

Deletion of Interleukin-1β Converting Enzyme Alters Mouse Cardiac Structure and Function

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Echocardiography
2.3. Heart Weight/Body Weight
2.4. Histological Analysis
2.5. Electron Microscopy
2.6. RNA Isolation and Northern Blot Analysis
2.7. Western Blot
2.8. Neonatal Cardiomyocyte Isolation
2.9. DNA Synthesis in the Cardiac Fibroblasts
2.10. Beat Rate Assay
2.11. Oxidative Stress in the Neonatal Cardiomyocytes
2.12. Apoptotic Nuclear Changes in the Neonatal Cardiomyocytes
2.13. Statistical Analysis
3. Results
3.1. Assessment of the ICE KO Cardiac Structure and Function Using Echocardiography
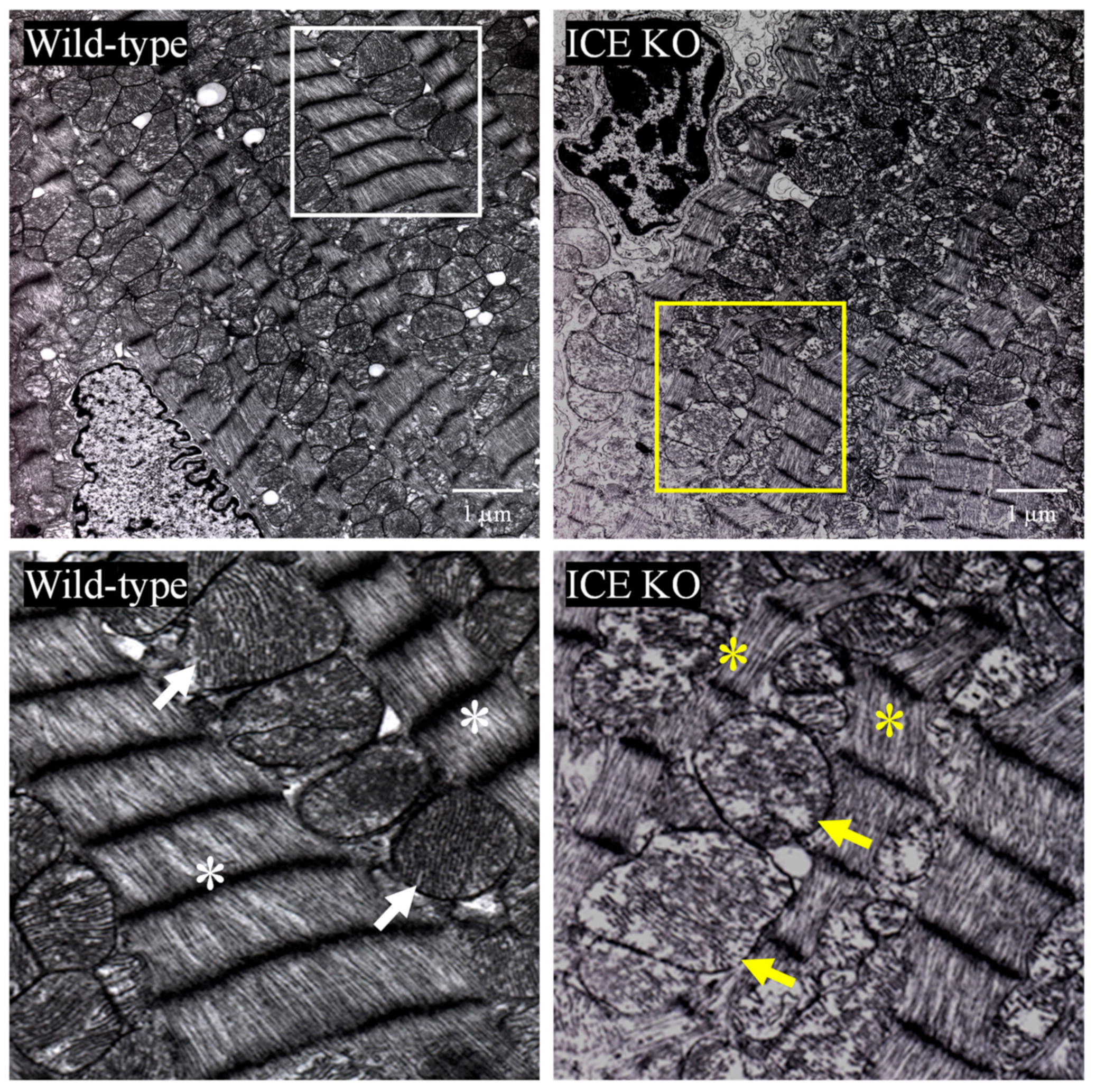
3.2. ICE Deletion Induces Cardiac Hypertrophy, Fibrosis, and Mitochondrial Damage
3.3. ICE-Deletion-Induced Expression of Markers of Cardiac Hypertrophy
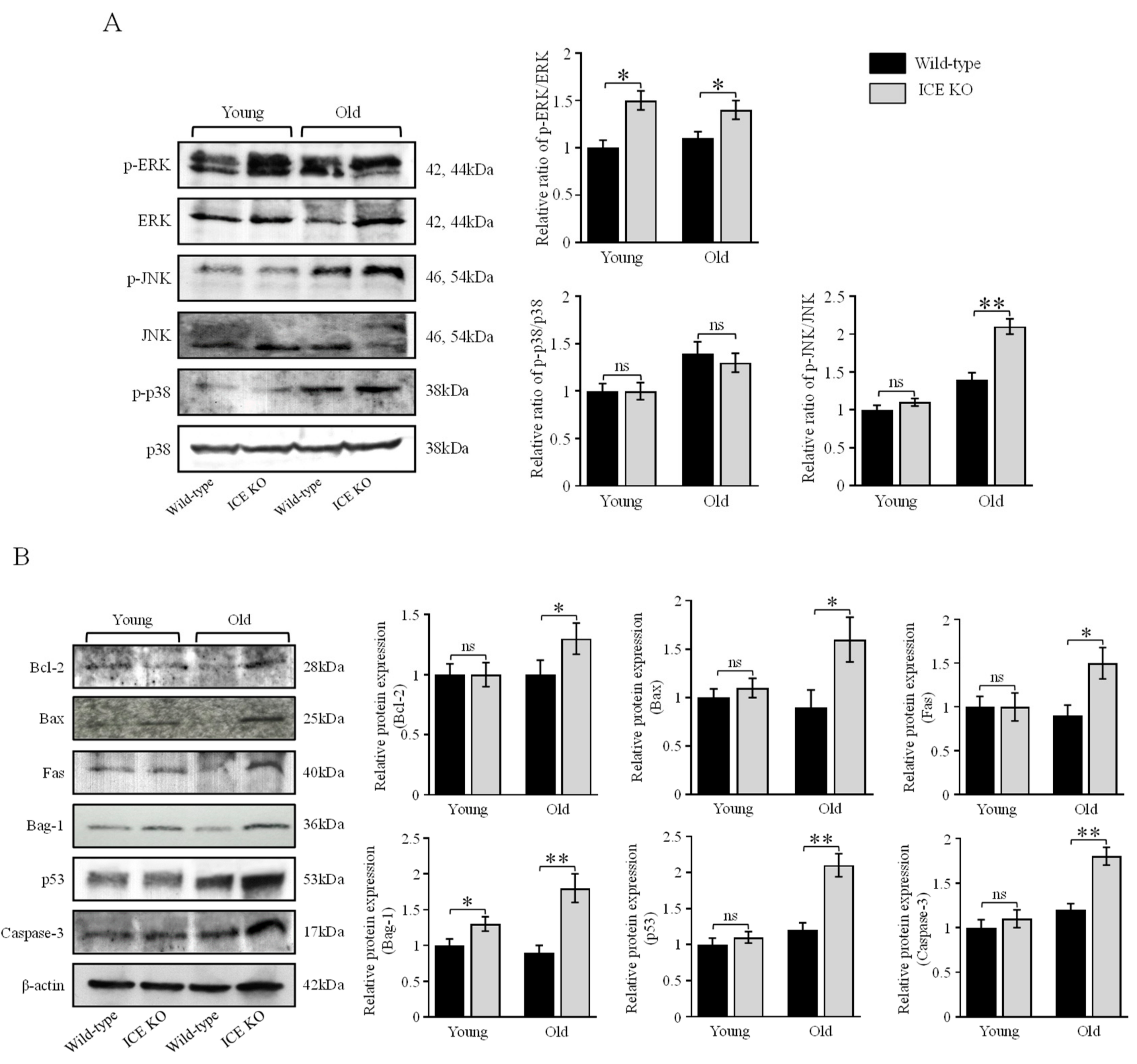
3.4. ICE Regulates the MAPK Signaling Pathway and Apoptotic Proteins during Cardiac Hypertrophy
3.5. Functional Analysis of the ICE KO Neonatal Cardiomyocytes under Oxidative Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaduganathan, M.; Mensah, G.A.; Turco, J.V.; Fuster, V.; Roth, G.A. The Global Burden of Cardiovascular Diseases and Risk. J. Am. Coll. Cardiol. 2022, 80, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics—2022 Update: A Report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Mechanisms of Physiological and Pathological Cardiac Hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Mann, D.L. Innate Immunity and the Failing Heart: The Cytokine Hypothesis Revisited. Circ Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [PubMed]
- Reina-Couto, M.; Pereira-Terra, P.; Quelhas-Santos, J.; Silva-Pereira, C.; Albino-Teixeira, A.; Sousa, T. Inflammation in Human Heart Failure: Major Mediators and Therapeutic Targets. Front. Physiol. 2021, 12, 746494. [Google Scholar] [CrossRef]
- Galea, J.; Armstrong, J.; Gadsdon, P.; Holden, H.; Francis, S.E.; Holt, C.M. Interleukin-1β in Coronary Arteries of Patients with Ischemic Heart Disease. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1000–1006. [Google Scholar] [CrossRef]
- Ørn, S.; Ueland, T.; Manhenke, C.; Sandanger, Ø.; Godang, K.; Yndestad, A.; Mollnes, T.E.; Dickstein, K.; Aukrust, P. Increased Interleukin-1β Levels Are Associated with Left Ventricular Hypertrophy and Remodelling Following Acute ST Segment Elevation Myocardial Infarction Treated by Primary Percutaneous Coronary Intervention. J. Intern. Med. 2012, 272, 267–276. [Google Scholar] [CrossRef]
- Bujak, M.; Frangogiannis, N.G. The Role of IL-1 in the Pathogenesis of Heart Disease. Arch. Immunol. Ther. Exp. 2009, 57, 165–176. [Google Scholar] [CrossRef]
- Sandanger, Ø.; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G.; et al. The NLRP3 Inflammasome Is Up-Regulated in Cardiac Fibroblasts and Mediates Myocardial Ischaemia–Reperfusion Injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome Activation of Cardiac Fibroblasts Is Essential for Myocardial Ischemia/Reperfusion Injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Deten, A. Cardiac Cytokine Expression Is Upregulated in the Acute Phase after Myocardial Infarction. Experimental Studies in Rats. Cardiovasc. Res. 2002, 55, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.A. Interleukin 1 and Interleukin 18 as Mediators of Inflammation and the Aging Process. Am. J. Clin. Nutr. 2006, 83, 447S–455S. [Google Scholar] [CrossRef]
- Seaman, J.E.; Julien, O.; Lee, P.S.; Rettenmaier, T.J.; Thomsen, N.D.; Wells, J.A. Cacidases: Caspases Can Cleave after Aspartate, Glutamate and Phosphoserine Residues. Cell Death Differ. 2016, 23, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Kronheim, S.R.; Cantrell, M.; Deeley, M.C.; March, C.J.; Prickett, K.S.; Wignall, J.; Conlon, P.J.; Cosman, D.; Hopp, T.P. Generation of Biologically Active Interleukin-1 Beta by Proteolytic Cleavage of the Inactive Precursor. J. Biol. Chem. 1988, 263, 9437–9442. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Lopez-Castejon, G.; Brough, D. Caspase-1: Is IL-1 Just the Tip of the ICEberg? Cell Death Dis. 2012, 3, e338. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Luc, G.; Ducimetière, P.; Arveiler, D.; Ferrières, J.; Amouyel, P.; Evans, A.; Cambien, F.; Tiret, L. Interleukin-18 and the Risk of Coronary Heart Disease in European Men: The Prospective Epidemiological Study of Myocardial Infarction (PRIME). Circulation 2003, 108, 2453–2459. [Google Scholar] [CrossRef]
- Mallat, Z. Increased Plasma Concentrations of Interleukin-18 in Acute Coronary Syndromes. Heart 2002, 88, 467–469. [Google Scholar] [CrossRef]
- Zeng, C.; Duan, F.; Hu, J.; Luo, B.; Huang, B.; Lou, X.; Sun, X.; Li, H.; Zhang, X.; Yin, S.; et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020, 34, 101523. [Google Scholar] [CrossRef]
- Frantz, S. Targeted Deletion of Caspase-1 Reduces Early Mortality and Left Ventricular Dilatation Following Myocardial Infarction. J. Mol. Cell. Cardiol. 2003, 35, 685–694. [Google Scholar] [CrossRef]
- Merkle, S.; Frantz, S.; Schön, M.P.; Bauersachs, J.; Buitrago, M.; Frost, R.J.A.; Schmitteckert, E.M.; Lohse, M.J.; Engelhardt, S. A Role for Caspase-1 in Heart Failure. Circ. Res. 2007, 100, 645–653. [Google Scholar] [CrossRef]
- Trentin-Sonoda, M.; Fratoni, F.M.; da Cruz Junho, C.V.; Silva, W.C.; Panico, K.; Carneiro-Ramos, M.S. Caspase-1 as molecular key in cardiac remodeling during Cardiorenal syndrome type 3 in the murine model. Curr. Mol. Med. 2019, 20, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Miura, M. Induction of Apoptosis in Fibroblasts by IL-1β-Converting Enzyme, a Mammalian Homolog of the C. Elegans Cell Death Gene Ced-3. Cell 1993, 75, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Aries, A.; Whitcomb, J.; Shao, W.; Komati, H.; Saleh, M.; Nemer, M. Caspase-1 Cleavage of Transcription Factor GATA4 and Regulation of Cardiac Cell Fate. Cell Death Dis. 2014, 5, e1566. [Google Scholar] [CrossRef] [PubMed]
- Putinski, C.; Abdul-Ghani, M.; Stiles, R.; Brunette, S.; Dick, S.A.; Fernando, P.; Megeney, L.A. Intrinsic-Mediated Caspase Activation Is Essential for Cardiomyocyte Hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, E4079–E4087. [Google Scholar] [CrossRef] [PubMed]
- Maulik, S.K.; Kumar, S. Oxidative Stress and Cardiac Hypertrophy: A Review. Toxicol. Mech. Methods 2012, 22, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Vascular Oxidative Stress, Nitric Oxide and Atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Strait, J.B.; Lakatta, E.G. Aging-Associated Cardiovascular Changes and Their Relationship to Heart Failure. Heart Fail. Clin. 2012, 8, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Sastre, J.; Pallardó, F.V.; Viña, J. Mitochondrial Oxidative Stress Plays a Key Role in Aging and Apoptosis. IUBMB Life 2000, 49, 427–435. [Google Scholar] [CrossRef]
- Mirza, M.; Strunets, A.; Shen, W.-K.; Jahangir, A. Mechanisms of Arrhythmias and Conduction Disorders in Older Adults. Clin. Geriatr. Med. 2012, 28, 555–573. [Google Scholar] [CrossRef]
- Marín-Aguilar, F.; Lechuga-Vieco, A.V.; Alcocer-Gómez, E.; Castejón-Vega, B.; Lucas, J.; Garrido, C.; Peralta-Garcia, A.; Pérez-Pulido, A.J.; Varela-López, A.; Quiles, J.L.; et al. NLRP3 Inflammasome Suppression Improves Longevity and Prevents Cardiac Aging in Male Mice. Aging Cell 2020, 19, e13050. [Google Scholar] [CrossRef]
- Narayan, P.; Trikantzopoulos, E.; Mezzaroma, E.; Mauro, A.G.; Vohra, H.; Abbate, A.; Toldo, S. The Interleukin-1 Receptor Type I Promotes the Development of Aging-Associated Cardiomyopathy in Mice. Cytokine 2022, 151, 155811. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Allen, H.; Banerjee, S.; Franklin, S.; Herzog, L.; Johnston, C.; McDowell, J.; Paskind, M.; Rodman, L.; Salfeld, J.; et al. Mice Deficient in IL-1β-Converting Enzyme Are Defective in Production of Mature IL-1β and Resistant to Endotoxic Shock. Cell 1995, 80, 401–411. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Azhar, G.; Furr, M.C.; Zhong, Y.; Wei, J.Y. Model of Functional Cardiac Aging: Young Adult Mice with Mild Overexpression of Serum Response Factor. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R552–R560. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.; Chai, J.; Azhar, G.; Sheridan, P.; Borras, A.M.; Furr, M.C.; Khrapko, K.; Lawitts, J.; Misra, R.P.; Wei, J.Y. Early Postnatal Cardiac Changes and Premature Death in Transgenic Mice Overexpressing a Mutant Form of Serum Response Factor. J. Biol. Chem. 2001, 276, 40033–40040. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Azhar, G.; Nagano, K.; Wei, J.Y. Differential Vulnerability to Oxidative Stress in Rat Cardiac Myocytes versus Fibroblasts. J. Am. Coll. Cardiol. 2001, 38, 2055–2062. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1β, Interleukin-18, and the Interleukin-1β Converting Enzymea. Ann. N. Y. Acad. Sci. 1998, 856, 1–11. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Sun, Q.; Scott, M.J. Caspase-1 as a Multifunctional Inflammatory Mediator: Noncytokine Maturation Roles. J. Leukoc. Biol. 2016, 100, 961–967. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Li, P.; Allen, H.; Banerjee, S.; Seshadri, T. Characterization of mice deficient in interleukin-1 beta converting enzyme. J. Cell. Biochem. 1997, 64, 27–32. [Google Scholar] [CrossRef]
- Syed, F.M.; Hahn, H.S.; Odley, A.; Guo, Y.; Vallejo, J.G.; Lynch, R.A.; Mann, D.L.; Bolli, R.; Dorn, G.W. Proapoptotic Effects of Caspase-1/Interleukin-Converting Enzyme Dominate in Myocardial Ischemia. Circ. Res. 2005, 96, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Kuida, K.; Lippke, J.A.; Ku, G.; Harding, M.W.; Livingston, D.J.; Su, M.S.-S.; Flavell, R.A. Altered Cytokine Export and Apoptosis in Mice Deficient in Interleukin-1β Converting Enzyme. Science 1995, 267, 2000–2003. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, B.J.; Reznikov, L.L.; Harken, A.H.; Dinarello, C.A. Inhibition of Caspase 1 Reduces Human Myocardial Ischemic Dysfunction via Inhibition of IL-18 and IL-1β. Proc. Natl. Acad. Sci. USA 2001, 98, 2871–2876. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Li, X.; Li, X.; Ding, Z.; Xu, R.; Yin, P.; Wang, S.; Ge, J.; Wu, J.; Zou, Y. Caspase-1 Abrogates the Salutary Effects of Hypertrophic Preconditioning in Pressure Overload Hearts via IL-1β and IL-18. Front. Mol. Biosci. 2021, 8, 641585. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-M.; Downey, J.M.; Cohen, M.V.; Housley, N.A.; Alvarez, D.F.; Audia, J.P. The Highly Selective Caspase-1 Inhibitor VX-765 Provides Additive Protection against Myocardial Infarction in Rat Hearts When Combined with a Platelet Inhibitor. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Shah, M.; Yellon, D.M.; Davidson, S.M. Role of Caspase 1 in Ischemia/Reperfusion Injury of the Myocardium. J. Cardiovasc. Pharmacol. 2019, 74, 194–200. [Google Scholar] [CrossRef]
- De Vasconcelos, N.M.; Lamkanfi, M. Recent Insights on Inflammasomes, Gasdermin Pores, and Pyroptosis. Cold Spring Harb. Perspect. Biol. 2020, 12, a036392. [Google Scholar] [CrossRef]
- Saxena, A.; Chen, W.; Su, Y.; Rai, V.; Uche, O.U.; Li, N.; Frangogiannis, N.G. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J. Immunol. 2013, 191, 4838–4848. [Google Scholar] [CrossRef]
- Magupalli, V.G.; Negro, R.; Tian, Y.; Hauenstein, A.V.; Di Caprio, G.; Skillern, W.; Deng, Q.; Orning, P.; Alam, H.B.; Maliga, Z.; et al. HDAC6 Mediates an Aggresome-like Mechanism for NLRP3 and Pyrin Inflammasome Activation. Science 2020, 369, eaas8995. [Google Scholar] [CrossRef]
- Rajamäki, K.; Mäyränpää, M.I.; Risco, A.; Tuimala, J.; Nurmi, K.; Cuenda, A.; Eklund, K.K.; Öörni, K.; Kovanen, P.T. P38δ MAPK: A Novel Regulator of NLRP3 Inflammasome Activation with Increased Expression in Coronary Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.; Marunouchi, T.; Tanonaka, K. Heat-Shock Protein 90 Modulates Cardiac Ventricular Hypertrophy via Activation of MAPK Pathway. J. Mol. Cell. Cardiol. 2019, 127, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Bueno, O.F.; Molkentin, J.D. Involvement of Extracellular Signal-Regulated Kinases 1/2 in Cardiac Hypertrophy and Cell Death. Circ. Res. 2002, 91, 776–781. [Google Scholar] [CrossRef]
- Titus, A.S.; Harikrishnan, V.; Kailasam, S. Coordinated Regulation of Cell Survival and Cell Cycle Pathways by DDR2-Dependent SRF Transcription Factor in Cardiac Fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1538–H1558. [Google Scholar] [CrossRef]
- Craige, S.M.; Chen, K.; Blanton, R.M.; Keaney, J.F.; Kant, S. JNK and Cardiometabolic Dysfunction. Biosci. Rep. 2019, 39, BSR20190267. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Georgakopoulos, D.; Kovacs, A.; Zheng, M.; Lerner, D.; Pu, H.; Saffitz, J.; Chien, K.; Xiao, R.P.; Kass, D.A.; et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc. Natl. Acad. Sci. USA 2001, 98, 12283–12288. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; You, J.; Dai, F.; Wang, S.; Yang, F.H.; Wang, X.; Ding, Z.; Huang, J.; Chen, L.; Abudureyimu, M.; et al. TAK1 Activation by NLRP3 Deficiency Confers Cardioprotection Against Pressure Overload-Induced Cardiomyocyte Pyroptosis and Hypertrophy. JACC Basic Transl. Sci. 2023, 8, 1555–1573. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, H.; Enninga, J.; Kälble, S.; Thiefes, A.; Dörrie, A.; Broemer, M.; Winzen, R.; Wilhelm, A.; Ninomiya-Tsuji, J.; Matsumoto, K.; et al. The MAPK Kinase Kinase TAK1 Plays a Central Role in Coupling the Interleukin-1 Receptor to Both Transcriptional and RNA-Targeted Mechanisms of Gene Regulation. J. Biol. Chem. 2001, 276, 3508–3516. [Google Scholar] [CrossRef]
- Zhang, J.H.; Xu, M. DNA Fragmentation in Apoptosis. Cell Res. 2000, 10, 205–211. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Wild-Type | ICE-KO | p Value | |
|---|---|---|---|---|
| HR (bpm) | 293.8 ± 74.3 | 216.7 ± 22.2 | 0.35 | |
| SV (ml/beat) | 33.15 ± 8.17 | 44.57 ± 10.91 | 0.42 | |
| CI (ml/min/g) | 348.9 ± 116.9 | 293.4 ± 95.6 | 0.72 | |
| EnFS (%) | 35.13 ± 16.20 | 44.57 ± 10.91 | 0.64 | |
| MW FS (%) | 20.30 ± 7.87 | 19.67 ± 1.81 | 0.94 | |
| Peak E | 0.58 ± 0.21 | 0.44 ± 0.05 | 0.53 | |
| Peak A | 0.30 ± 0.07 | 0.29 ± 0.04 | 0.81 | |
| E/A | 1.46 ± 0.09 | 1.45 ± 0.13 | 0.94 | |
| PWs (mm) | 1.02 ± 0.17 | 1.55 ± 0.14 | 0.04 | * |
| PWd (mm) | 0.58 ± 0.08 | 0.86 ± 0.09 | 0.008 | ** |
| AWs (mm) | 0.98 ± 0.15 | 1.36 ± 0.05 | 0.0430 | * |
| AWd (mm) | 0.58 ± 0.07 | 0.94 ± 0.02 | 0.048 | * |
| LVDs (mm) | 2.37 ± 0.32 | 2.36 ± 0.21 | 0.97 | |
| LVDd (mm) | 3.65 ± 0.17 | 3.85 ± 0.31 | 0.58 | |
| LV mass (g) | 0.07 ± 0.01 | 0.13 ± 0.01 | 0.0003 | *** |
| BW (g) | 28.0 ± 4.3 | 33.3 ± 0.5 | 0.25 | |
| LV mass/BW | 2.39 ± 0.29 | 3.91 ± 0.47 | 0.02 | * |
| epi-D (mm) | 4.80 ± 0.22 | 5.66 ± 0.21 | 0.02 | * |
| epi-V (mm3) | 111.3 ± 14.4 | 181.5 ± 20.8 | 0.02 | * |
| Vols (mm3) | 15.5 ± 10.4 | 13.4 ± 3.7 | 0.85 | |
| Vold (mm3) | 48.7 ± 6.8 | 58.0 ± 14.6 | 0.57 |
| Genes | Fold Amount of mRNA Levels (ICE KO/Wild-Type) | p Value | |
|---|---|---|---|
| Cardiac α-Actin | 1.47 | 0.006 | ** |
| Skeletal α-Actin | 3.88 | 0.034 | * |
| Myosin Heavy Chain Beta (MHC-β) | 7.41 | 0.039 | * |
| Atrial natriuretic peptide (ANP) | 3.72 | 0.021 | * |
| Brain natriuretic peptide (BNP) | 7.66 | 0.036 | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azhar, G.; Nagano, K.; Patyal, P.; Zhang, X.; Verma, A.; Wei, J.Y. Deletion of Interleukin-1β Converting Enzyme Alters Mouse Cardiac Structure and Function. Biology 2024, 13, 172. https://doi.org/10.3390/biology13030172
Azhar G, Nagano K, Patyal P, Zhang X, Verma A, Wei JY. Deletion of Interleukin-1β Converting Enzyme Alters Mouse Cardiac Structure and Function. Biology. 2024; 13(3):172. https://doi.org/10.3390/biology13030172
Chicago/Turabian StyleAzhar, Gohar, Koichiro Nagano, Pankaj Patyal, Xiaomin Zhang, Ambika Verma, and Jeanne Y. Wei. 2024. "Deletion of Interleukin-1β Converting Enzyme Alters Mouse Cardiac Structure and Function" Biology 13, no. 3: 172. https://doi.org/10.3390/biology13030172
APA StyleAzhar, G., Nagano, K., Patyal, P., Zhang, X., Verma, A., & Wei, J. Y. (2024). Deletion of Interleukin-1β Converting Enzyme Alters Mouse Cardiac Structure and Function. Biology, 13(3), 172. https://doi.org/10.3390/biology13030172