
Muscle Transcriptome Analysis of Mink at Different Growth Stages Using RNA-Seq

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples Selection and Preparation
2.2. RNA Extraction and Quality Control
2.3. Transcriptome Library Construction and Sequencing
2.4. Reads Mapping to the Reference Genome
2.5. Gene Expression Analysis
2.6. Quantitative RT-PCR Analysis
3. Results
3.1. Overview of the Mink Transcriptome
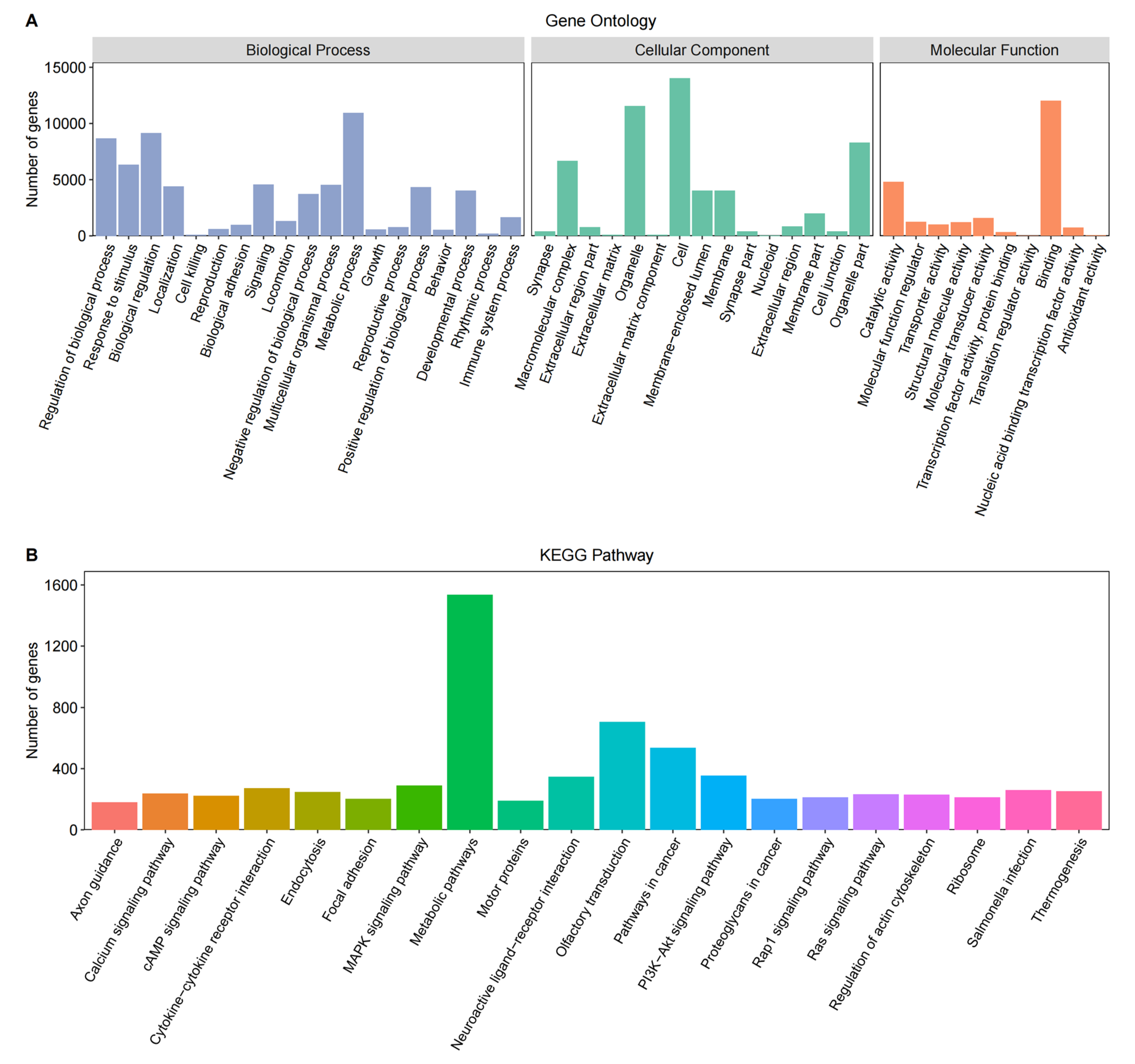
3.2. Gene Functional Annotation
3.3. Identifying the Differentially Expressed Genes
3.4. Functional Analysis of the Differentially Expressed Genes
3.5. qRT-PCR Validation of DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thirstrup, J.P.; Jensen, J.; Lund, M.S. Genetic parameters for fur quality graded on live animals and dried pelts of American mink (Neovison vison). J. Anim. Breed. Genet. 2017, 134, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Valipour, S.; Karimi, K.; Barrett, D.; Do, D.N.; Hu, G.; Sargolzaei, M.; Wang, Z.; Miar, Y. Genetic and Phenotypic Parameters for Pelt Quality and Body Length and Weight Traits in American Mink. Animals 2022, 12, 3184. [Google Scholar] [CrossRef] [PubMed]
- Cendron, F.; Cassandro, M.; Penasa, M. Genome-wide investigation to assess copy number variants in the Italian local chicken population. J. Anim. Sci. Biotechnol. 2024, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Arikawa, L.M.; Mota, L.F.M.; Schmidt, P.I.; Frezarim, G.B.; Fonseca, L.F.S.; Magalhaes, A.F.B.; Silva, D.A.; Carvalheiro, R.; Chardulo, L.A.L.; Albuquerque, L.G. Genome-wide scans identify biological and metabolic pathways regulating carcass and meat quality traits in beef cattle. Meat Sci. 2024, 209, 109402. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sun, G.; Mu, X.; Li, X.; Wang, J.; Zhao, M.; Zhang, G.; Ji, R.; Chen, C.; Gao, G. Genome-wide selective signatures mining the candidate genes for egg laying in goose. BMC Genom. 2023, 24, 750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Yuan, M.; Zhan, F.; Song, M.; Shang, P.; Yang, F.; Li, X.; Qiao, R.; Han, X.; et al. Genome-Wide Association Studies and Runs of Homozygosity to Identify Reproduction-Related Genes in Yorkshire Pig Population. Genes 2023, 14, 2133. [Google Scholar] [CrossRef] [PubMed]
- Liufu, S.; Lan, Q.; Liu, X.; Chen, B.; Xu, X.; Ai, N.; Li, X.; Yu, Z.; Ma, H. Transcriptome Analysis Reveals the Age-Related Developmental Dynamics Pattern of the Longissimus Dorsi Muscle in Ningxiang Pigs. Genes 2023, 14, 1050. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Petersen, B.; Sahana, G.; Madsen, L.B.; Larsen, K.; Thomsen, B.; Bendixen, C.; Lund, M.S.; Guldbrandtsen, B.; Panitz, F. The first draft reference genome of the American mink (Neovison vison). Sci. Rep. 2017, 7, 14564. [Google Scholar] [CrossRef] [PubMed]
- Karimi, K.; Do, D.N.; Wang, J.; Easley, J.; Borzouie, S.; Sargolzaei, M.; Plastow, G.; Wang, Z.; Miar, Y. A chromosome-level genome assembly reveals genomic characteristics of the American mink (Neogale vison). Commun. Biol. 2022, 5, 1381. [Google Scholar] [CrossRef]
- Shang, P.; Wang, Z.; Chamba, Y.; Zhang, B.; Zhang, H.; Wu, C. A comparison of prenatal muscle transcriptome and proteome profiles between pigs with divergent growth phenotypes. J. Cell Biochem. 2019, 120, 5277–5286. [Google Scholar] [CrossRef]
- Wang, X.; Xiao, Y.; Yang, H.; Lu, L.; Liu, X.; Lyu, W. Transcriptome Analysis Reveals the Genes Involved in Growth and Metabolism in Muscovy Ducks. BioMed Res. Int. 2021, 2021, 6648435. [Google Scholar] [CrossRef]
- Tang, J.; Shen, X.; Ouyang, H.; Luo, W.; Huang, Y.; Tian, Y.; Zhang, X. Transcriptome analysis of pituitary gland revealed candidate genes and gene networks regulating the growth and development in goose. Anim. Biotechnol. 2022, 33, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Ouyang, H.; Chen, X.; Jiang, D.; Tian, Y.; Huang, Y.; Shen, X. Comparative Transcriptome Analyses of Leg Muscle during Early Growth between Geese (Anser cygnoides) Breeds Differing in Body Size Characteristics. Genes 2023, 14, 1048. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Li, S.; Bao, G.; Wang, J.; Liu, X.; Hu, J.; Zhao, F.; Zhao, Z.; Shi, B.; Luo, Y. Comparative Transcriptome Analysis Reveals the Mechanism Associated With Dynamic Changes in Meat Quality of the Longissimus Thoracis Muscle in Tibetan Sheep at Different Growth Stages. Front. Vet. Sci. 2022, 9, 926725. [Google Scholar] [CrossRef] [PubMed]
- Rong, M.; Tu, J.; Xu, J. Comparison of growth and development and study on fit-ting of growth curves in different colored minks. Heilongjiang Anim. Sci. Vet. Med. 2018, 9, 184–186. [Google Scholar] [CrossRef]
- Kawai, Y.; Imada, K.; Akamatsu, S.; Zhang, F.; Seiler, R.; Hayashi, T.; Leong, J.; Beraldi, E.; Saxena, N.; Kretschmer, A.; et al. Paternally Expressed Gene 10 (PEG10) Promotes Growth, Invasion, and Survival of Bladder Cancer. Mol. Cancer Ther. 2020, 19, 2210–2220. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gui, W.; Tan, B.; Du, Y.; Zhou, J.; Wu, F.; Li, H.; Lin, X. IGF2 deficiency causes mitochondrial defects in skeletal muscle. Clin. Sci. 2021, 135, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Denda, K.; Nakao-Wakabayashi, K.; Okamoto, N.; Kitamura, N.; Ryu, J.Y.; Tagawa, Y.I.; Ichisaka, T.; Yamanaka, S.; Komada, M. Nrk, an X-linked protein kinase in the germinal center kinase family, is required for placental development and fetoplacental induction of labor. J. Biol. Chem. 2011, 286, 28802–28810. [Google Scholar] [CrossRef] [PubMed]
- Kanai-Azuma, M.; Kanai, Y.; Okamoto, M.; Hayashi, Y.; Yonekawa, H.; Yazaki, K. Nrk: A murine X-linked NIK (Nck-interacting kinase)-related kinase gene expressed in skeletal muscle. Mech. Dev. 1999, 89, 155–159. [Google Scholar] [CrossRef]
- Monk, D.; Sanches, R.; Arnaud, P.; Apostolidou, S.; Hills, F.A.; Abu-Amero, S.; Murrell, A.; Friess, H.; Reik, W.; Stanier, P.; et al. Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum. Mol. Genet. 2006, 15, 1259–1269. [Google Scholar] [CrossRef]
- Zanou, N.; Gailly, P. Skeletal muscle hypertrophy and regeneration: Interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell Mol. Life Sci. 2013, 70, 4117–4130. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Xu, H.; Lu, Y.; He, Q.; Yan, C.; Zhao, X.; Tian, Y.; Yang, C.; Zhang, Z.; Qiu, M.; et al. MUSTN1 is an indispensable factor in the proliferation, differentiation and apoptosis of skeletal muscle satellite cells in chicken. Exp. Cell Res. 2021, 407, 112833. [Google Scholar] [CrossRef]
- Ayuso, M.; Fernandez, A.; Nunez, Y.; Benitez, R.; Isabel, B.; Fernandez, A.I.; Rey, A.I.; Gonzalez-Bulnes, A.; Medrano, J.F.; Canovas, A.; et al. Developmental Stage, Muscle and Genetic Type Modify Muscle Transcriptome in Pigs: Effects on Gene Expression and Regulatory Factors Involved in Growth and Metabolism. PLoS ONE 2016, 11, e0167858. [Google Scholar] [CrossRef] [PubMed]
- Lucifero, D.; Chaillet, J.; Trasler, J. Potential significance of genomic imprinting defects for reproduction and assisted reproductive technology. Hum. Reprod. Update 2004, 10, 3–18. [Google Scholar] [CrossRef]
- Xu, F.; Jiang, W.; Zhang, T.; Jiang, Q.; Zhang, R.; Bi, H. Fibrillin-2 gene mutations associated with hereditary con-nective tissue diseases. Hereditas 2019, 41, 919–927. [Google Scholar] [CrossRef]
- Li, D.; Pan, Z.; Zhang, K.; Yu, M.; Yu, D.; Lu, Y.; Wang, J.; Zhang, J.; Zhang, K.; Du, W. Identification of the Differentially Expressed Genes of Muscle Growth and Intramuscular Fat Metabolism in the Development Stage of Yellow Broilers. Genes 2020, 11, 244. [Google Scholar] [CrossRef]
- Ito, N.; Kii, I.; Shimizu, N.; Tanaka, H.; Takeda, S. Direct reprogramming of fibroblasts into skeletal muscle progenitor cells by transcription factors enriched in undifferentiated subpopulation of satellite cells. Sci. Rep. 2017, 7, 8097. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Glaser, G.; Wernig, A.; Wegner, M.; Rosorius, O. Sox8 is a specific marker for muscle satellite cells and inhibits myogenesis. J. Biol. Chem. 2003, 278, 29769–29775. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Yun, Y.; Kim, M.J.; Kim, I.S. Myogenin is a positive regulator of MEGF10 expression in skeletal muscle. Biochem. Biophys. Res. Commun. 2014, 450, 1631–1637. [Google Scholar] [CrossRef]
- Bi, P.; McAnally, J.R.; Shelton, J.M.; Sanchez-Ortiz, E.; Bassel-Duby, R.; Olson, E.N. Fusogenic micropeptide Myomixer is essential for satellite cell fusion and muscle regeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 3864–3869. [Google Scholar] [CrossRef]
- Xu, T.S.; Gu, L.H.; Sun, Y.; Zhang, X.H.; Ye, B.G.; Liu, X.L.; Hou, S.S. Characterization of MUSTN1 gene and its relationship with skeletal muscle development at postnatal stages in Pekin ducks. Genet Mol. Res. 2015, 14, 4448–4460. [Google Scholar] [CrossRef]
- Shi, B.; Shi, X.; Zuo, Z.; Zhao, S.; Zhao, Z.; Wang, J.; Zhou, H.; Luo, Y.; Hu, J.; Hickford, J.G.H. Identification of differentially expressed genes at different post-natal development stages of longissimus dorsi muscle in Tianzhu white yak. Gene 2022, 823, 146356. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, T.; Zhuang, J.; Dai, Y.; Zhang, X.; Bai, S.; Zhao, B.; Tang, X.; Wu, X.; Chen, Y. Effects of feeding restriction on skeletal muscle development and functional analysis of TNNI1 in New Zealand white rabbits. Anim. Biotechnol. 2023, 34, 4435–4447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Suh, Y.; Choi, Y.M.; Chen, P.R.; Davis, M.E.; Lee, K. Differential Expression of Cell Cycle Regulators During Hyperplastic and Hypertrophic Growth of Broiler Subcutaneous Adipose Tissue. Lipids 2015, 50, 965–976. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Lee, J.; Pilch, P.F. The insulin receptor: Structure, function, and signaling. Am. J. Physiol. 1994, 266, C319–C334. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Zhang, W.Y.; Bai, J.B.; Zhang, H.X.; Zhao, Y.Y.; Li, X.Y.; Zhao, S.H. The NF-kappaB-modulated microRNAs miR-195 and miR-497 inhibit myoblast proliferation by targeting Igf1r, Insr and cyclin genes. J. Cell Sci. 2016, 129, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Florini, J.R.; Ewton, D.Z.; Coolican, S.A. Growth hormone and the insulin-like growth factor system in myogenesis. Endocr. Rev. 1996, 17, 481–517. [Google Scholar] [CrossRef]
- Saito, T.; Akutsu, S.; Urushiyama, T.; Ishibashi, K.; Nakagawa, Y.; Shuler, C.F.; Yamane, A. Changes in the mRNA expressions of insulin-like growth factors, their receptors, and binding proteins during the postnatal development of rat masseter muscle. Zool. Sci. 2003, 20, 441–447. [Google Scholar] [CrossRef][Green Version]
- Xue, Q.; Zhang, G.; Li, T.; Ling, J.; Zhang, X.; Wang, J. Transcriptomic profile of leg muscle during early growth in chicken. PLoS ONE 2017, 12, e0173824. [Google Scholar] [CrossRef]
- Zhan, S.Y.; Chen, L.; Li, L.; Wang, L.J.; Zhong, T.; Zhang, H.P. Molecular characterization and expression patterns of insulin-like growth factor-binding protein genes in postnatal Nanjiang brown goats. Genet. Mol. Res. 2015, 14, 12547–12560. [Google Scholar] [CrossRef] [PubMed]
- Aboalola, D.; Han, V.K.M. Insulin-Like Growth Factor Binding Protein-6 Promotes the Differentiation of Placental Mesenchymal Stem Cells into Skeletal Muscle Independent of Insulin-Like Growth Factor Receptor-1 and Insulin Receptor. Stem. Cells Int. 2019, 2019, 9245938. [Google Scholar] [CrossRef] [PubMed]
- deLapeyriere, O.; Ollendorff, V.; Planche, J.; Ott, M.O.; Pizette, S.; Coulier, F.; Birnbaum, D. Expression of the Fgf6 gene is restricted to developing skeletal muscle in the mouse embryo. Development 1993, 118, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Kastner, S.; Elias, M.C.; Rivera, A.J.; Yablonka-Reuveni, Z. Gene expression patterns of the fibroblast growth factors and their receptors during myogenesis of rat satellite cells. J. Histochem. Cytochem. 2000, 48, 1079–1096. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Description | Primers Sequence (from 5′ to 3′) |
|---|---|---|
| CDC27 | Cell division cycle 27 | F: TCTCCACAATCACACCTCAGATCC R: TTCACGAAGAAGGCTCATCAAACC |
| IGF2 | Insulin like growth factor 2 | F: GCCCTTCTGGAGACCTACTGTG R: AGGTGTCGTATTGGAAGAACTTGC |
| MEGF10 | Multiple EGF like domains 10 | F: TTCCGAGGCACCACTTGTCAG R: CCAGGCAGGCAGTCACAGAG |
| MUSTN1 | Musculoskeletal | F: GCCAAGAACCAGGAGATCAAGTC R: TCGGCTGCCACTGAACACC |
| PEG10 | Paternally expressed 10 | F: GATGGACATGGACGATCACTCTATG R: TGCGGCGGCGGATACTG |
| TNNI1 | Troponin I1, slow skeletal type | F: GTGGAGGTGGTGGATGAGGAG R: CCCGACGCAGTGGTGGAC |
| MYH3 | Myosin heavy chain 3 | F: CGTCCTGGATGATCTACACCTACTC R: TTCTTGCCTCGGTAGCCTTCC |
| Samples | Raw Reads | Clean Reads | Clean Bases (Gb) | Total Mapped | Uniquely Mapped |
|---|---|---|---|---|---|
| Y45_1 | 46,417,512 | 45,271,560 | 6.79 | 87.79% | 84.94% |
| Y45_2 | 55,331,762 | 54,289,360 | 8.14 | 90.09% | 87.11% |
| Y45_3 | 50,207,396 | 49,355,610 | 7.4 | 89.09% | 86.20% |
| Y90_1 | 50,046,636 | 49,083,374 | 7.36 | 88.26% | 84.96% |
| Y90_2 | 48,711,358 | 47,736,736 | 7.16 | 88.75% | 85.48% |
| Y90_3 | 50,268,714 | 48,472,740 | 7.27 | 87.95% | 85.02% |
| Y120_1 | 48,166,278 | 47,430,618 | 7.11 | 90.19% | 87.36% |
| Y120_2 | 48,847,066 | 47,665,894 | 7.15 | 87.26% | 83.75% |
| Y120_3 | 49,488,234 | 48,537,954 | 7.28 | 89.67% | 86.8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rong, M.; Xing, X.; Zhang, R. Muscle Transcriptome Analysis of Mink at Different Growth Stages Using RNA-Seq. Biology 2024, 13, 283. https://doi.org/10.3390/biology13050283
Rong M, Xing X, Zhang R. Muscle Transcriptome Analysis of Mink at Different Growth Stages Using RNA-Seq. Biology. 2024; 13(5):283. https://doi.org/10.3390/biology13050283
Chicago/Turabian StyleRong, Min, Xiumei Xing, and Ranran Zhang. 2024. "Muscle Transcriptome Analysis of Mink at Different Growth Stages Using RNA-Seq" Biology 13, no. 5: 283. https://doi.org/10.3390/biology13050283
APA StyleRong, M., Xing, X., & Zhang, R. (2024). Muscle Transcriptome Analysis of Mink at Different Growth Stages Using RNA-Seq. Biology, 13(5), 283. https://doi.org/10.3390/biology13050283