15193–15199. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Kim, Y.J.; Park, S.H.; Yun, M.R.; Yang, J.S.; Kang, H.J.; Han, Y.W.; Lee, H.S.; Kim, H.M.; Kim, H.; et al. Variations in Spike Glycoprotein Gene of MERS-CoV, South Korea, 2015. Emerg. Infect. Dis. 2016, 22, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Cheon, S.; Min, C.K.; Sohn, K.M.; Kang, Y.J.; Cha, Y.J.; Kang, J.I.; Han, S.K.; Ha, N.Y.; Kim, G.; et al. Spread of Mutant Middle East Respiratory Syndrome Coronavirus with Reduced Affinity to Human CD26 during the South Korean Outbreak. mBio 2016, 7, e00019. [Google Scholar] [CrossRef] [PubMed]
- Omrani, A.S.; Al-Tawfiq, J.A.; Memish, Z.A. Middle East respiratory syndrome coronavirus (MERS-CoV): Animal to human interaction. Pathog. Glob. Health 2015, 109, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Amer, H.; Alqahtani, A.S.; Alzoman, H.; Aljerian, N.; Memish, Z.A. Unusual presentation of Middle East respiratory syndrome coronavirus leading to a large outbreak in Riyadh during 2017. Am. J. Infect. Control 2018, 46, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Assiri, A.M.; Midgley, C.M.; Abedi, G.R.; Bin Saeed, A.; Almasri, M.M.; Lu, X.; Al-Abdely, H.M.; Abdalla, O.; Mohammed, M.; Algarni, H.S.; et al. Epidemiology of a Novel Recombinant Middle East Respiratory Syndrome Coronavirus in Humans in Saudi Arabia. J. Infect. Dis. 2016, 214, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining host jump of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Dudas, G.; Carvalho, L.M.; Rambaut, A.; Bedford, T. MERS-CoV spillover at the camel-human interface. Elife 2018, 7, e31257. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.; Jia, N.; Zhang, Y.W.; Shum, M.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef]
- Borucki, M.K.; Lao, V.; Hwang, M.; Gardner, S.; Adney, D.; Munster, V.; Bowen, R.; Allen, J.E. Middle East Respiratory Syndrome Coronavirus Intra-Host Populations Are Characterized by Numerous High Frequency Variants. PLoS ONE 2016, 11, e0146251. [Google Scholar] [CrossRef]
- Cotten, M.; Watson, S.J.; Zumla, A.I.; Makhdoom, H.Q.; Palser, A.L.; Ong, S.H.; Al Rabeeah, A.A.; Alhakeem, R.F.; Assiri, A.; Al-Tawfiq, J.A.; et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio 2014, 5, e01062-13. [Google Scholar] [CrossRef] [PubMed]
- Tonkin-Hill, G.; Martincorena, I.; Amato, R.; Lawson, A.R.J.; Gerstung, M.; Johnston, I.; Jackson, D.K.; Park, N.; Lensing, S.V.; Quail, M.A.; et al. Patterns of within-host genetic diversity in SARS-CoV-2. Elife 2021, 10, e66857. [Google Scholar] [CrossRef] [PubMed]
- Varabyou, A.; Pockrandt, C.; Salzberg, S.L.; Pertea, M. Rapid detection of inter-clade recombination in SARS-CoV-2 with Bolotie. Genetics 2021, 218, iyab074. [Google Scholar] [CrossRef] [PubMed]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.; McBroome, J.; Ayala, N.; Ye, C.; Smith, K.; De Maio, N.; Haussler, D.; Lanfear, R.; et al. Pandemic-scale phylogenomics reveals the SARS-CoV-2 recombination landscape. Nature 2022, 609, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Preska Steinberg, A.; Silander, O.K.; Kussell, E. Correlated substitutions reveal SARS-like coronaviruses recombine frequently with a diverse set of structured gene pools. Proc. Natl. Acad. Sci. USA 2023, 120, e2206945119. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Accession Number | Virus | Country | Year | % Identity | |
|---|---|---|---|---|---|---|
| NT | AA | |||||
| 1 | NC_045512 | SARS-CoV-2 | China | 2019 | 99.9 | 99.9 |
| 2 | LC534419 | SARS-CoV-2 | Japan | 2020 | 99.9 | 99.9 |
| 3 | MT020781 | SARS-CoV-2 | Finland | 2020 | 99.9 | 99.8 |
| 4 | MT192759 | SARS-CoV-2 | Taiwan | 2020 | 99.9 | 99.9 |
| 5 | MT126808 | SARS-CoV-2 | Brazil | 2020 | 99.9 | 99.9 |
| 6 | MT093571 | SARS-CoV-2 | Sweden | 2020 | 99.9 | 99.8 |
| 7 | MT007544 | SARS-CoV-2 | Australia | 2020 | 99.9 | 98.8 |
| 8 | MW463458 | SARS-CoV-2 | Tunisia | 2021 | 99.9 | 99.9 |
| 9 | MW454390 | SARS-CoV-2 | USA | 2020 | 99.9 | 99.8 |
| 10 | MT731346 | SARS-CoV-2 | Bahrain | 2020 | 99.9 | 99.9 |
| 11 | MT762944 | SARS-CoV-2 | Bangladesh | 2020 | 99.9 | 99.9 |
| 12 | MT919769 | SARS-CoV-2 | Philippines | 2020 | 99.9 | 99.9 |
| 13 | MW315213 | SARS-CoV-2 | Mexico | 2020 | 99.9 | 99.8 |
| 14 | MW491251 | SARS-CoV-2 | Myanmar | 2020 | 99.8 | 99.8 |
| 15 | MW527392 | SARS-CoV-2 | Iraq | 2021 | 99.9 | 99.8 |
| 16 | OM984850 | SARS-CoV-2 | Thailand | 2022 | 99.0 | 97.6 |
| 17 | OL405084 | SARS-CoV-2 | France | 2022 | 99.5 | 99.2 |
| 18 | OM064634 | SARS-CoV-2 | Brazil | 2021 | 99.8 | 99.5 |
| 19 | NC_004718 | SARS-CoV-1 | Canada | 2003 | 72.9 | 75.5 |
| 20 | AP006561 | SARS-CoV-1 | Taiwan | 2003 | 72.9 | 75.6 |
| 21 | AY864806 | SARS-CoV-1 | China | 2004 | 73.0 | 75.7 |
| 22 | KF514423 | SARS-CoV-1 | USA | 2009 | 72.9 | 75.6 |
| 23 | AY291315 | SARS-CoV-1 | Germany | 2003 | 73.0 | 75.5 |
| 24 | JQ316196 | SARS-CoV-1 | UK | 2003 | 72.9 | 75.6 |
| 25 | AY427439 | SARS-CoV-1 | Italy | 2003 | 72.9 | 75.6 |
| 26 | AB263618 | SARS-CoV-1 | Japan | 2006 | 72.9 | 75.3 |
| 27 | NC_038294 | MERS-CoV | England | 2012 | 45.9 | 25.8 |
| 28 | KJ614529 | MERS-CoV | Jordon | 2012 | 45.8 | 25.9 |
| 29 | KF192507 | MERS-CoV | UAE | 2013 | 45.8 | 25.8 |
| 30 | KR912196 | MERS-CoV | KSA | 2014 | 45.8 | 25.8 |
| 31 | KT006149 | MERS-CoV | China | 2015 | 45.9 | 25.8 |
| 32 | MH978888 | MERS-CoV | South Korea | 2018 | 45.8 | 25.8 |
| 33 | MK280984 | MERS-CoV | Qatar | 2015 | 46.0 | 25.8 |
| 34 | KP223131 | MERS-CoV | USA | 2014 | 45.9 | 25.9 |
| 35 | KJ782549 | MERS-CoV | Greece/KSA | 2014 | 45.9 | 25.9 |
| 36 | KT156561 | MERS-CoV | Oman | 2013 | 45.9 | 25.9 |
| 37 | MH822886 | MERS-CoV | England/KSA | 2018 | 45.8 | 25.8 |
| 38 | NC_006213 | HCoV-OC43 | USA | 2004 | 40.9 | 26.3 |
| 39 | KX344031 | HCoV-OC43 | Mexico | 2011 | 40.8 | 26.4 |
| 40 | KF963244 | HCoV-OC43 | France | 2014 | 40.7 | 26.1 |
| 41 | MN026164 | HCoV-OC43 | Kenya | 2018 | 40.7 | 26.2 |
| 42 | MG197723 | HCoV-OC43 | China | 2016 | 40.8 | 26.2 |
| 43 | KX538979 | HCoV-OC43 | Malaysia | 2013 | 40.8 | 26.4 |
| 44 | LC315649 | HCoV-OC43 | Japan | 2016 | 40.7 | 26.2 |
| 45 | AY903458 | HCoV-OC43 | Belgium | 2004 | 40.5 | 26.3 |
| 46 | OK662397 | HCoV-OC43 | Finland | 2021 | 40.7 | 26.0 |
| 47 | OL770288 | HCoV-OC43 | Australia | 2019 | 40.7 | 26.4 |
| 48 | NC_005831 | HCoV-NL63 | Netherlands | 2004 | 37.5 | 19.8 |
| 49 | LC488388 | HCoV-NL63 | Japan | 2018 | 37.3 | 19.9 |
| 50 | JQ771059 | HCoV-NL63 | USA | 2010 | 37.7 | 19.6 |
| 51 | KX179496 | HCoV-NL63 | Haiti | 2015 | 37.5 | 19.6 |
| 52 | KM055650 | HCoV-NL63 | China | 2008 | 37.6 | 19.6 |
| 53 | MG428706 | HCoV-NL63 | Kenya | 2010 | 37.6 | 19.6 |
| 54 | MG772808 | HCoV-NL63 | S. Korea | 2014 | 37.5 | 20.1 |
| 55 | NC_006577 | HCoV-HKU1 | China | 2004 | 40.2 | 25.1 |
| 56 | LC315651 | HCoV-HKU1 | Japan | 2016 | 40.6 | 25.2 |
| 57 | MH940245 | HCoV-HKU1 | Thailand | 2017 | 40.7 | 25.2 |
| 58 | HM034837 | HCoV-HKU1 | France | 2005 | 40.1 | 25.0 |
| 59 | KF686346 | HCoV-HKU1 | USA | 2010 | 40.2 | 25.0 |
| 60 | KF430197 | HCoV-HKU1 | Brazil | 2007 | 40.7 | 25.1 |
| 61 | NC_002645 | HCoV-229E | Germany | 2000 | 32.4 | 19.9 |
| 62 | KY369914 | HCoV-229E | USA | 2016 | 32.3 | 19.8 |
| 63 | KM055560 | HCoV-229E | China | 2009 | 32.5 | 19.8 |
| 64 | DQ243986 | HCoV-229E | Australia | 2005 | 32.6 | 19.8 |
| 65 | AB691767 | HCoV-229E | Japan | 2008 | 32.5 | 19.8 |
| 66 | KF293666 | HCoV-229E | Sweden | 2013 | 32.4 | 19.8 |
| 67 | MH048989 | HCoV-229E | France | 2014 | 32.5 | 19.8 |
| 68 | JX503061 | HCoV-229E | Italy | 2009 | 32.6 | 19.9 |
| 69 | MF542265 | HCoV-229E | Haiti | 2016 | 32.4 | 19.8 |
| 70 | OK662398 | HCoV-229E | Finland | 2021 | 32.4 | 19.9 |
| S. No. | Accession Number | Virus | Country | Year | % Identity | |
|---|---|---|---|---|---|---|
| NT | AA | |||||
| 1 | KF958702 | MERS-CoV | KSA | 2013 | 99.7 | 99.9 |
| 2 | KM027292 | MERS-CoV | KSA | 2014 | 99.7 | 99.8 |
| 3 | KT806055 | MERS-CoV | KSA | 2015 | 99.5 | 99.7 |
| 4 | MH306207 | MERS-CoV | KSA | 2016 | 99.6 | 99.9 |
| 5 | MN723542 | MERS-CoV | KSA | 2017 | 99.5 | 99.8 |
| 6 | MK483839 | MERS-CoV | KSA | 2018 | 99.3 | 99.7 |
| 7 | MN120514 | MERS-CoV | KSA | 2019 | 99.3 | 99.7 |
| 8 | KJ829365 | MERS-CoV | US/KSA | 2014 | 99.7 | 99.7 |
| 9 | KC776174 | MERS-CoV | Jordan | 2012 | 99.7 | 99.7 |
| 10 | KT861633 | MERS-CoV | Jordan | 2014 | 99.7 | 99.8 |
| 11 | MK052676 | MERS-CoV | Jordan | 2014 | 99.7 | 99.8 |
| 12 | MF741825 | MERS-CoV | Jordan | 2015 | 99.6 | 99.9 |
| 13 | KY688114 | MERS-CoV | Oman | 2015 | 99.6 | 99.7 |
| 14 | KJ477102 | MERS-CoV | Egypt | 2013 | 99.2 | 99.0 |
| 15 | KT182957 | MERS-CoV | S. Korea | 2015 | 99.5 | 99.7 |
| 16 | MK129253 | MERS-CoV | S. Korea | 2018 | 99.5 | 99.9 |
| 17 | KT036374 | MERS-CoV | China | 2015 | 99.5 | 99.9 |
| 18 | KT003530 | MERS-CoV | China | 2015 | 99.4 | 99.7 |
| 19 | KP209312 | MERS-CoV | UAE | 2013 | 99.6 | 99.7 |
| 20 | KP209313 | MERS-CoV | UAE | 2014 | 99.6 | 99.7 |
| 21 | KU201953 | MERS-CoV | Nigeria | 2015 | 99.5 | 99.4 |
| 22 | KF811036 | MERS-CoV | Tunisia | 2013 | 99.7 | 99.7 |
| 23 | KT225476 | MERS-CoV | Thailand | 2015 | 99.6 | 99.8 |
| 24 | KJ361500 | MERS-CoV | France | 2013 | 99.7 | 99.7 |
| 25 | KF192507 | MERS-CoV | Germany | 2013 | 99.7 | 99.8 |
| 26 | KJ813439 | MERS-CoV | USA | 2014 | 99.7 | 99.8 |
| 27 | KY983588 | HCoV-OC43 | USA | 2015 | 46.1 | 28.8 |
| 28 | MH121121 | HCoV-OC43 | USA | 2016 | 46.1 | 28.8 |
| 29 | KF530093 | HCoV-OC43 | USA | 1983 | 46.2 | 28.7 |
| 30 | MW202339 | HCoV-OC43 | USA | 2018 | 45.7 | 28.5 |
| 31 | KF530083 | HCoV-OC43 | USA | 1987 | 46.1 | 28.7 |
| 32 | KF923925 | HCoV-OC43 | China | 2010 | 46.1 | 28.8 |
| 33 | KF572872 | HCoV-OC43 | China | 2008 | 46.0 | 28.9 |
| 34 | KU745548 | HCoV-OC43 | China | 2014 | 46.2 | 28.8 |
| 35 | OK500335 | HCoV-OC43 | China | 2021 | 45.8 | 28.5 |
| 36 | OK500334 | HCoV-OC43 | China | 2021 | 45.8 | 28.5 |
| 37 | AY585229 | HCoV-OC43 | France | 2004 | 46.0 | 28.6 |
| 38 | KF963233 | HCoV-OC43 | France | 2002 | 46.1 | 28.6 |
| 39 | KF963229 | HCoV-OC43 | France | 1967 | 46.0 | 28.6 |
| 40 | MK303625 | HCoV-OC43 | France | 2018 | 45.8 | 28.5 |
| 41 | KX538964 | HCoV-OC43 | Malaysia | 2012 | 46.1 | 28.8 |
| 42 | AY903460 | HCoV-OC43 | Belgium | 2004 | 45.8 | 28.9 |
| 43 | FJ211861 | HCoV-NL63 | USA | 2008 | 38.8 | 18.3 |
| 44 | KY829118 | HCoV-NL63 | USA | 2015 | 39.0 | 18.3 |
| 45 | JQ765575 | HCoV-NL63 | USA | 2005 | 38.9 | 18.3 |
| 46 | KY554971 | HCoV-NL63 | USA | 2016 | 39.0 | 18.3 |
| 47 | MN306040 | HCoV-NL63 | USA | 2019 | 39.0 | 18.3 |
| 48 | MK334047 | HCoV-NL63 | China | 2018 | 38.7 | 18.3 |
| 49 | OK073076 | HCoV-NL63 | China | 2017 | 39.0 | 18.4 |
| 50 | JX524171 | HCoV-NL63 | China | 2009 | 39.0 | 18.4 |
| 51 | JX104161 | HCoV-NL63 | China | 2008 | 39.0 | 18.4 |
| 52 | DQ445911 | HCoV-NL63 | Netherlands | 2006 | 38.7 | 18.3 |
| 53 | DQ445912 | HCoV-NL63 | Netherland | 2006 | 38.8 | 18.3 |
| 54 | LC720428 | HCoV-NL63 | Japan | 2019 | 39.0 | 18.3 |
| 55 | OK625405 | HCoV-NL63 | S. Korea | 2017 | 38.9 | 18.3 |
| 56 | MZ682627 | HCoV-NL63 | S. Korea | 2017 | 38.9 | 18.3 |
| 57 | MN026166 | HCoV-NL63 | Kenya | 2018 | 38.8 | 18.3 |
| 58 | DQ437619 | HCoV-HKU1 | China | 2006 | 45.7 | 28.1 |
| 59 | KT779556 | HCoV-HKU1 | China | 2009 | 45.6 | 27.9 |
| 60 | AY884001 | HCoV-HKU1 | China | 2005 | 45.5 | 28.0 |
| 61 | DQ339101 | HCoV-HKU1 | China | 2005 | 45.5 | 28.1 |
| 62 | DQ415900 | HCoV-HKU1 | China | 2006 | 45.7 | 2.8.1 |
| 63 | MH557024 | HCoV-HKU1 | China | 2011 | 45.3 | 27.9 |
| 64 | MK167038 | HCoV-HKU1 | USA | 2017 | 45.5 | 28.0 |
| 65 | KF686340 | HCoV-HKU1 | USA | 2009 | 45.7 | 28.1 |
| 66 | KF514433 | HCoV-229E | USA | 1993 | 34.7 | 18.2 |
| 67 | MN306046 | HCoV-229E | USA | 2019 | 34.7 | 18.2 |
| 68 | AF344189 | HCoV-229E | USA | 2001 | 34.7 | 18.1 |
| 69 | AF344186 | HCoV-229E | USA | 2001 | 34.7 | 18.2 |
| 70 | KF514429 | HCoV-229E | USA | 1989 | 34.7 | 18.2 |
| 71 | KM055559 | HCoV-229E | China | 2005 | 34.9 | 18.3 |
| 72 | MW532107 | HCoV-229E | China | 2009 | 34.7 | 18.2 |
| 73 | MW532103 | HCoV-229E | China | 2017 | 34.7 | 18.1 |
| 74 | KM055544 | HCoV-229E | China | 2011 | 34.8 | 18.2 |
| 75 | KM055552 | HCoV-229E | China | 2007 | 34.8 | 18.2 |
| 76 | DQ243963 | HCoV-229E | Australia | 2005 | 34.7 | 18.2 |
| 77 | DQ243979 | HCoV-229E | Australia | 2002 | 34.8 | 18.3 |
| 78 | DQ243968 | HCoV-229E | Australia | 2005 | 34.6 | 18.2 |
| 79 | KU291448 | HCoV-229E | Germany | 2015 | 34.8 | 18.1 |
| 80 | AB691766 | HCoV-229E | Japan | 2004 | 34.7 | 18.2 |
| 81 | KF293664 | HCoV-229E | Sweden | 2013 | 34.7 | 18.3 |
| S. No. | Accession Number | Virus | Country | Year | % Identity | |
|---|---|---|---|---|---|---|
| NT | AA | |||||
| 1 | MW181733 | SARS-CoV-2 | China | 2020 | 99.9 | 99.9 |
| 2 | MW181764 | SARS-CoV-2 | China | 2020 | 99.9 | 99.9 |
| 3 | MN908947 | SARS-CoV-2 | China | 2019 | 99.9 | 99.9 |
| 4 | MT049951 | SARS-CoV-2 | China | 2020 | 99.9 | 99.8 |
| 5 | MN996531 | SARS-CoV-2 | China | 2019 | 99.9 | 99.9 |
| 6 | MT512645 | SARS-CoV-2 | USA | 2020 | 99.9 | 99.9 |
| 7 | MW035459 | SARS-CoV-2 | USA | 2020 | 99.9 | 99.9 |
| 8 | MW310427 | SARS-CoV-2 | USA | 2020 | 99.9 | 99.9 |
| 9 | MW374912 | SARS-CoV-2 | USA | 2020 | 99.9 | 99.8 |
| 10 | MW486391 | SARS-CoV-2 | USA | 2021 | 99.9 | 99.9 |
| 11 | MW251511 | SARS-CoV-2 | Tunisia | 2020 | 99.9 | 99.8 |
| 12 | MW404672 | SARS-CoV-2 | Tunisia | 2020 | 99.9 | 99.8 |
| 13 | OM984824 | SARS-CoV-2 | Thailand | 2022 | 99.0 | 97.6 |
| 14 | OM984776 | SARS-CoV-2 | Thailand | 2022 | 98.5 | 96.9 |
| 15 | OP684303 | SARS-CoV-2 | Vietnam | 2022 | 98.8 | 97.4 |
| 16 | ON025123 | SARS-CoV-2 | Vietnam | 2022 | 98.6 | 96.8 |
| 17 | AY291451 | SARS-CoV1 | Taiwan | 2003 | 72.9 | 75.6 |
| 18 | AY274119 | SARS-CoV1 | Canada | 2003 | 72.9 | 75.5 |
| 19 | AY463060 | SARS-CoV1 | China | 2003 | 72.9 | 75.4 |
| 20 | AY278489 | SARS-CoV1 | China | 2003 | 72.9 | 75.7 |
| 21 | AY390556 | SARS-CoV1 | China | 2003 | 73.0 | 75.8 |
| 22 | AY508724 | SARS-CoV1 | China | 2003 | 72.9 | 75.5 |
| 23 | AY282752 | SARS-CoV1 | China | 2003 | 72.9 | 75.6 |
| 24 | FJ882963 | SARS-CoV1 | USA | 2004 | 72.9 | 75.6 |
| 25 | GU553365 | SARS-CoV1 | USA | 2003 | 72.9 | 75.6 |
| 26 | AY714217 | SARS-CoV1 | USA | 2004 | 72.9 | 75.5 |
| 27 | MK062182 | SARS-CoV1 | USA | 2017 | 72.9 | 75.5 |
| 28 | MK062184 | SARS-CoV1 | USA | 2017 | 72.9 | 75.5 |
| 29 | AY348314 | SARS-CoV1 | Taiwan | 2003 | 72.9 | 75.6 |
| 30 | AY338175 | SARS-CoV1 | Taiwan | 2003 | 72.9 | 75.6 |
| 31 | AY310120 | SARS-CoV1 | Germany | 2003 | 72.9 | 75.5 |
| 32 | AY323977 | SARS-CoV1 | Italy | 2003 | 72.9 | 75.6 |
| 33 | KY983588 | HCoV-OC43 | USA | 2015 | 42.4 | 26.4 |
| 34 | MH121121 | HCoV-OC43 | USA | 2016 | 42.4 | 26.4 |
| 35 | KF530093 | HCoV-OC43 | USA | 1983 | 42.7 | 26.6 |
| 36 | MW202339 | HCoV-OC43 | USA | 2018 | 42.7 | 26.5 |
| 37 | KF530083 | HCoV-OC43 | USA | 1987 | 42.7 | 26.6 |
| 38 | KF923925 | HCoV-OC43 | China | 2010 | 42.4 | 26.5 |
| 39 | KF572872 | HCoV-OC43 | China | 2008 | 42.5 | 26.5 |
| 40 | KU745548 | HCoV-OC43 | China | 2014 | 42.4 | 26.4 |
| 41 | OK500335 | HCoV-OC43 | China | 2021 | 42.6 | 26.0 |
| 42 | OK500334 | HCoV-OC43 | China | 2021 | 42.6 | 26.0 |
| 43 | AY585229 | HCoV-OC43 | France | 2004 | 42.7 | 26.5 |
| 44 | KF963233 | HCoV-OC43 | France | 2002 | 42.8 | 26.5 |
| 45 | KF963229 | HCoV-OC43 | France | 1967 | 42.7 | 26.5 |
| 46 | MK303625 | HCoV-OC43 | France | 2018 | 42.8 | 26.5 |
| 47 | KX538964 | HCoV-OC43 | Malaysia | 2012 | 42.4 | 26.4 |
| 48 | AY903460 | HCoV-NL63 | Belgium | 2004 | 42.4 | 26.4 |
| 49 | FJ211861 | HCoV-NL63 | USA | 2008 | 38.2 | 18.7 |
| 50 | KY829118 | HCoV-NL63 | USA | 2015 | 38.5 | 18.6 |
| 51 | JQ765575 | HCoV-NL63 | USA | 2005 | 38.3 | 18.6 |
| 52 | KY554971 | HCoV-NL63 | USA | 2016 | 38.6 | 18.6 |
| 53 | MN306040 | HCoV-NL63 | USA | 2019 | 38.1 | 18.5 |
| 54 | MK334047 | HCoV-NL63 | China | 2018 | 38.2 | 18.5 |
| 55 | OK073076 | HCoV-NL63 | China | 2017 | 38.3 | 18.6 |
| 56 | JX524171 | HCoV-NL63 | China | 2009 | 38.2 | 18.6 |
| 57 | JX104161 | HCoV-NL63 | China | 2008 | 38.3 | 18.6 |
| 58 | DQ445911 | HCoV-NL63 | Netherlands | 2006 | 38.4 | 18.7 |
| 59 | DQ445912 | HCoV-NL63 | Netherland | 2006 | 38.5 | 18.7 |
| 60 | LC720428 | HCoV-NL63 | Japan | 2019 | 38.2 | 18.6 |
| 61 | OK625405 | HCOV-NL63 | S. Korea | 2017 | 38.3 | 18.7 |
| 62 | MZ682627 | HCoV-NL63 | S. Korea | 2017 | 38.3 | 18.7 |
| 63 | MN026166 | HCoV-NL63 | Kenya | 2018 | 38.4 | 18.7 |
| 64 | DQ437619 | HCoV-HKU1 | China | 2006 | 42.2 | 24.8 |
| 65 | KT779556 | HCoV-HKU1 | China | 2009 | 42.2 | 24.8 |
| 66 | AY884001 | HCoV-HKU1 | China | 2005 | 42.2 | 25.0 |
| 67 | DQ339101 | HCoV-HKU1 | China | 2005 | 42.3 | 25.1 |
| 68 | DQ415900 | HCoV-HKU1 | China | 2006 | 42.2 | 24.8 |
| 69 | MH557024 | HCoV-HKU1 | China | 2011 | 42.2 | 25.0 |
| 70 | MK167038 | HCoV-HKU1 | USA | 2017 | 42.0 | 25.0 |
| 71 | KF686340 | HCoV-HKU1 | USA | 2009 | 42.2 | 24.8 |
| 72 | KF514433 | HCoV-229E | USA | 1993 | 33.0 | 19.6 |
| 73 | MN306046 | HCoV-229E | USA | 2019 | 32.7 | 19.6 |
| 74 | AF344189 | HCoV-229E | USA | 2001 | 32.8 | 19.6 |
| 75 | AF344186 | HCoV-229E | USA | 2001 | 32.9 | 19.5 |
| 76 | KF514429 | HCoV-229E | USA | 1989 | 33.0 | 19.6 |
| 77 | KM055559 | HCoV-229E | China | 2005 | 32.9 | 19.6 |
| 78 | MW532107 | HCoV-229E | China | 2009 | 32.8 | 19.6 |
| 79 | MW532103 | HCoV-229E | China | 2017 | 32.8 | 19.6 |
| 80 | KM055544 | HCoV-229E | China | 2011 | 32.9 | 19.6 |
| 81 | KM055552 | HCoV-229E | China | 2007 | 32.9 | 19.6 |
| 82 | DQ243963 | HCoV-229E | Australia | 2005 | 32.9 | 19.5 |
| 83 | DQ243979 | HCoV-229E | Australia | 2002 | 32.9 | 19.6 |
| 84 | DQ243968 | HCoV-229E | Australia | 2005 | 32.9 | 19.6 |
| 85 | KU291448 | HCoV-229E | Germany | 2015 | 32.8 | 19.5 |
| 86 | AB691766 | HCoV-229E | Japan | 2004 | 32.8 | 19.6 |
| 87 | KF293664 | HCoV-229E | Sweden | 2013 | 32.8 | 19.4 |
| S. No. | Accession Number | Virus | Country | Year | % Identity | |
|---|---|---|---|---|---|---|
| NT | AA | |||||
| 1 | KF958702 | MERS-CoV | KSA | 2013 | 99.7 | 99.9 |
| 2 | KM027292 | MERS-CoV | KSA | 2014 | 99.7 | 99.8 |
| 3 | KT806055 | MERS-CoV | KSA | 2015 | 99.5 | 99.7 |
| 4 | MH306207 | MERS-CoV | KSA | 2016 | 99.6 | 99.9 |
| 5 | MN723542 | MERS-CoV | KSA | 2017 | 99.5 | 99.8 |
| 6 | MK483839 | MERS-CoV | KSA | 2018 | 99.3 | 99.7 |
| 7 | MN120514 | MERS-CoV | KSA | 2019 | 99.3 | 99.7 |
| 8 | KJ829365 | MERS-CoV | USA/KSA | 2014 | 99.7 | 99.7 |
| 9 | KC776174 | MERS-CoV | Jordan | 2012 | 99.7 | 99.7 |
| 10 | KT861633 | MERS-CoV | Jordan | 2014 | 99.7 | 99.8 |
| 11 | MK052676 | MERS-CoV | Jordan | 2014 | 99.7 | 99.8 |
| 12 | MF741825 | MERS-CoV | Jordan | 2015 | 99.6 | 99.9 |
| 13 | KY688114 | MERS-CoV | Oman | 2015 | 99.6 | 99.7 |
| 14 | KJ477102 | MERS-CoV | Egypt | 2013 | 99.2 | 99.0 |
| 15 | KT182957 | MERS-CoV | S. Korea | 2015 | 99.5 | 99.7 |
| 16 | MK129253 | MERS-CoV | S. Korea | 2018 | 99.5 | 99.9 |
| 17 | KT036374 | MERS-CoV | China | 2015 | 99.5 | 99.9 |
| 18 | KT003530 | MERS-CoV | China | 2015 | 99.4 | 99.7 |
| 19 | KP209312 | MERS-CoV | UAE | 2013 | 99.6 | 99.7 |
| 20 | KP209313 | MERS-CoV | UAE | 2014 | 99.6 | 99.7 |
| 21 | KU201953 | MERS-CoV | Nigeria | 2015 | 99.5 | 99.4 |
| 22 | KF811036 | MERS-CoV | Tunisia | 2013 | 99.7 | 99.7 |
| 23 | KT225476 | MERS-CoV | Thailand | 2015 | 99.6 | 99.8 |
| 24 | KJ361500 | MERS-CoV | France | 2013 | 99.7 | 99.7 |
| 25 | KF192507 | MERS-CoV | Germany | 2013 | 99.7 | 99.8 |
| 26 | KJ813439 | MERS-CoV | USA | 2014 | 99.7 | 99.8 |
| 27 | MW837148 | SARS-CoV-2 | KSA | 2020 | 44.2 | 26.7 |
| 28 | NC_045512 | SARS-CoV-2 | China | 2019 | 44.2 | 26.7 |
| 29 | LC534419 | SARS-CoV-2 | Japan | 2020 | 44.2 | 26.7 |
| 30 | MT020781 | SARS-CoV-2 | Finland | 2020 | 44.2 | 26.7 |
| 31 | MT192759 | SARS-CoV-2 | Taiwan | 2020 | 44.2 | 26.7 |
| 32 | MT126808 | SARS-CoV-2 | Brazil | 2020 | 44.2 | 26.7 |
| 33 | MT093571 | SARS-CoV-2 | Sweden | 2020 | 44.2 | 26.6 |
| 34 | MT007544 | SARS-CoV-2 | Australia | 2020 | 44.2 | 26.7 |
| 35 | MW463458 | SARS-CoV-2 | Tunisia | 2021 | 44.2 | 26.7 |
| 36 | MW454390 | SARS-CoV-2 | USA | 2020 | 44.2 | 26.7 |
| 37 | MT731346 | SARS-CoV-2 | Bahrain | 2020 | 44.2 | 26.7 |
| 38 | MT762944 | SARS-CoV-2 | Bangladesh | 2020 | 44.2 | 26.7 |
| 39 | MT919769 | SARS-CoV-2 | Philippines | 2020 | 44.2 | 26.7 |
| 40 | MW315213 | SARS-CoV-2 | Mexico | 2020 | 44.2 | 26.7 |
| 41 | MW491251 | SARS-CoV-2 | Myanmar | 2020 | 44.2 | 26.7 |
| 42 | MW527392 | SARS-CoV-2 | Iraq | 2021 | 44.2 | 26.7 |
| 43 | OM984850 | SARS-CoV-2 | Thailand | 2022 | 44.0 | 26.6 |
| 44 | OL405084 | SARS-CoV-2 | France | 2022 | 44.0 | 26.6 |
| 45 | OM064634 | SARS-CoV-2 | Brazil | 2021 | 44.1 | 26.6 |
| 46 | NC_004718 | SARS-CoV1 | Canada | 2003 | 44.8 | 26.5 |
| 47 | AP006561 | SARS-CoV1 | Taiwan | 2003 | 44.9 | 26.5 |
| 48 | AY864806 | SARS-CoV1 | China | 2004 | 44.8 | 26.4 |
| 49 | KF514423 | SARS-CoV1 | USA | 2009 | 44.8 | 26.5 |
| 50 | AY291315 | SARS-CoV1 | Germany | 2003 | 44.9 | 26.6 |
| 51 | JQ316196 | SARS-CoV1 | UK | 2003 | 44.9 | 26.5 |
| 52 | AY427439 | SARS-CoV1 | Italy | 2003 | 44.9 | 26.5 |
| 53 | AB263618 | SARS-CoV1 | Japan | 2006 | 44.9 | 26.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sohrab, S.S.; Alsaqaf, F.; Hassan, A.M.; Tolah, A.M.; Bajrai, L.H.; Azhar, E.I. Genomic Diversity and Recombination Analysis of the Spike Protein Gene from Selected Human Coronaviruses. Biology 2024, 13, 282. https://doi.org/10.3390/biology13040282
Sohrab SS, Alsaqaf F, Hassan AM, Tolah AM, Bajrai LH, Azhar EI. Genomic Diversity and Recombination Analysis of the Spike Protein Gene from Selected Human Coronaviruses. Biology. 2024; 13(4):282. https://doi.org/10.3390/biology13040282
Chicago/Turabian StyleSohrab, Sayed Sartaj, Fatima Alsaqaf, Ahmed Mohamed Hassan, Ahmed Majdi Tolah, Leena Hussein Bajrai, and Esam Ibraheem Azhar. 2024. "Genomic Diversity and Recombination Analysis of the Spike Protein Gene from Selected Human Coronaviruses" Biology 13, no. 4: 282. https://doi.org/10.3390/biology13040282
APA StyleSohrab, S. S., Alsaqaf, F., Hassan, A. M., Tolah, A. M., Bajrai, L. H., & Azhar, E. I. (2024). Genomic Diversity and Recombination Analysis of the Spike Protein Gene from Selected Human Coronaviruses. Biology, 13(4), 282. https://doi.org/10.3390/biology13040282