
New Genetic Variants of RUNX2 in Mexican Families Cause Cleidocranial Dysplasia

 , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
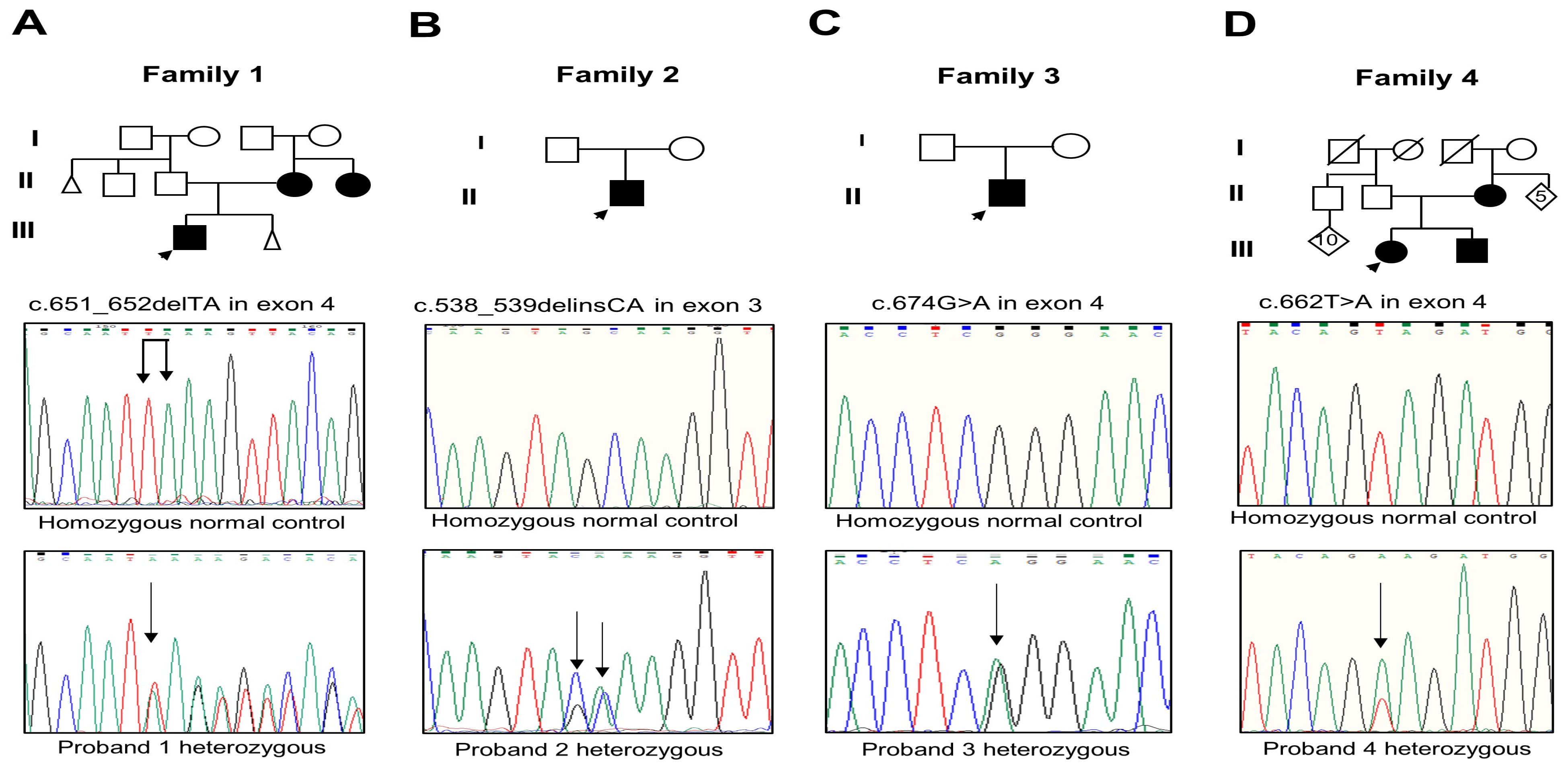
2.1. Cases Studied
2.2. Direct Sequencing of the RUNX2 Gene
2.3. In Silico Prediction Analysis
2.4. Ethical Aspects
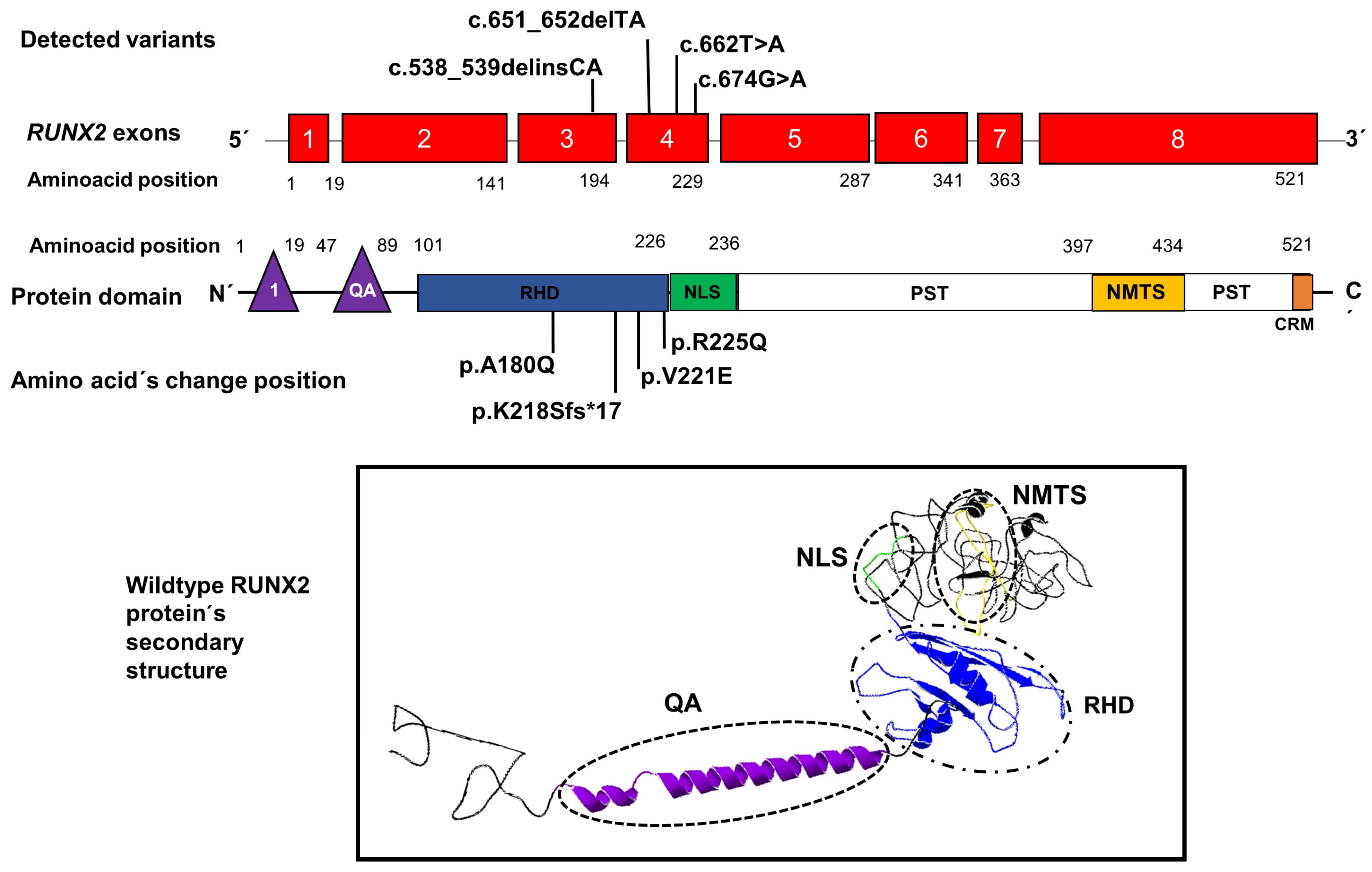
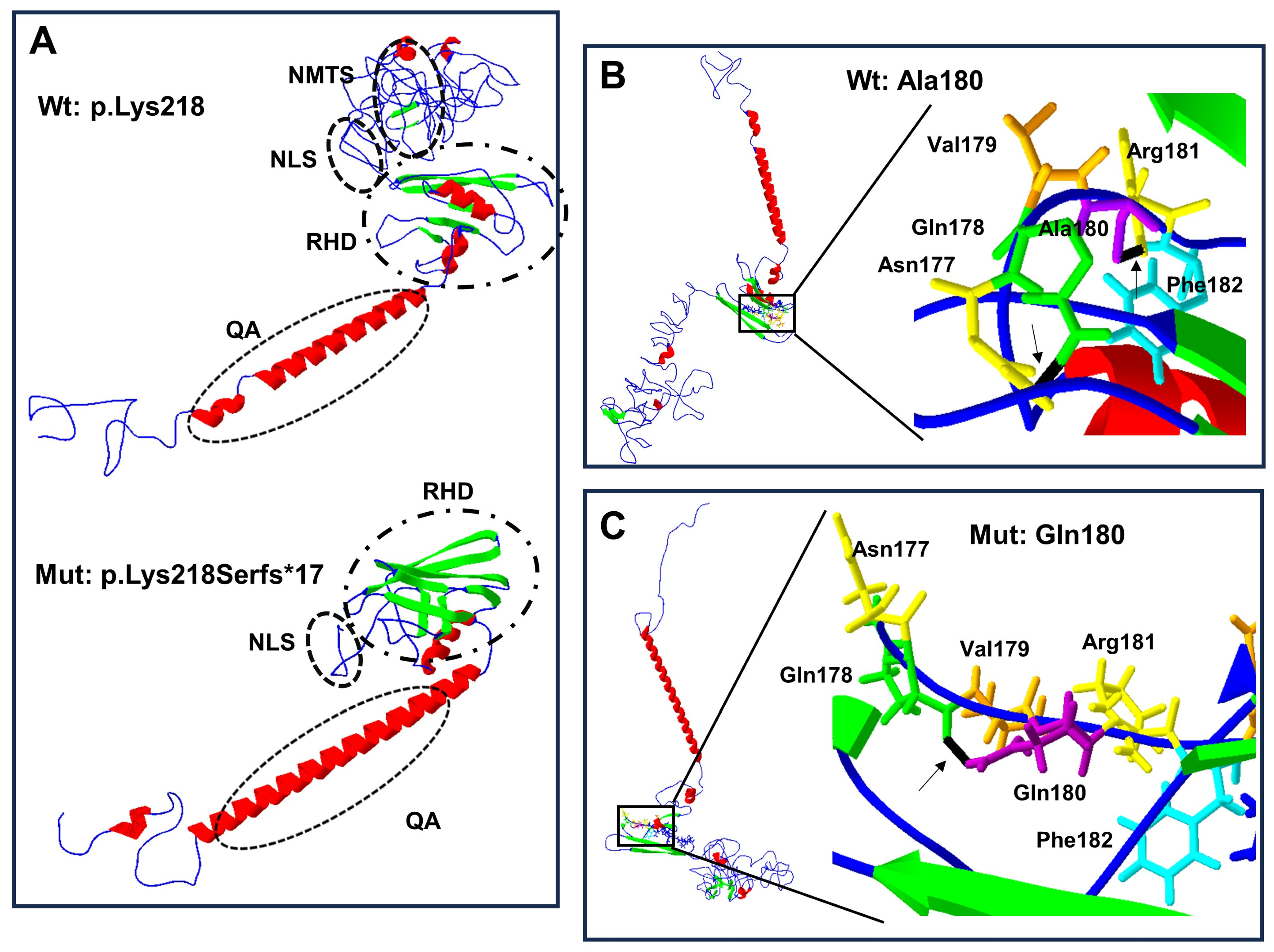
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CCD | Cleidocranial dysplasia |
| RUNX2 | Runt-related transcription factor 2 gene |
| Q/A | Polyglutamine/alanine domains |
| RHD | Runt homologous domain |
| NLS | Nuclear localization signal |
| PST-rich region | Proline/serine/threonine-rich region |
| NMTS | Nuclear matrix-targeting sequence domain |
| PEBP2b | Polyomavirus enhancer-binding protein 2b |
| CBFβ | Core binding factor β |
References
- Farrow, E.; Nicot, R.; Wiss, A.; Laborde, A.; Ferri, J. Cleidocranial Dysplasia: A Review of Clinical, Radiological, Genetic Implications and a Guidelines Proposal. J. Craniofac. Surg. 2018, 29, 382–389. [Google Scholar] [CrossRef]
- Mundlos, S. Cleidocranial dysplasia: Clinical and molecular genetics. J. Med. Genet. 1999, 36, 177–182. [Google Scholar]
- Jaruga, A.; Hordyjewska, E.; Kandzierski, G.; Tylzanowski, P. Cleidocranial dysplasia and RUNX2-clinical phenotype-genotype correlation. Clin. Genet. 2016, 90, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; Chen, L.F.; Ogawa, E.; Kim, W.Y.; Ito, Y. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Mol. Cell. Biol. 1998, 18, 2444–2454. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Bae, H.S.; Ryoo, H.M.; Baek, S.H. A novel RUNX2 mutation in exon 8, G462X, in a patient with Cleidocranial Dysplasia. J. Cell. Biochem. 2018, 119, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Lengner, C.J.; Gaur, T.; Lou, Y.; Hussain, S.; Jones, M.D.; Borodic, B.; Colby, J.L.; Steinman, H.A.; Van Wijnen, A.J.; et al. RUNX2 protein expression utilizes the Runx2 P1 promoter to establish osteoprogenitor cell number for normal bone formation. J. Biol. Chem. 2011, 286, 30057–30070. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Lee, S.W.; Park, M.H.; Bae, Y.C.; Shin, H.I.; Nam, S.; Kim, Y.J.; Kim, H.J.; Ryoo, H.M. Spatio-temporal expression patterns of Runx2 isoforms in early skeletogenesis. Exp. Mol. Med. 2002, 34, 426–433. [Google Scholar] [CrossRef]
- Stein, G.S.; Lian, J.B.; Van Wijnen, A.J.; Stein, J.L.; Montecino, M.; Javed, A.; Zaidi, S.K.; Young, D.W.; Choi, J.Y.; Pockwinse, S.M. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene 2004, 23, 4315–4329. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Whole Aspect of Runx2 Functions in Skeletal Development. Int. J. Mol. Sci. 2022, 23, 5776. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, Y.A.; Duz, M.B.; Seven, M. Rare Findings in Cleidocranial Dysplasia Caused by RUNX Mutation. Glob. Med. Genet. 2021, 9, 23–28. [Google Scholar] [CrossRef] [PubMed]
- HGMD® Professional 2020.4 [Internet]—The Human Gene Mutation Database, QIAGEN Digital Insights. Redwood, US: QIAGEN; 2020. Available online: https://portal.biobaseinternational.com/hgmd/pro/start.php (accessed on 18 May 2021).
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Zhou, G.; Chen, Y.; Zhou, L.; Thirunavukkarasu, K.; Hecht, J.; Chitayat, D.; Gelb, B.D.; Pirinen, S.; Berry, S.A.; Greenberg, C.R.; et al. CBFA1 mutation analysis and functional correlation with phenotypic variability in cleidocranial dysplasia. Hum. Mol. Genet. 1999, 8, 2311–2316. [Google Scholar] [CrossRef] [PubMed]
- Quack, I.; Vonderstrass, B.; Stock, M.; Aylsworth, A.S.; Becker, A.; Brueton, L.; Lee, P.J.; Majewski, F.; Mulliken, J.B.; Suri, M.; et al. Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am. J. Hum. Genet. 1999, 65, 1268–1278. [Google Scholar] [CrossRef]
- Källberg, M.; Margaryan, G.; Wang, S.; Ma, J.; Xu, J. Raptor X server: A resource for template-based protein structure modeling. Methods Mol. Biol. 2014, 1137, 17–27. [Google Scholar]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Mexican Standard NOM-253-SSA1-2012 Guidelines. Available online: https://www.gob.mx/cnts/documentos/norma-oficial-mexicana-nom-253-ssa1-2012-para-la-disposicion-de-sangre-humana-y-sus-componentes-con-fines-terapeuticos (accessed on 17 May 2023).
- Yoshida, T.; Yoshida, T.; Kanegane, H.; Osato, M.; Yanagida, M.; Miyawaki, T.; Ito, Y.; Shigesada, K. Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations. Am. J. Hum. Genet. 2002, 71, 724–738. [Google Scholar] [CrossRef]
- Wang, G.X.; Sun, R.P.; Song, F.L. A novel RUNX2 mutation (T420I) in Chinese patients with cleidocranial dysplasia. Genet. Mol. Res. 2010, 9, 41–47. [Google Scholar] [CrossRef]
- Chen, T.; Hou, J.; Hu, L.L.; Gao, J.; Wu, B.L. A novel small deletion mutation in RUNX2 gene in one Chinese family with cleidocranial dysplasia. Int. J. Clin. Exp. Pathol. 2014, 7, 2490–2495. [Google Scholar] [PubMed]
- Matsushita, M.; Kitoh, H.; Kaneko, H.; Mishima, K.; Itoh, Y.; Tokita, Y.; Ishiguro, N. A novel in-frame deletion of the RUNX2 gene causes a classic form of cleidocranial dysplasia. J. Bone Miner. Metab. 2014, 32, 96–99. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Wang, X.; Sun, X.; Zhang, C.; Zheng, S. Analysis of novel RUNX2 mutations in Chinese patients with cleidocranial dysplasia. PLoS ONE 2017, 12, e0181653. [Google Scholar] [CrossRef]
- Ott, C.E.; Leschik, G.; Trotier, F.; Brueton, L.; Brunner, H.G.; Brussel, W.; Guillen, E.; Haase, C.; Kohlhase, J.; Kotzot, D.; et al. Deletions of the RUNX2 gene are present in about 10% of individuals with cleidocranial dysplasia. Hum. Mutat. 2010, 31, 1587–1593. [Google Scholar] [CrossRef]
- Nagata, T.; Werner, M.H. Functional mutagenesis of AML1/RUNX1 and PEBP2 beta/CBF beta define distinct, non-overlapping sites for DNA recognition and heterodimerization by the Runt domain. J. Mol. Biol. 2001, 308, 191–203. [Google Scholar] [CrossRef]
- Tessa, A.; Salvi, S.; Casali, C.; Garavelli, L.; Digilio, M.C.; Dotti, M.T.; Di Giandomenico, S.; Valoppi, M.; Grieco, G.S.; Comanducci, G.; et al. Six novel mutations of the RUNX2 gene in Italian patients with cleidocranial dysplasia. Hum. Mutat. 2003, 22, 104. [Google Scholar] [CrossRef]
- Gong, L.; Odilov, B.; Han, F.; Liu, F.; Sun, Y.; Zhang, N.; Zuo, X.; Yang, J.; Wang, S.; Hou, X.; et al. Identification a novel de novo RUNX2 frameshift mutation associated with cleidocranial dysplasia. Genes. Genom. 2022, 44, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Ye, G.; Yu, X.; Xu, J.; Li, Z. Identification of a Novel Mutation in the Runt-Related Transcription Factor 2 Gene in a Chinese Family With Cleidocranial Dysplasia. J. Craniofac. Surg. 2021, 32, 541–544. [Google Scholar] [CrossRef]
- Tahirov, T.H.; Inoue-Bungo, T.; Morii, H. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFbeta. Cell 2001, 104, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Gebhardt, M.; Golovchenko, S. Dual pathways to endochondral osteoblasts: A novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol. Open 2015, 4, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Zhou, X.; Kunkel, G. The novel zinc finger-containing transcription factor Osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef]
- Liu, T.M.; Lee, E.H. Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng. Part B Rev. 2013, 19, 254–263. [Google Scholar] [CrossRef]
- Suda, N.; Hattori, M.; Kosaki, K. Correlation between genotype and supernumerary tooth formation in cleidocranial dysplasia. Orthod. Craniofac. Res. 2010, 13, 197–202. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Case 1 | Case 2 | Case 3 | Case 4 |
|---|---|---|---|---|
| Age (y) | 12 | 1 | 7 | 29 |
| Sex | M | M | M | F |
| Cranial abnormalities | Low hair implantation; Wormian bones; mild open fontanelle/sutures. | Prominence frontal and parietal; broad fontanelle and sutures. | Frontal prominent; broad sutures and fontanelles. | Brachycephaly; broad forehead with medium cleft; cranial bone thickening; Wormian bones; headache. |
| Facial abnormalities | Populated eyelashes/eyebrows; strabismus; hypertelorism; eyelids turned down; anteverted nostrils; long philtrum; wide mouth. | Hypertelorism. | Midface hypoplasia; hypertelorism; wide/low nasal bridge; bulbous nasal tip; long philtrum. | Midface asymmetry and hypoplasia; nasal deviation; hypertelorism. |
| Oral-dental abnormalities | Oligodontia and supernumerary teeth. | Central and lateral teeth present apparently normal. | Yellow and misaligned teeth. | Molars absent; supernumerary teeth. |
| Clavicle abnormalities | Hypoplasia. | Agenesis. | Agenesis. | Hypoplasia. |
| Thorax and spine abnormalities | Pectum excavatum and thoraco-lumbar scoliosis. | Absent. | Pectum excavatum and thoraco-lumbar scoliosis. | Short and wide chest; thoracic scoliosis. |
| Limbs abnormalities | Absent. | Absent. | Absent. | Brachydactyly 4th phalanx left foot. Cubitus valgus; genu varum. |
| Others abnormalities | Cataract, detachment of retina. | Absent. | Short stature; short neck. | Short stature; short neck with limited rotation. |
| Phenotype expression | Severe. | Moderate. | Severe. | Severe. |
| Family history | (+) | (−) | (−) | (+) |
| DNA change | c.651_652delTA; | c.538,539delinsCA | c.674G>A | c.662T>A |
| Exon location | 4 | 3 | 4 | 4 |
| Protein change | p.Lys218Serfs*17 | p.Ala180Gln | p.Arg225Gln | p.Val221Glu |
| Domain location | RHD/NLS. | RHD | RHD | RHD |
| Reference | Novel. | Novel. | [13,14] | Novel. |
| Exon Number | Primers | Size | Tm |
|---|---|---|---|
| 2 | F 5′-GGCCACTTCGCTAACTTGTG-3′ R 5′-GTAGCCTCTTACCTTGAAGG-3′ | 422 pb | 56 °C |
| 3 | F 5′-GGACTAGAACACTAAGTCCTG-3′ R 5′-CACTCAACTTCATCTGGATG-3′ | 419 pb | 60 °C |
| 4 | F 5′-CATTGCCTCCTTAGAGATGC-3′ R 5′-ATTCCTCATAGGGTCTCTGG-3′ | 280 pb | 60 °C |
| 5 | F 5′-GCAT GGTCAATTGTTCAGCT-3′ R 5′-CTGCCAGCGTCTATGCAAG-3′ | 273 pb | 60 °C |
| 6 | F 5′-GGCTGCAATGGTTGCTATAC-3′ R 5′-TGTGAGCATGGATGAGACAG-3′ | 289 pb | 60 °C |
| 7 | F 5′-CATAGAACATTAGAGCTGGAAGG-3′ R 5′-CTCACAAAATCGGACAGTAAC-3′ | 199 pb | 60 °C |
| 8 | F 5′-GGTGCATTTGAAGGTCTGTC-3′ R 5′-ATTGATACGTGTGGGATGTGG-3′ | 677 pb | 60 °C |
| Protein/Amino Acid ID | PolyPhen2/MutationTaster/PROVEAN/SIFT (Score) | Log of Force Field Energy (KJ/mol) | I-Mutant 2.0 | Secondary Structure Changes | |||
|---|---|---|---|---|---|---|---|
| Structural Molding | # of α-Helix | # of β-Leaf | # of Hydrogen Bonds | ||||
| RUNX2 wild-type | - | 14.48 | - | - | 6 | 7 | NA |
| RUNX2 p.Gln180 | NA | 18.45 | NA | Folded | 5 | 11 | NA |
| RUNX2 p.Glu221 | NA | 15.00 | NA | Folded | 4 | 11 | NA |
| RUNX2 p.Gln225 | NA | 15.40 | NA | Folded | 7 | 9 | NA |
| p.Lys218Serfs*17 | 1.000/disease causing (1.0)/NA/NA | 12.51 | NA | Truncated | 3 | 8 | NA |
| Ala180 | - | 5.53 | - | NA | NA | NA | 0 |
| Gln180 | 1.000/disease causing (0.99)/deleterious (−4.66)/damaging (0.00) | 6.24 | −1.72 | NA | NA | NA | 1 |
| Val221 | - | 2.59 | - | NA | NA | NA | 2 |
| Glu221 | 1.000/disease causing (0.99)/deleterious (−5.85)/damaging (0.00) | 7.99 | −1.69 | NA | NA | NA | 0, Gain of a covalent bond |
| Arg225 | - | 7.99 | - | NA | NA | NA | 2 |
| Gln225 | 0.980/disease causing (0.99)/deleterious (−3.8)/damaging (0.03) | 6.46 | −1.03 | NA | NA | NA | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toral López, J.; Gómez Martinez, S.; Rivera Vega, M.d.R.; Hernández-Zamora, E.; Cuevas Covarrubias, S.; Ibarra Castrejón, B.A.; González Huerta, L.M. New Genetic Variants of RUNX2 in Mexican Families Cause Cleidocranial Dysplasia. Biology 2024, 13, 173. https://doi.org/10.3390/biology13030173
Toral López J, Gómez Martinez S, Rivera Vega MdR, Hernández-Zamora E, Cuevas Covarrubias S, Ibarra Castrejón BA, González Huerta LM. New Genetic Variants of RUNX2 in Mexican Families Cause Cleidocranial Dysplasia. Biology. 2024; 13(3):173. https://doi.org/10.3390/biology13030173
Chicago/Turabian StyleToral López, Jaime, Sandra Gómez Martinez, María del Refugio Rivera Vega, Edgar Hernández-Zamora, Sergio Cuevas Covarrubias, Belem Arely Ibarra Castrejón, and Luz María González Huerta. 2024. "New Genetic Variants of RUNX2 in Mexican Families Cause Cleidocranial Dysplasia" Biology 13, no. 3: 173. https://doi.org/10.3390/biology13030173
APA StyleToral López, J., Gómez Martinez, S., Rivera Vega, M. d. R., Hernández-Zamora, E., Cuevas Covarrubias, S., Ibarra Castrejón, B. A., & González Huerta, L. M. (2024). New Genetic Variants of RUNX2 in Mexican Families Cause Cleidocranial Dysplasia. Biology, 13(3), 173. https://doi.org/10.3390/biology13030173