
Maternal Obesity Programs the Premature Aging of Rat Offspring Liver Mitochondrial Electron Transport Chain Genes in a Sex-Dependent Manner
 , ,
, ,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
2.3. Care and Maintenance of Offspring to Study for Developmental Programming and Aging Interactions
2.4. Offspring Tissue Collection
2.5. RNA Extraction and cDNA Library Preparation and Sequencing
2.6. Bioinformatic Analysis
2.7. KEGG Pathway Analysis
2.8. Liver Immunohistochemical (IHC) Analysis
2.9. Statistical Analysis
3. Results
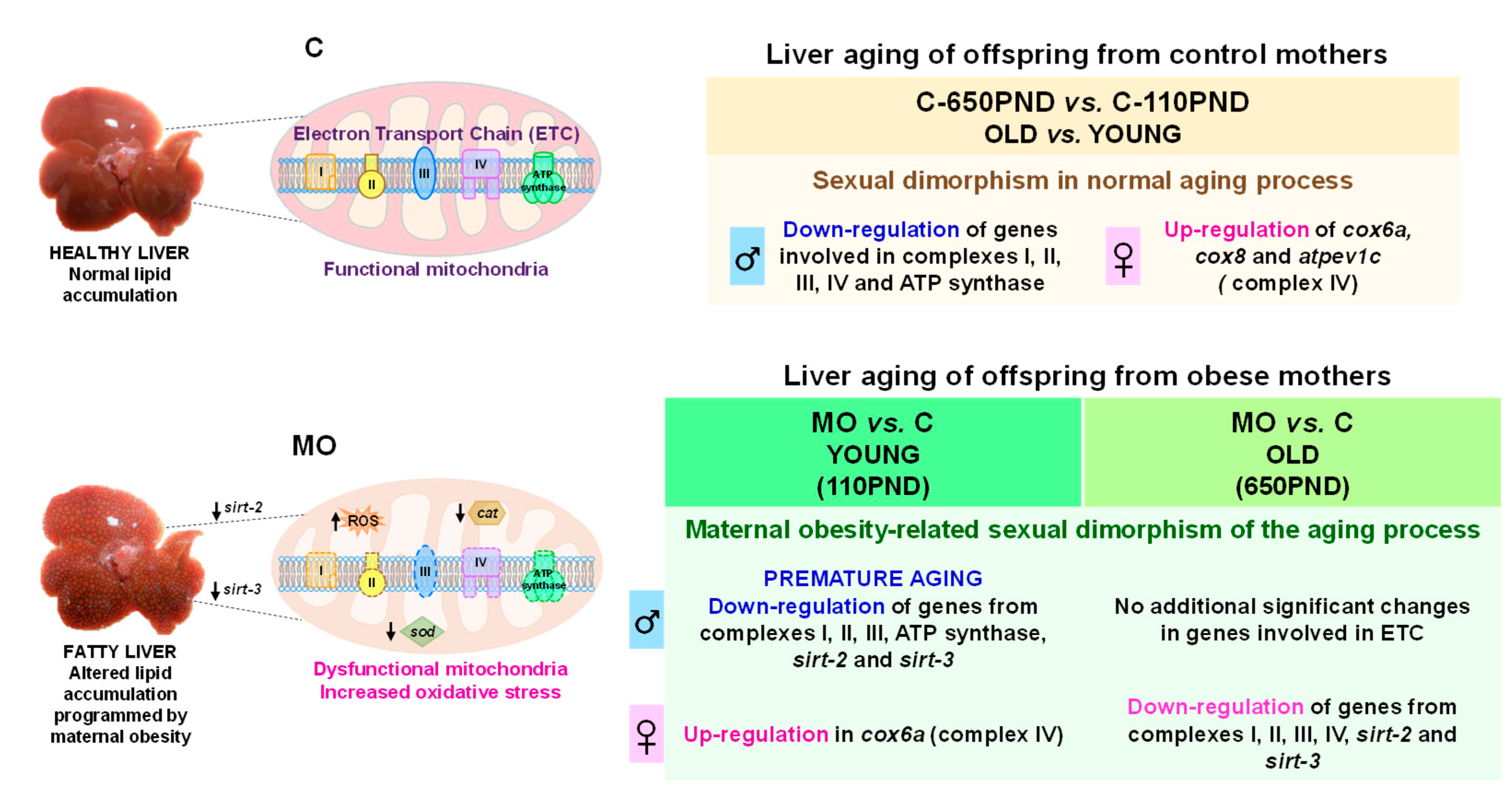
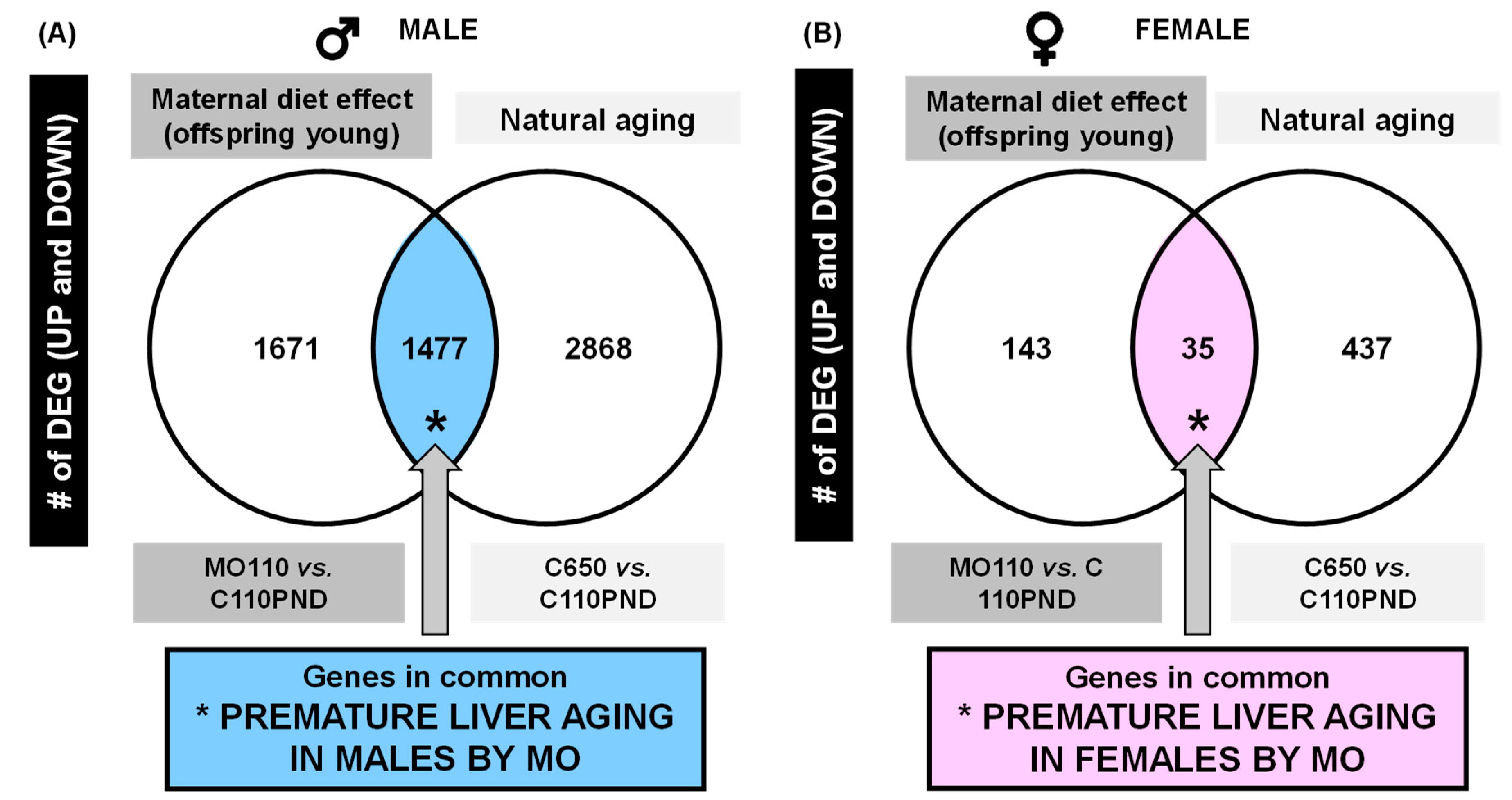
3.1. Liver Differentially Expressed Genes (DEGs)
3.2. KEGG Pathway Analysis for Prematurely Aging Genes in Males and Females
3.3. Pathway Analysis Related to Mitochondria
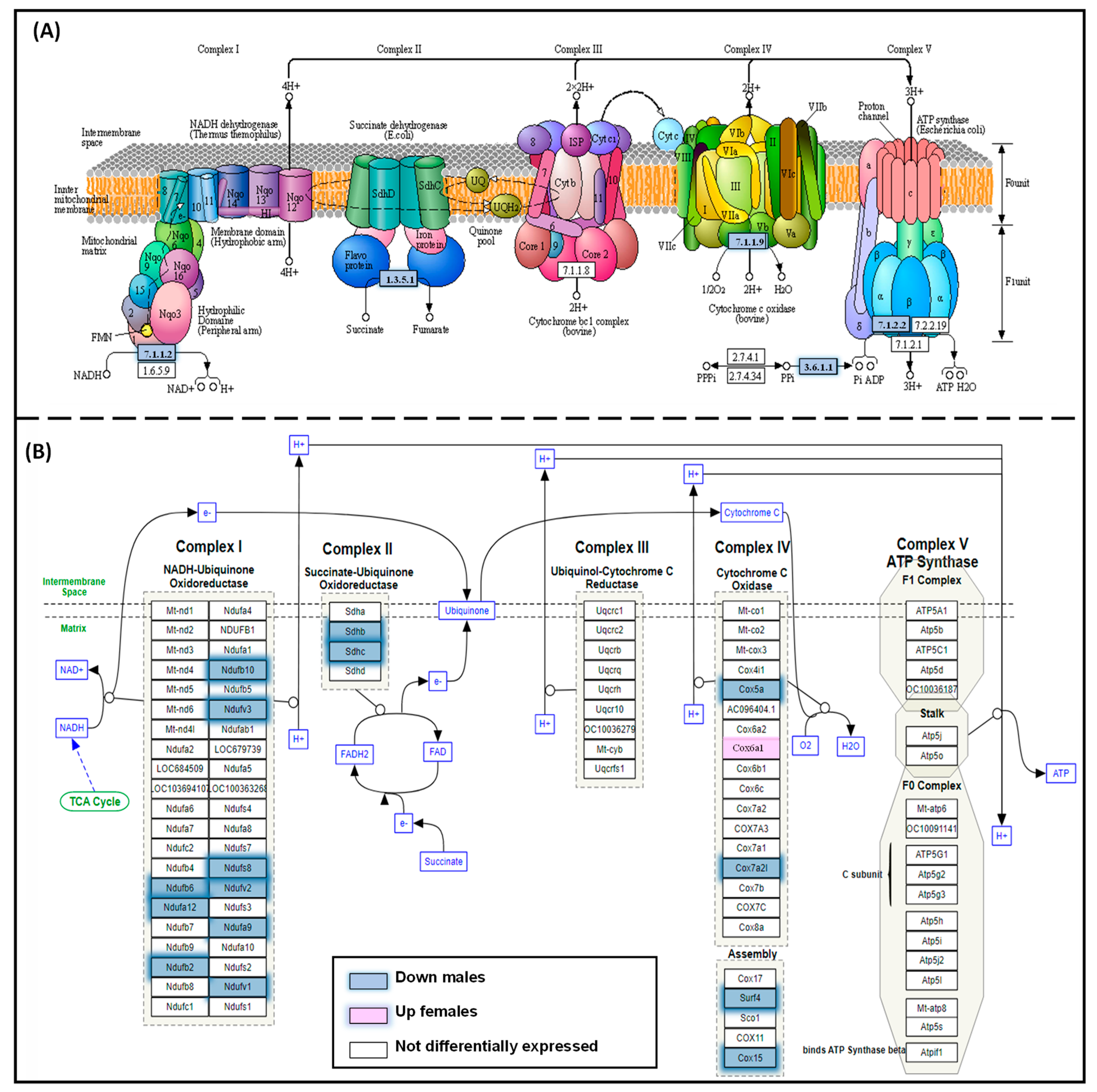
3.4. Oxidative Phosphorylation KEGG Pathway
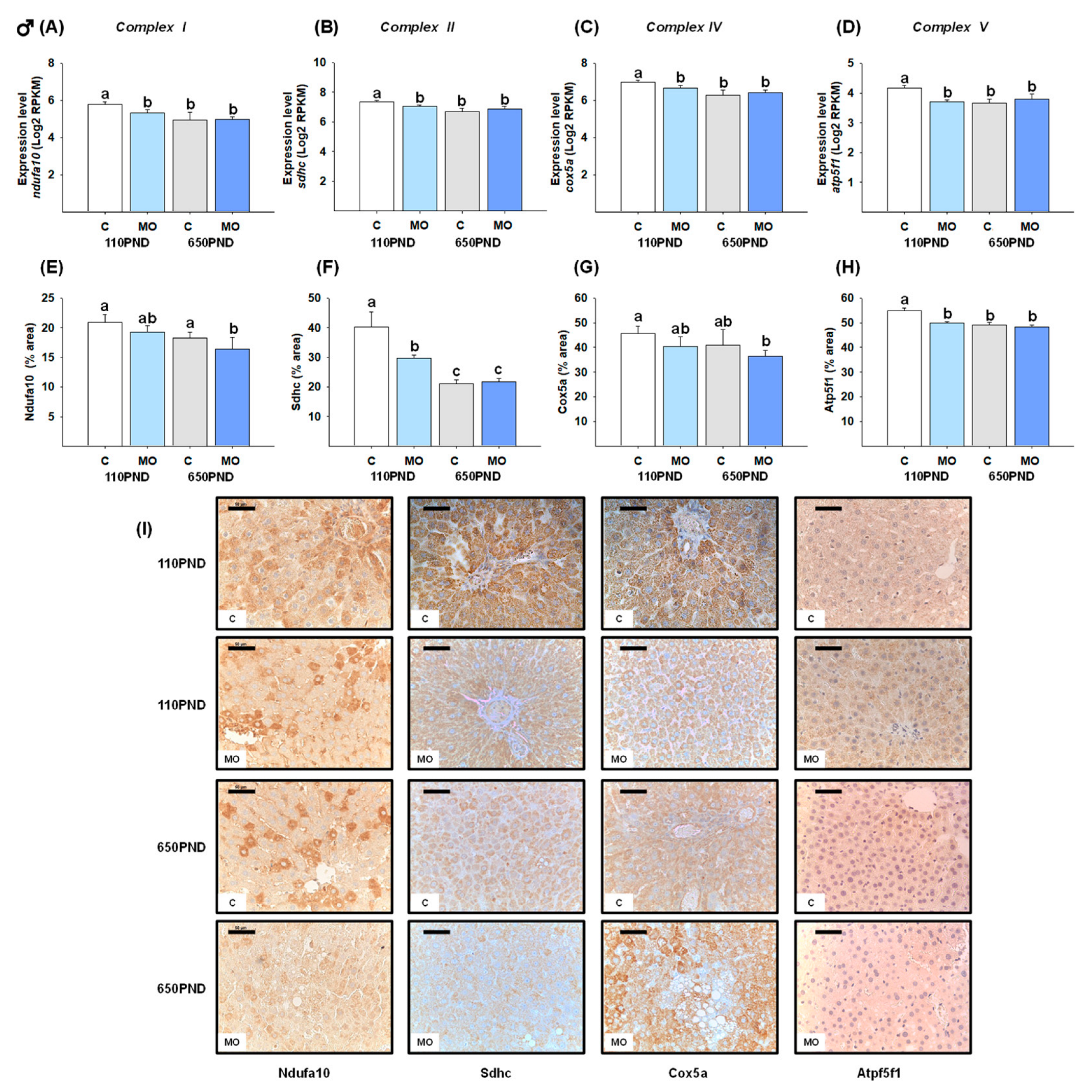
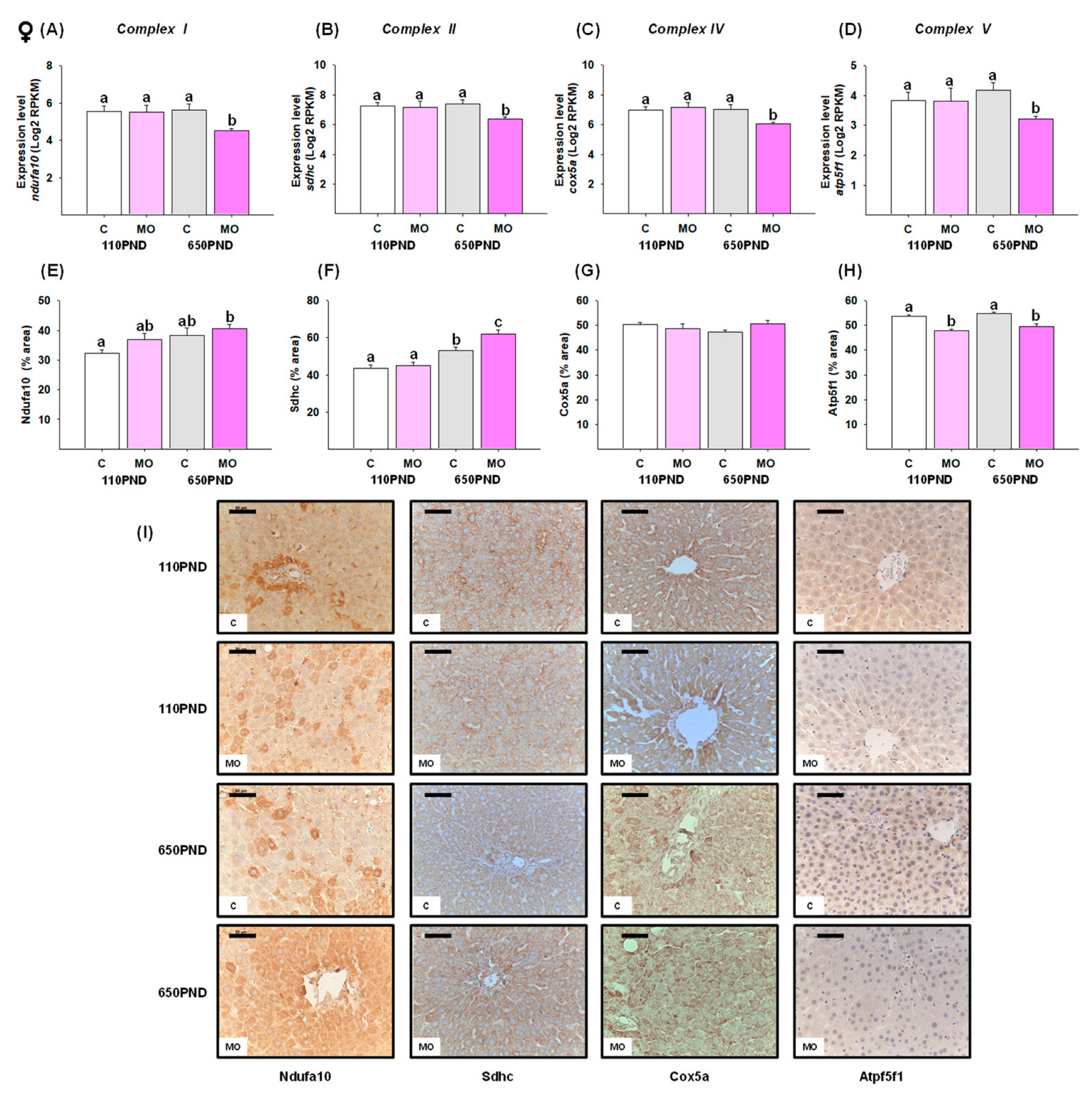
3.5. Male and Female Liver Oxidative Phosphorylation Complexes
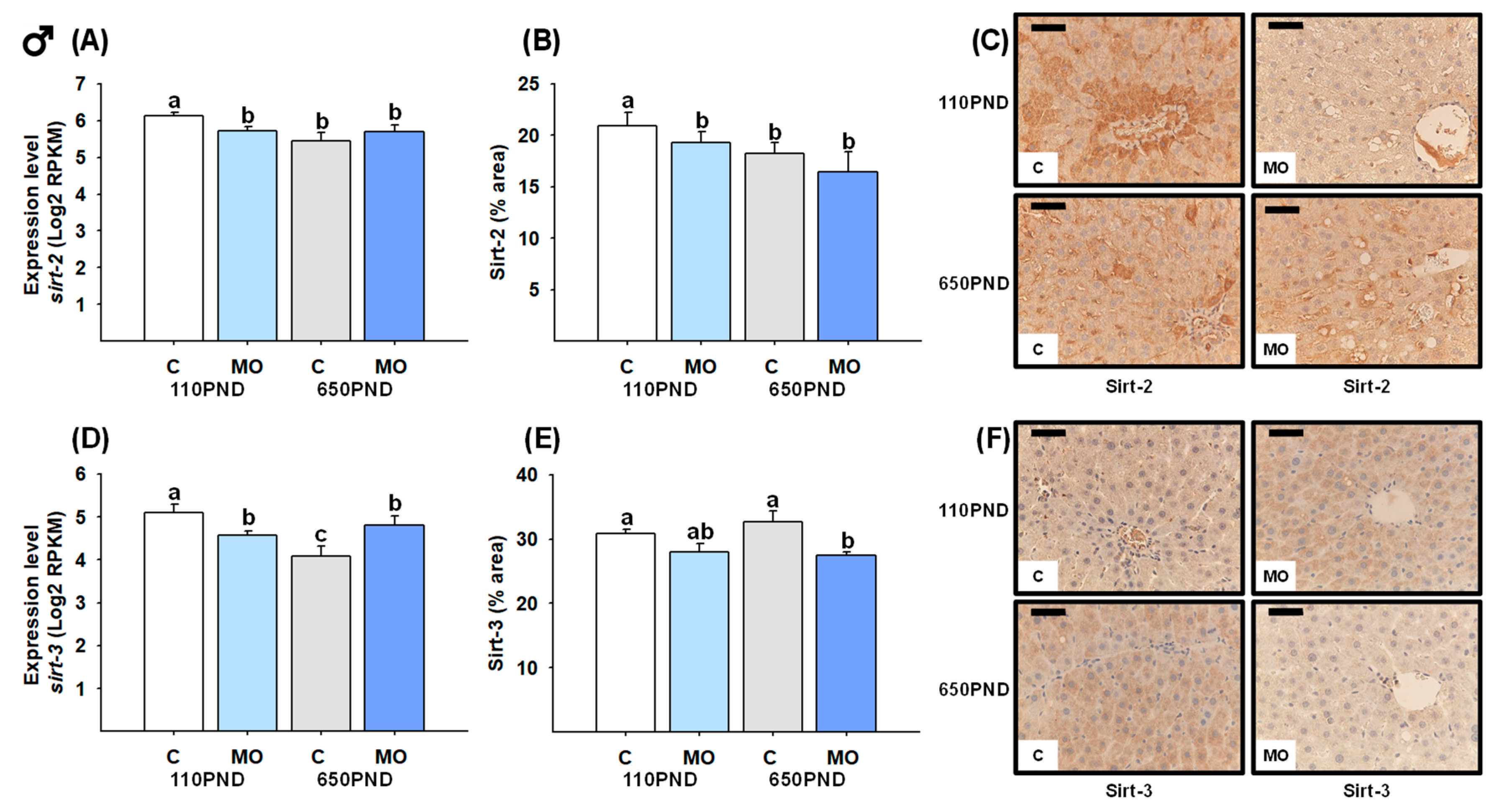
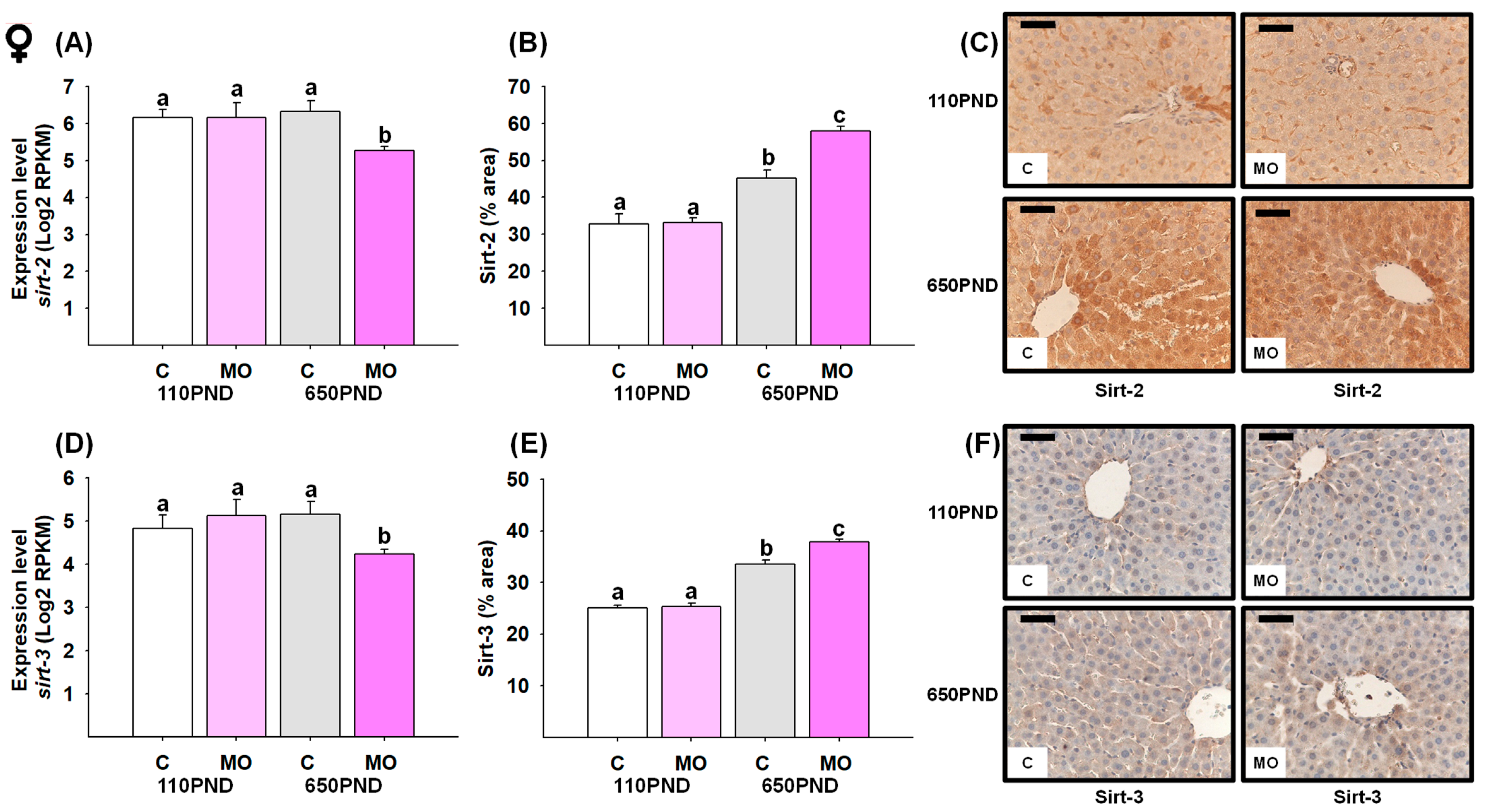
3.6. Male and Female Liver Sirtuins
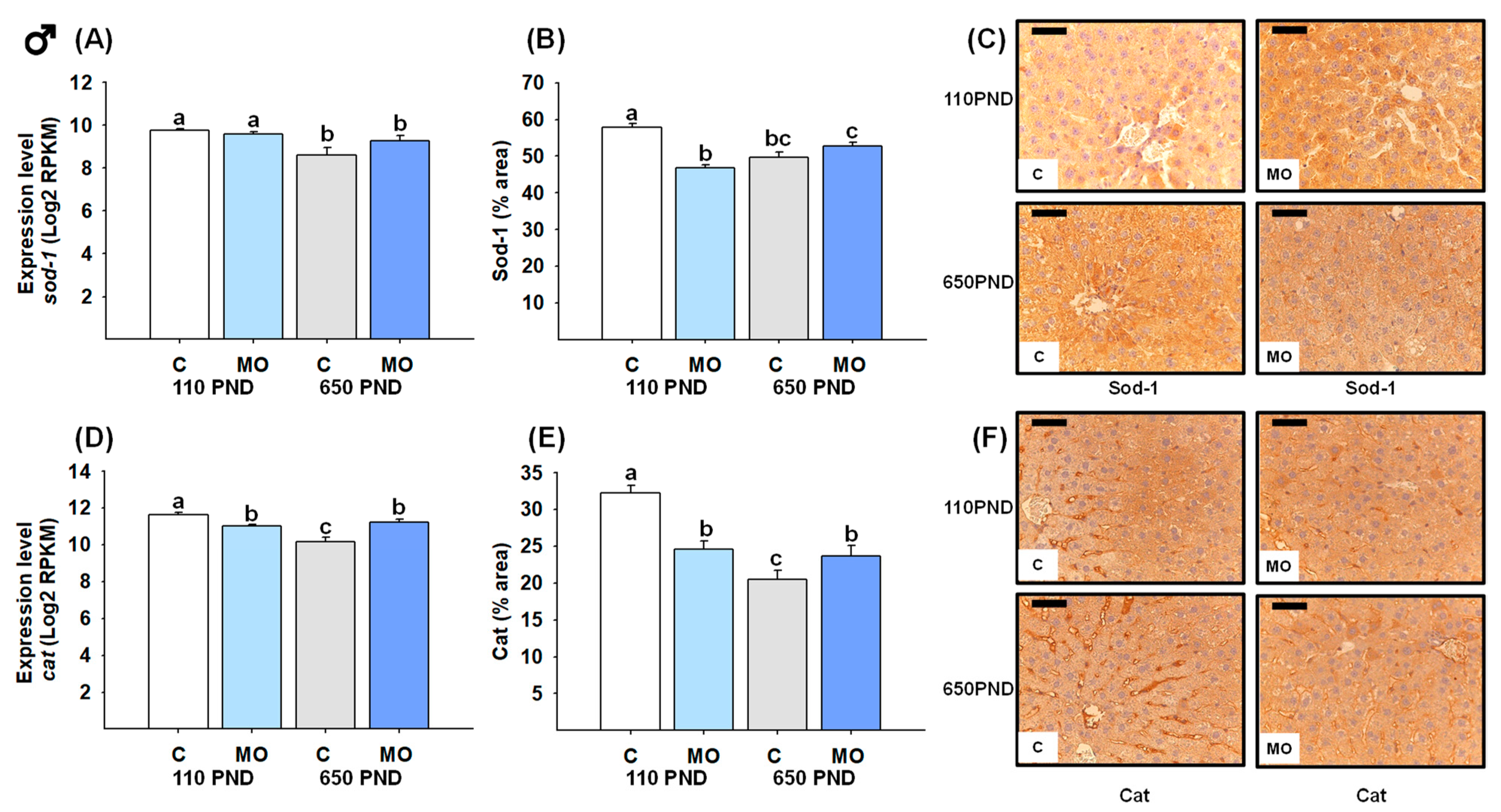
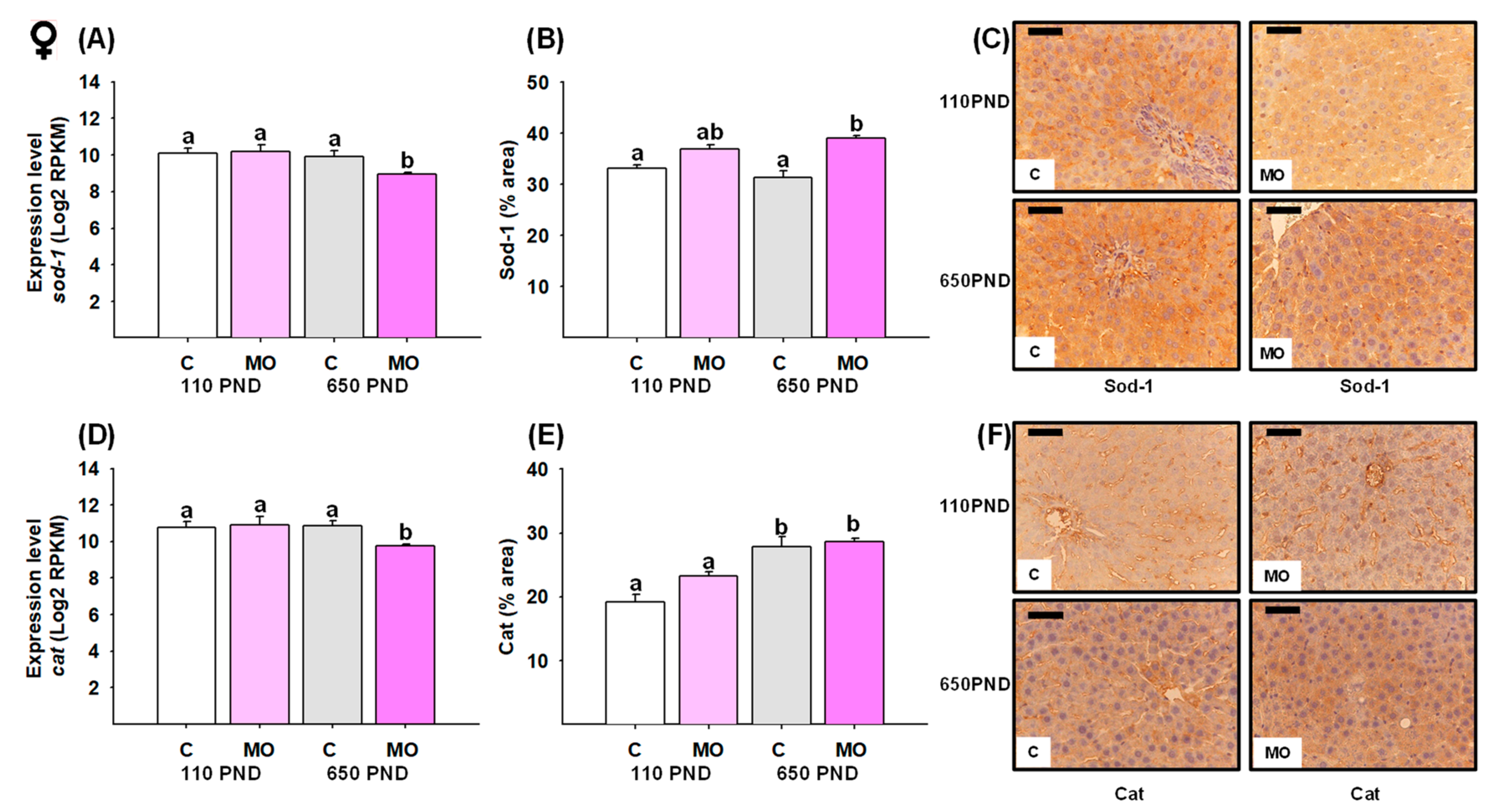
3.7. Male and Female Liver Sod-1 and Catalase
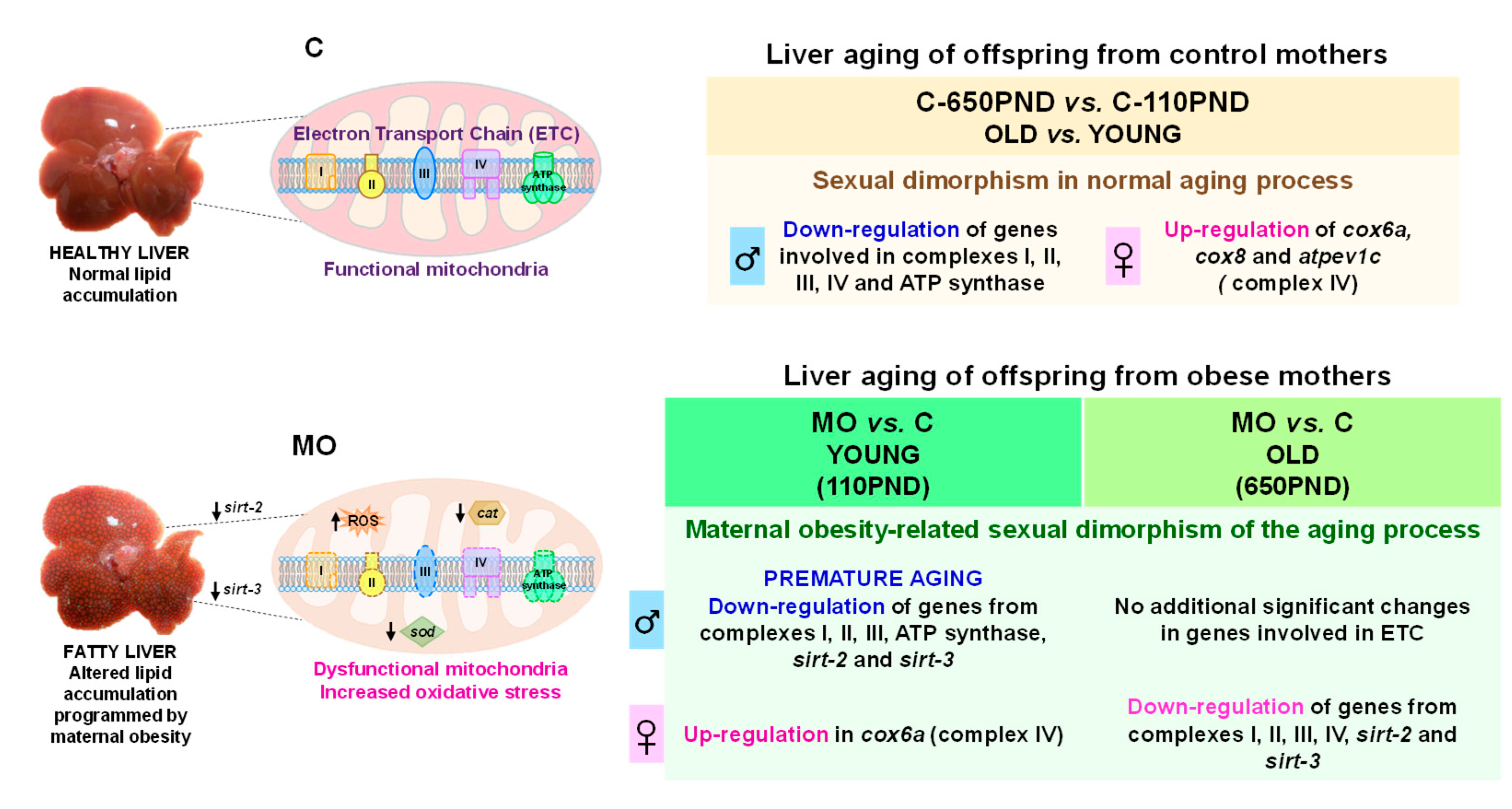
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tiwari, A.; Balasundaram, P. Public Health Considerations Regarding Obesity; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Reichetzeder, C. Overweight and obesity in pregnancy: Their impact on epigenetics. Eur. J. Clin. Nutr. 2021, 75, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Snelgrove-Clarke, E.; Macdonald, D.; Helwig, M.; Alsius, A. Women’s experiences of living with obesity during pregnancy, birthing, and postpartum: A qualitative systematic review protocol. JBI Evid. Synth. 2021, 19, 3183–3189. [Google Scholar] [CrossRef]
- Lomas-Soria, C.; Reyes-Castro, L.A.; Rodriguez-Gonzalez, G.L.; Ibanez, C.A.; Bautista, C.J.; Cox, L.A.; Nathanielsz, P.W.; Zambrano, E. Maternal obesity has sex-dependent effects on insulin, glucose and lipid metabolism and the liver transcriptome in young adult rat offspring. J. Physiol. 2018, 596, 4611–4628. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, E.; Rodriguez-Gonzalez, G.L.; Reyes-Castro, L.A.; Bautista, C.J.; Castro-Rodriguez, D.C.; Juarez-Pilares, G.; Ibanez, C.A.; Hernandez-Rojas, A.; Nathanielsz, P.W.; Montano, S.; et al. DHA Supplementation of Obese Rats throughout Pregnancy and Lactation Modifies Milk Composition and Anxiety Behavior of Offspring. Nutrients. 2021, 13, 4243. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gonzalez, G.L.; De Los Santos, S.; Mendez-Sanchez, D.; Reyes-Castro, L.A.; Ibanez, C.A.; Canto, P.; Zambrano, E. High-fat diet consumption by male rat offspring of obese mothers exacerbates adipose tissue hypertrophy and metabolic alterations in adult life. Br. J. Nutr. 2022, 130, 783–792. [Google Scholar] [CrossRef]
- Ibanez, C.A.; Lira-Leon, G.; Reyes-Castro, L.A.; Rodriguez-Gonzalez, G.L.; Lomas-Soria, C.; Hernandez-Rojas, A.; Bravo-Flores, E.; Solis-Paredes, J.M.; Estrada-Gutierrez, G.; Zambrano, E. Programming Mechanism of Adipose Tissue Expansion in the Rat Offspring of Obese Mothers Occurs in a Sex-Specific Manner. Nutrients 2023, 15, 2245. [Google Scholar] [CrossRef]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef] [PubMed]
- Chavira-Suarez, E.; Reyes-Castro, L.A.; Lopez, T., II; Vargas-Hernandez, L.; Rodriguez-Gonzalez, G.L.; Chavira, R.; Zarate-Segura, P.; Dominguez-Lopez, A.; Vadillo-Ortega, F.; Zambrano, E. Sex-differential RXRalpha gene methylation effects on mRNA and protein expression in umbilical cord of the offspring rat exposed to maternal obesity. Front. Cell Dev. Biol. 2022, 10, 892315. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalez, G.L.; Reyes-Castro, L.A.; Bautista, C.J.; Beltran, A.A.; Ibanez, C.A.; Vega, C.C.; Lomas-Soria, C.; Castro-Rodriguez, D.C.; Elias-Lopez, A.L.; Nathanielsz, P.W.; et al. Maternal obesity accelerates rat offspring metabolic ageing in a sex-dependent manner. J. Physiol. 2019, 597, 5549–5563. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalez, G.L.; Vargas-Hernandez, L.; Reyes-Castro, L.A.; Ibanez, C.A.; Bautista, C.J.; Lomas-Soria, C.; Itani, N.; Estrada-Gutierrez, G.; Espejel-Nunez, A.; Flores-Pliego, A.; et al. Resveratrol Supplementation in Obese Pregnant Rats Improves Maternal Metabolism and Prevents Increased Placental Oxidative Stress. Antioxidants. 2022, 11, 1871. [Google Scholar] [CrossRef]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.; Lawlor, D.A.; Fraser, A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef]
- Vos, M.B.; Abrams, S.H.; Barlow, S.E.; Caprio, S.; Daniels, S.R.; Kohli, R.; Mouzaki, M.; Sathya, P.; Schwimmer, J.B.; Sundaram, S.S.; et al. NASPGHAN Clinical Practice Guideline for the Diagnosis and Treatment of Nonalcoholic Fatty Liver Disease in Children: Recommendations from the Expert Committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN). J. Pediatr. Gastroenterol. Nutr. 2017, 64, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.P.; Feldman, H.S.; Chambers, C.D.; Wilson, L.; Behling, C.; Clark, J.M.; Molleston, J.P.; Chalasani, N.; Sanyal, A.J.; Fishbein, M.H.; et al. Low and High Birth Weights Are Risk Factors for Nonalcoholic Fatty Liver Disease in Children. J. Pediatr. 2017, 187, 141–146.e141. [Google Scholar] [CrossRef]
- Serafim, T.L.; Cunha-Oliveira, T.; Deus, C.M.; Sardao, V.A.; Cardoso, I.M.; Yang, S.; Odhiambo, J.F.; Ghnenis, A.B.; Smith, A.M.; Li, J.; et al. Maternal obesity in sheep impairs foetal hepatic mitochondrial respiratory chain capacity. Eur. J. Clin. Investig. 2021, 51, e13375. [Google Scholar] [CrossRef]
- Reynolds, R.M.; Allan, K.M.; Raja, E.A.; Bhattacharya, S.; McNeill, G.; Hannaford, P.C.; Sarwar, N.; Lee, A.J.; Bhattacharya, S.; Norman, J.E. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: Follow-up of 1 323 275 person years. BMJ 2013, 347, f4539. [Google Scholar] [CrossRef] [PubMed]
- Mennitti, L.V.; Carpenter, A.A.M.; Loche, E.; Pantaleao, L.C.; Fernandez-Twinn, D.S.; Schoonejans, J.M.; Blackmore, H.L.; Ashmore, T.J.; Pisani, L.P.; Tadross, J.A.; et al. Effects of maternal diet-induced obesity on metabolic disorders and age-associated miRNA expression in the liver of male mouse offspring. Int. J. Obes. 2022, 46, 269–278. [Google Scholar] [CrossRef]
- Odhiambo, J.F.; Pankey, C.L.; Ghnenis, A.B.; Ford, S.P. A Review of Maternal Nutritionduring Pregnancy and Impact on the Off-spring through Development: Evidencefrom Animal Models of Over-and Undernutrition. Int. J. Environ. Res. Public Health 2020, 17, 6926. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Feng, X.; Cao, X.; Miao, R.; Sun, Y.; Li, R.; Ye, J.; Zhong, B. Age patterns of nonalcoholic fatty liver disease incidence: Heterogeneous associations with metabolic changes. Diabetol. Metab. Syndr. 2022, 14, 181. [Google Scholar] [CrossRef]
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: From molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef]
- He, F.; Huang, Y.; Song, Z.; Zhou, H.J.; Zhang, H.; Perry, R.J.; Shulman, G.I.; Min, W. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J. Exp. Med. 2021, 218, e20201416. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Schmauck-Medina, T.; Moliere, A.; Lautrup, S.; Zhang, J.; Chlopicki, S.; Madsen, H.B.; Cao, S.; Soendenbroe, C.; Mansell, E.; Vestergaard, M.B.; et al. New hallmarks of ageing: A 2022 Copenhagen ageing meeting summary. Aging 2022, 14, 6829–6839. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Peleg, S. Biphasic Modeling of Mitochondrial Metabolism Dysregulation during Aging. Trends Biochem. Sci. 2017, 42, 702–711. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. BioMed Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef]
- Wang, Y.; Hekimi, S. Mitochondrial dysfunction and longevity in animals: Untangling the knot. Science 2015, 350, 1204–1207. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Bezsonov, E.E.; Orekhov, A.N. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed. Pharmacother. 2021, 142, 112041. [Google Scholar] [CrossRef]
- Dornas, W.; Schuppan, D. Mitochondrial oxidative injury: A key player in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef]
- Fromenty, B.; Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 2023, 78, 415–429. [Google Scholar] [CrossRef]
- Baker, P.R., 2nd; Friedman, J.E. Mitochondrial role in the neonatal predisposition to developing nonalcoholic fatty liver disease. J. Clin. Investig. 2018, 128, 3692–3703. [Google Scholar] [CrossRef]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Bayol, S.A.; Simbi, B.H.; Fowkes, R.C.; Stickland, N.C. A maternal “junk food” diet in pregnancy and lactation promotes nonalcoholic Fatty liver disease in rat offspring. Endocrinology 2010, 151, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Alfaradhi, M.Z.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Musial, B.; Fowden, A.; Ozanne, S.E. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R26–R34. [Google Scholar] [CrossRef] [PubMed]
- Puppala, S.; Li, C.; Glenn, J.P.; Saxena, R.; Gawrieh, S.; Quinn, A.; Palarczyk, J.; Dick, E.J., Jr.; Nathanielsz, P.W.; Cox, L.A. Primate fetal hepatic responses to maternal obesity: Epigenetic signalling pathways and lipid accumulation. J. Physiol. 2018, 596, 5823–5837. [Google Scholar] [CrossRef]
- Wesolowski, S.R.; Kasmi, K.C.; Jonscher, K.R.; Friedman, J.E. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar] [CrossRef]
- Thompson, M.D. Developmental Programming of NAFLD by Parental Obesity. Hepatol. Commun. 2020, 4, 1392–1403. [Google Scholar] [CrossRef]
- Valentini, F.; Rocchi, G.; Vespasiani-Gentilucci, U.; Guarino, M.P.L.; Altomare, A.; Carotti, S. The Origins of NAFLD: The Potential Implication of Intrauterine Life and Early Postnatal Period. Cells 2022, 11, 562. [Google Scholar] [CrossRef]
- Grundy, D. Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J. Physiol. 2015, 593, 2547–2549. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthi, I.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Vet. Clin. Pathol. 2012, 41, 27–31. [Google Scholar] [CrossRef]
- Vega, C.C.; Reyes-Castro, L.A.; Bautista, C.J.; Larrea, F.; Nathanielsz, P.W.; Zambrano, E. Exercise in obese female rats has beneficial effects on maternal and male and female offspring metabolism. Int. J. Obes. 2015, 39, 712–719. [Google Scholar] [CrossRef]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic. Acids. Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Brumbaugh, D.E.; Friedman, J.E. Developmental origins of nonalcoholic fatty liver disease. Pediatr. Res. 2014, 75, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhu, F.; Huang, X.; Yan, P.; Xu, K.; Shen, F.; Sun, J.; Yang, Z.; Jin, G.; Teng, Y. Maternal obesity accelerated non-alcoholic fatty liver disease in offspring mice by reducing autophagy. Exp. Ther. Med. 2021, 22, 716. [Google Scholar] [CrossRef] [PubMed]
- Thorn, S.R.; Baquero, K.C.; Newsom, S.A.; El Kasmi, K.C.; Bergman, B.C.; Shulman, G.I.; Grove, K.L.; Friedman, J.E. Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates. Diabetes 2014, 63, 2702–2713. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, H.; Simon, T.G.; Roelstraete, B.; Stephansson, O.; Soderling, J.; Ludvigsson, J.F. Maternal obesity increases the risk and severity of NAFLD in offspring. J. Hepatol. 2021, 75, 1042–1048. [Google Scholar] [CrossRef]
- Nathanielsz, P.W.; Huber, H.F.; Li, C.; Clarke, G.D.; Kuo, A.H.; Zambrano, E. The nonhuman primate hypothalamo-pituitary-adrenal axis is an orchestrator of programming-aging interactions: Role of nutrition. Nutr. Rev. 2020, 78, 48–61. [Google Scholar] [CrossRef]
- Zambrano, E.; Reyes-Castro, L.A.; Rodriguez-Gonzalez, G.L.; Chavira, R.; Nathanielsz, P.W. Aging Endocrine and Metabolic Phenotypes Are Programmed by Mother’s Age at Conception in a Sex-Dependent Fashion in the Rat. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2304–2307. [Google Scholar] [CrossRef]
- Aiken, C.E.; Tarry-Adkins, J.L.; Spiroski, A.M.; Nuzzo, A.M.; Ashmore, T.J.; Rolfo, A.; Sutherland, M.J.; Camm, E.J.; Giussani, D.A.; Ozanne, S.E. Chronic gestational hypoxia accelerates ovarian aging and lowers ovarian reserve in next-generation adult rats. FASEB J. 2019, 33, 7758–7766. [Google Scholar] [CrossRef]
- Nathanielsz, P.W. Life before Birth: The Challenges of Fetal Development, 1st ed.; Life Course Health Press, LLC.: San Antonio, TX, USA, 2022; p. 351. [Google Scholar]
- Lomas-Soria, C.; Cox, L.A.; Nathanielsz, P.W.; Zambrano, E. Sexual dimorphism in liver cell cycle and senescence signalling pathways in young and old rats. J. Physiol. 2021, 599, 4309–4320. [Google Scholar] [CrossRef]
- Cheng, C.J.; Gelfond, J.A.L.; Strong, R.; Nelson, J.F. Genetically heterogeneous mice exhibit a female survival advantage that is age- and site-specific: Results from a large multi-site study. Aging Cell 2019, 18, e12905. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, M.; Lonardo, A.; Mussi, C.; Baldelli, E.; Pellegrini, E.; Ballestri, S.; Romagnoli, D.; Loria, P. Nonalcoholic fatty liver disease and aging: Epidemiology to management. World J. Gastroenterol. 2014, 20, 14185–14204. [Google Scholar] [CrossRef] [PubMed]
- Capel, F.; Delmotte, M.H.; Brun, M.; Lonchampt, M.; De Fanti, B.; Xuereb, L.; Baschet, L.; Rolland, G.; Galizzi, J.P.; Lockhart, B.; et al. Aging and obesity induce distinct gene expression adaptation in the liver of C57BL/6J mice. J. Nutrigenet. Nutr. 2011, 4, 154–164. [Google Scholar] [CrossRef]
- Chishti, M.A.; Kaya, N.; Binbakheet, A.B.; Al-Mohanna, F.; Goyns, M.H.; Colak, D. Induction of cell proliferation in old rat liver can reset certain gene expression levels characteristic of old liver to those associated with young liver. Age 2013, 35, 719–732. [Google Scholar] [CrossRef]
- White, R.R.; Milholland, B.; MacRae, S.L.; Lin, M.; Zheng, D.; Vijg, J. Comprehensive transcriptional landscape of aging mouse liver. BMC Genom. 2015, 16, 899. [Google Scholar] [CrossRef] [PubMed]
- Bochkis, I.M.; Przybylski, D.; Chen, J.; Regev, A. Changes in nucleosome occupancy associated with metabolic alterations in aged mammalian liver. Cell Rep. 2014, 9, 996–1006. [Google Scholar] [CrossRef]
- Bacalini, M.G.; Franceschi, C.; Gentilini, D.; Ravaioli, F.; Zhou, X.; Remondini, D.; Pirazzini, C.; Giuliani, C.; Marasco, E.; Gensous, N.; et al. Molecular Aging of Human Liver: An Epigenetic/Transcriptomic Signature. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1–8. [Google Scholar] [CrossRef]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- Lozada-Delgado, J.G.; Torres-Ramos, C.A.; Ayala-Peña, S. Aging, oxidative stress, mitochondrial dysfunction, and the liver. In Aging; Preedy, V.R., Patel, V.B., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 37–46. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, Y.; Li, Q.; Li, J. Understanding the Unique Microenvironment in the Aging Liver. Front. Med. 2022, 9, 842024. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef]
- Goedeke, L.; Murt, K.N.; Di Francesco, A.; Camporez, J.P.; Nasiri, A.R.; Wang, Y.; Zhang, X.M.; Cline, G.W.; de Cabo, R.; Shulman, G.I. Sex- and strain-specific effects of mitochondrial uncoupling on age-related metabolic diseases in high-fat diet-fed mice. Aging Cell 2022, 21, e13539. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.M.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Emanuele, E.; Lucia, A.; Galvez, B.G. ‘Adipaging’: Ageing and obesity share biological hallmarks related to a dysfunctional adipose tissue. J. Physiol. 2016, 594, 3187–3207. [Google Scholar] [CrossRef] [PubMed]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Sultanova, R.F.; Schibalski, R.; Yankelevich, I.A.; Stadler, K.; Ilatovskaya, D.V. Sex differences in renal mitochondrial function: A hormone-gous opportunity for research. Am. J. Physiol. Renal. Physiol. 2020, 319, F1117–F1124. [Google Scholar] [CrossRef]
- Von Schulze, A.M.C.; Onyekere, C.; Allen, J.; Geiger, P.; Dorn, G.W., 2nd; Morris, E.M.; Thyfault, J.P. Hepatic mitochondrial adaptations to physical activity: Impact of sexual dimorphism, PGC1α and BNIP3-mediated mitophagy. J. Physiol. 2018, 596, 6157–6171. [Google Scholar] [CrossRef]
- Arnold, P.K.; Finley, L.W.S. Regulation and function of the mammalian tricarboxylic acid cycle. J. Biol. Chem. 2023, 299, 102838. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Murata, D.; Kleiner, D.E.; Anders, R.; Rosenberg, A.Z.; Kaplan, J.; Hamilton, J.P.; Aghajan, M.; Levi, M.; Wang, N.Y.; et al. Prevention and regression of megamitochondria and steatosis by blocking mitochondrial fusion in the liver. iScience 2022, 25, 103996. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Ziolkowska, S.; Binienda, A.; Jablkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 1128. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Guarente, L. Sirtuins at a glance. J. Cell Sci. 2011, 124, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.; Fleming Outeiro, T.; Cavadas, C. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends. Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Lemos, V.; de Oliveira, R.M.; Naia, L.; Szego, E.; Ramos, E.; Pinho, S.; Magro, F.; Cavadas, C.; Rego, A.C.; Costa, V.; et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum. Mol. Genet. 2017, 26, 4105–4117. [Google Scholar] [CrossRef]
- Ren, H.; Hu, F.; Wang, D.; Kang, X.; Feng, X.; Zhang, L.; Zhou, B.; Liu, S.; Yuan, G. Sirtuin 2 Prevents Liver Steatosis and Metabolic Disorders by Deacetylation of Hepatocyte Nuclear Factor 4alpha. Hepatology. 2021, 74, 723–740. [Google Scholar] [CrossRef]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschop, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef]
- Choudhury, M.; Jonscher, K.R.; Friedman, J.E. Reduced mitochondrial function in obesity-associated fatty liver: SIRT3 takes on the fat. Aging 2011, 3, 175–178. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Walsh, M.; Bokov, A.; Ikeno, Y.; Jang, Y.C.; Perez, V.I.; Van Remmen, H.; Richardson, A. Liver specific expression of Cu/ZnSOD extends the lifespan of Sod1 null mice. Mech. Ageing Dev. 2016, 154, 1–8. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A). MALE. | |||
|---|---|---|---|
| Id Pathway | Name | Size | p-Value |
| rno01100 | Metabolic pathways | 1380 | <2.2 × 10−16 |
| rno00280 | Valine, leucine, and isoleucine degradation | 56 | 1.7 × 10−6 |
| rno00640 | Propanoate metabolism | 32 | 1.0 × 10−5 |
| rno00190 | Oxidative phosphorylation | 143 | 4.1 × 10−5 |
| rno04932 | Non-alcoholic fatty liver disease (NAFLD) | 159 | 9.4 × 10−5 |
| rno04714 | Thermogenesis | 243 | 9.5 × 10−5 |
| rno00260 | Glycine, serine, and threonine metabolism | 40 | 1.0 × 10−4 |
| rno00310 | Lysine degradation | 61 | 1.1 × 10−4 |
| rno00140 | Steroid hormone biosynthesis | 84 | 1.1 × 10−4 |
| rno01200 | Carbon metabolism | 127 | 1.4 × 10−4 |
| rno00270 | Cysteine and methionine metabolism | 49 | 1.7 × 10−4 |
| rno00630 | Glyoxylate and dicarboxylate metabolism | 30 | 2.2 × 10−4 |
| rno03022 | Basal transcription factors | 45 | 1.3 × 10−3 |
| rno00760 | Nicotinate and nicotinamide metabolism | 32 | 1.8 × 10−3 |
| rno00380 | Tryptophan metabolism | 47 | 1.9 × 10−3 |
| rno04122 | Sulfur relay system | 9 | 2.8 × 10−3 |
| rno04142 | Lysosome | 129 | 2.8 × 10−3 |
| rno00510 | N-Glycan biosynthesis | 51 | 3.6 × 10−3 |
| rno03420 | Nucleotide excision repair | 47 | 6.6 × 10−3 |
| rno04144 | Endocytosis | 275 | 6.6 × 10−3 |
| rno04217 | Necroptosis | 161 | 7.5 × 10−3 |
| rno04120 | Ubiquitin mediated proteolysis | 141 | 7.6 × 10−3 |
| rno00670 | One carbon pool by folate | 18 | 8.3 × 10−3 |
| rno00830 | Retinol metabolism | 85 | 9.1 × 10−3 |
| rno03060 | Protein export | 26 | 1.0 × 10−2 |
| rno00053 | Ascorbate and aldarate metabolism | 27 | 1.2 × 10−2 |
| rno00563 | Glycosylphosphatidylinositol (GPI)-anchor biosynthesis | 27 | 1.2 × 10−2 |
| rno00650 | Butanoate metabolism | 28 | 1.5 × 10−2 |
| rno04141 | Protein processing in the endoplasmic reticulum | 164 | 1.8 × 10−2 |
| rno00071 | Fatty acid degradation | 47 | 2.0 × 10−2 |
| rno00350 | Tyrosine metabolism | 40 | 2.5 × 10−2 |
| rno04146 | Peroxisome | 88 | 2.8 × 10−2 |
| rno00410 | Beta-Alanine metabolism | 33 | 3.2 × 10−2 |
| rno00730 | Thiamine metabolism | 17 | 3.2 × 10−2 |
| rno00920 | Sulfur metabolism | 10 | 3.3 × 10−2 |
| rno00330 | Arginine and proline metabolism | 52 | 3.5 × 10−2 |
| rno00010 | Glycolysis/Gluconeogenesis | 72 | 3.8 × 10−2 |
| rno00980 | Metabolism of xenobiotics by cytochrome P450 | 74 | 4.5 × 10−2 |
| rno03040 | Spliceosome | 138 | 4.7 × 10−2 |
| (B). FEMALE | |||
| Id pathway | Name | Size | p-Value |
| rno04064 | NF-kappa B signaling pathway | 97 | 5.4 × 10−3 |
| rno00230 | Purine metabolism | 182 | 1.8 × 10−2 |
| rno04060 | Cytokine–cytokine receptor interaction | 269 | 3.8 × 10−2 |
| (A). KEGG Pathway | ||||
|---|---|---|---|---|
| Comparison | p-Value | Genes Down | Genes Up | |
| MO-110PND vs. C-110PND | Oxidative phosphorylation | 7.8 × 10−5 | 39 | 0 |
| Maternal diet effect (young) | Lysosome | 1.7 × 10−4 | 37 | 0 |
| Ribosome | 3.0 × 10−3 | 41 | 0 | |
| Peroxisome | 9.5 × 10−3 | 23 | 0 | |
| Citrate cycle (TCA cycle) | 1.1 × 10−2 | 11 | 0 | |
| C-650PND vs. C-110PND | Peroxisome | <2.2 × 10−16 | 55 | 0 |
| Aging effect in controls | Oxidative phosphorylation | 2.9 × 10−14 | 68 | 0 |
| Mitophagy | 1.6 × 10−4 | 27 | 0 | |
| Lysosome | 8.6 × 10−4 | 43 | 0 | |
| Citrate cycle (TCA cycle) | 7.2 × 10−2 | 11 | 0 | |
| (B). Wikipathway | ||||
| Comparison | p-Value | Genes Down | Genes Up | |
| MO-110PND vs. C-110PND | Oxidative phosphorylation | 1.0 × 10−3 | 22 | 0 |
| Maternal diet effect (young) | Electron Transport Chain | 1.5 × 10−3 | 30 | 0 |
| TCA Cycle | 1.7 × 10−2 | 10 | 0 | |
| Oxidative Stress | 1.9 × 10−2 | 12 | 0 | |
| C-650PND vs. C-110PND | Electron Transport Chain | 1.0 × 10−10 | 51 | 0 |
| Aging effect in controls | Mitochondrial LC-Fatty Acid Beta-Oxidation | 1.5 × 10−7 | 14 | 0 |
| Oxidative phosphorylation | 8.1 × 10−7 | 32 | 0 | |
| Oxidative stress | 1.4 × 10−5 | 20 | 0 | |
| TCA Cycle | 7.3 × 10−2 | 10 | 0 | |
| (C). Reactome | ||||
| Comparison | p-Value | Genes Down | Genes Up | |
| MO-110PND vs. C-110PND | Mitochondrial translation termination | 5.2 × 10−14 | 46 | 0 |
| Maternal diet effect (young) | Mitochondrial translation | 9.5 × 10−14 | 46 | 0 |
| The citric acid (TCA) cycle and respiratory electron transport | 1.3 × 10−6 | 46 | 0 | |
| Respiratory electron transport | 7.1 × 10−5 | 25 | 0 | |
| Citric acid cycle (TCA cycle) | 9.8 × 10−4 | 10 | 0 | |
| Pyruvate metabolism and Citric Acid (TCA) cycle | 1.3 × 10−3 | 17 | 0 | |
| Peroxisomal protein import | 1.9 × 10−3 | 18 | 0 | |
| C-650PND vs. C-110PND | Mitochondrial translation | <2.2 × 10−16 | 66 | 0 |
| Aging effect in controls | Mitochondrial translation termination | <2.2 × 10−16 | 66 | 0 |
| Mitochondrial translation elongation | <2.2 × 10−16 | 65 | 0 | |
| Peroxisomal protein import | 2.5 × 10−10 | 47 | 0 | |
| Respiratory electron transport | 3.4 × 10−10 | 39 | 0 |
| Comparison | Genes, Male | p-Value | Genes, Female | p-Value |
|---|---|---|---|---|
| (1) MO-110PND vs. C-110PND Maternal diet effect (young) | atp5d, atp5g2, atp5i, atp5o, atp6v0a1, atp6v1f, cox15, cox5b, cox7a2l, cyc1, lhpp, ndufa10l1, ndufa11, ndufa12, ndufa9, ndufb10, ndufb11, ndufb2, ndufb3, ndufb6, ndufb8, ndufc2, ndufs1, ndufs2, ndufs7, ndufs8, ndufv1, ndufv2, ndufv3, ppa2, sdha, sdhb, tcirg1, uqcr11, uqcrc1, uqcrc2, | 7.8 × 10−5 | cox6a | --- |
| (2) MO-650PND vs. C-650PND Maternal diet effect (old) | atp6v0a4, atp6v0a2 | ---- | atp5f1a, atp5f1b, atp5f1c, atp5f1c, atp5f1d, atp5f1e, atp5mc1, atp5mc2, atp5me, atp5mf, atp5mg, atp5pb, atp5pd, atp5pf, atp5po, atp6ap1, atp6v0a1, atp6v0a2, atp6v0a2, atp6v0c, atp6v0d1, atp6v0d2, atp6v0e1, atp6v1a, atp6v1b2, atp6v1c1, atp6v1c2, atp6v1d, atp6v1e1, atp6v1f, atp6v1g1, atp6v1h, cox15, cox17, cox4i1, cox5a, cox5b, cox6a1, cox6b1, cox6c, cox7a2, cox7a2l, cox7a2l2, cox7b, cox7c, cox8a, cox8b, cyc1, lhpp, ndufa1, ndufa10, ndufa10l1, ndufa11, ndufa12, ndufa13, ndufa2, ndufa4, ndufa5, ndufa6, ndufa7, ndufa8, ndufa9, ndufab1, ndufb10, ndufb11, ndufb2, ndufb3, ndufb4, ndufb5, ndufb6, ndufb7, ndufb8, ndufb9, ndufc2, ndufs1, ndufs2, ndufs3, ndufs4, ndufs5, ndufs6, ndufs7, ndufs8, ndufv1, ndufv2, ndufv3, ppa1, ppa2, sdha, sdhb, sdhc, sdhd, tcirg1, uqcr10, uqcr11, uqcrb, uqcrc2, uqcrfs1, uqcrh, uqcrq | 1.3 × 10−8 |
| (3) C-650PND vs. C-110 PND Aging effect in controls | atp5f1c, atp5mc1, atp5me, atp5mf, atp5mg, atp5pb, atp5pd, atp5pf, atp5po, atp6v0a2, atp6v0c, atp6v0d1, atp6v0e1, atp6v1a, atp6v1f, atp6v1g1, atp6v1h, cox15, cox17, cox4i1, cox5a, cox5b, cox6a1, cox6b1, cox6c, cox7a2, cox7a2l, cox7a2l2, cox7b, cox7c, cox8a, ndufa1, ndufa10l1, ndufa11, ndufa12, ndufa13, ndufa2, ndufa4, ndufa5, ndufa6, ndufa7, ndufa8, ndufa9, ndufb10, ndufb11, ndufb2, ndufb3, ndufb4, ndufb5, ndufb6, ndufb7, ndufb9, ndufs3, ndufs5, ndufs6, ndufs8, ndufv1, ndufv2, ndufv3, ppa1, ppa2, sdhb, sdhc, sdhd, tcirg1, uqcr10, uqcrb, uqcrfs1, uqcrh, uqcrq | 2.42 × 10−14 | cox6a, cox8, atpev1c | --- |
| (4) MO-650PND vs. MO-110PND Aging effect in MO | Cox8 | --- | ap2s1, apaf1, atp5f1c, atp5f1d, atp5f1e, atp5mc1, atp5mc2, atp5pb, atp5pd, atp5pf, atp5po, bax, casp3, cox4i1, cox5a, cox5b, cox6a1, cox6b1, cox6c, cox7a2, cox7a2l, cox7a2l2, cox7b, cox7c, cox8a, cox8b, crebbp, creb3l1, cycs, cyct, cyct, dlg4, dnah1, gpx1, hdac2, ndufa1, ndufa10, ndufa11, ndufa12, ndufa13, ndufa2, ndufa4, ndufa5, ndufa6, ndufa7, ndufa9, ndufab1, ndufb10, ndufb11, ndufb2, ndufb3, ndufb4, ndufb5, ndufb6, ndufb7, ndufb9, ndufc2, ndufs3, ndufs4, ndufs5, ndufs6, ndufs7, ndufs8, ndufv2, ndufv3, plcb1, polr2f, polr2g, polr2h, polr2i, polr2j, polr2k, pparg, sdhd, slc25a5, sod1, sod2, uqcr10, uqcr11, uqcr11, uqcrb, uqcrfs1, uqcrh, uqcrq, vdac3 | <2.2 × 10 −16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lomas-Soria, C.; Rodríguez-González, G.L.; Ibáñez, C.A.; Reyes-Castro, L.A.; Nathanielsz, P.W.; Zambrano, E. Maternal Obesity Programs the Premature Aging of Rat Offspring Liver Mitochondrial Electron Transport Chain Genes in a Sex-Dependent Manner. Biology 2023, 12, 1166. https://doi.org/10.3390/biology12091166
Lomas-Soria C, Rodríguez-González GL, Ibáñez CA, Reyes-Castro LA, Nathanielsz PW, Zambrano E. Maternal Obesity Programs the Premature Aging of Rat Offspring Liver Mitochondrial Electron Transport Chain Genes in a Sex-Dependent Manner. Biology. 2023; 12(9):1166. https://doi.org/10.3390/biology12091166
Chicago/Turabian StyleLomas-Soria, Consuelo, Guadalupe L. Rodríguez-González, Carlos A. Ibáñez, Luis A. Reyes-Castro, Peter W. Nathanielsz, and Elena Zambrano. 2023. "Maternal Obesity Programs the Premature Aging of Rat Offspring Liver Mitochondrial Electron Transport Chain Genes in a Sex-Dependent Manner" Biology 12, no. 9: 1166. https://doi.org/10.3390/biology12091166
APA StyleLomas-Soria, C., Rodríguez-González, G. L., Ibáñez, C. A., Reyes-Castro, L. A., Nathanielsz, P. W., & Zambrano, E. (2023). Maternal Obesity Programs the Premature Aging of Rat Offspring Liver Mitochondrial Electron Transport Chain Genes in a Sex-Dependent Manner. Biology, 12(9), 1166. https://doi.org/10.3390/biology12091166