
Cellular and Molecular Pathophysiology of Traumatic Brain Injury: What Have We Learned So Far?
 , , ,
, , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
1.1. Brain Edema
1.2. Types and Localization of Traumatic Lesions following Head Injury
1.3. Inflammation
1.4. Excitotoxicity
1.5. Oxidative Stress
1.6. Metabolic Disturbances
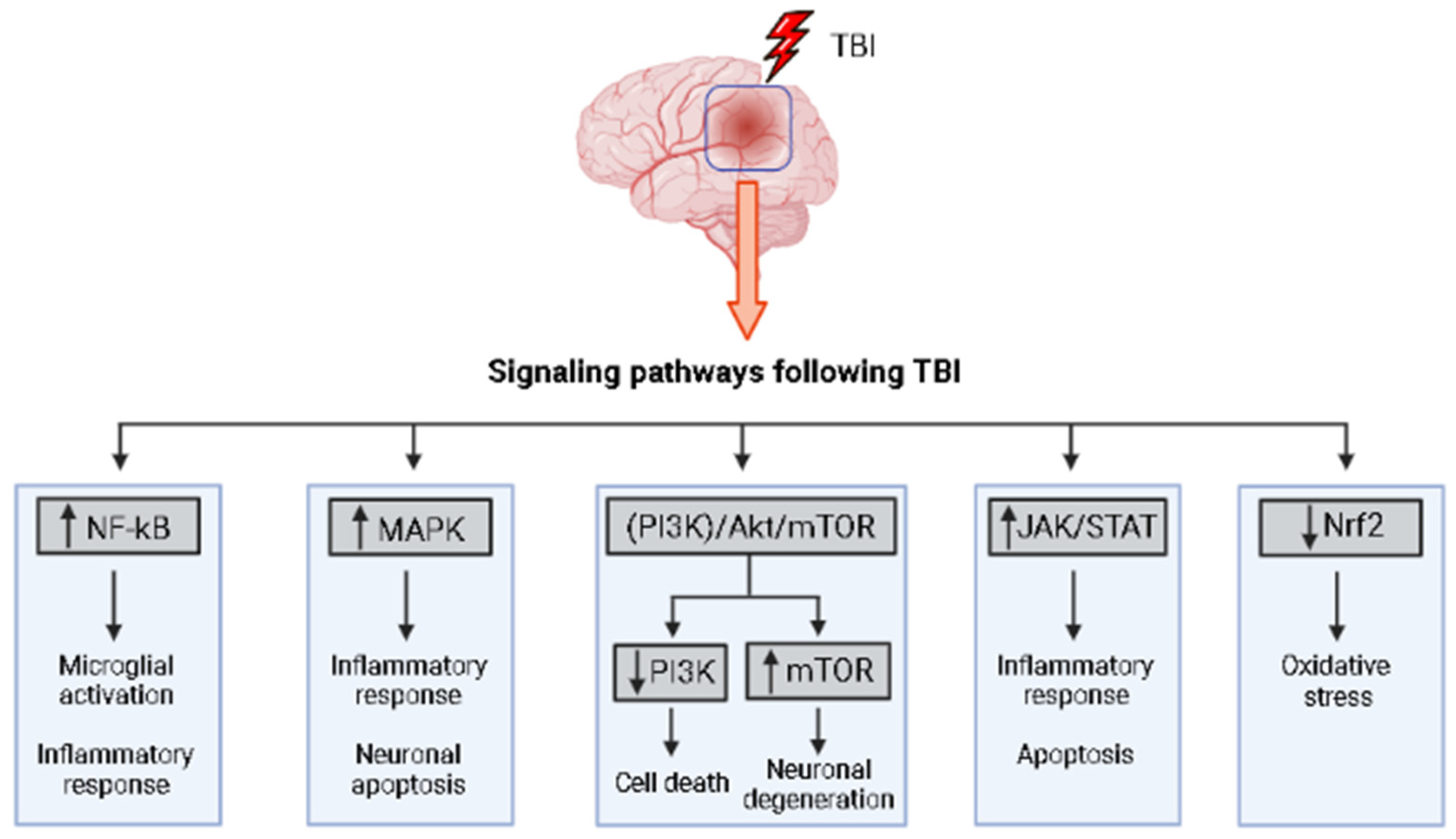
1.7. Signaling Pathways
2. Biomarkers following TBI
3. Inflammasomes in the Context of Neuroinflammation and TBI Pathophysiology
4. Translational Approach of Data Obtained in Animal Models of TBI and Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An overview of epidemiology, pathophysiology, and medical management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef]
- Feigin, V.L.; Forouzanfar, M.H.; Krishnamurthi, R.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.; Truelsen, T.; et al. Global and regional burden of stroke during 1990–2010: Findings from the Global Burden of Disease study 2010. Lancet 2014, 383, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef]
- Meaney, D.F.; Morrison, B.; Bass, C.D. The mechanics of traumatic brain injury: A review of what we know and what we need to know for reducing its societal burden. J. Biomech. Eng. 2014, 136, 021008. [Google Scholar] [CrossRef]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic Brain Injury: Oxidative Stress and Neuroprotection. Antioxid. Redox Signal 2013, 19, 836–853. [Google Scholar] [CrossRef]
- James, S.L.; Theadom, A.; Ellenbogen, R.G.; Bannick, M.S.; Montjoy-Venning, W.; Lucchesi, L.R.; Abbasi, N.; Abdulkader, R.; Abraha, H.N.; Adsuar, J.C.; et al. Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990–2016: A systematic analysis for the Global Burden of Disease Study. Lancet Neurol. 2019, 18, 56–87. [Google Scholar] [CrossRef] [PubMed]
- Soendergaard, P.L.; Siert, L.; Poulsen, I.; Wood, R.L.; Norup, A. Measuring neurobehavioral disabilities among severe brain injury survivors: Reports of survivors and proxies in the chronic phase. Front. Neurol. 2019, 10, 51. [Google Scholar] [CrossRef]
- Ma, V.; Chan, L.; Carruthers, K. Incidence, prevalence, costs, and impact on disability of common conditions requiring rehabilitation in the United States: Stroke, spinal cord injury, traumatic brain injury, multiple sclerosis, osteoarthritis, rheumatoid arthritis, limb loss, and back pain. Arch. Phys. Med. Rehabil. 2014, 95, 986–995. [Google Scholar] [CrossRef]
- Carson, H.J. Brain trauma in head injuries presenting with and without concurrent skull fractures. J. Forensic Leg. Med. 2009, 16, 115–120. [Google Scholar] [CrossRef]
- Santiago, L.A.; Oh, B.C.; Dash, P.K.; Holcomb, J.B.; Wade, C.E. A clinical comparison of penetrating and blunt traumatic brain injuries. Brain Inj. 2012, 26, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.R.; Fertig, S.J.; Desrocher, R.E.; Koroshetz, W.J.; Pancrazio, J.J. Neurological Effects of Blast Injury. J. Trauma Acute Care Surg. 2010, 68, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Fehily, B.; Fitzgerald, M. Repeated Mild Traumatic Brain Injury. Cell Transpl. 2017, 26, 1131–1155. [Google Scholar] [CrossRef]
- Kaur, P.; Sharma, S. Recent Advances in Pathophysiology of Traumatic Brain Injury. Curr. Neuropharmacol. 2018, 16, 1224–1238. [Google Scholar] [CrossRef] [PubMed]
- Bramlett, H.M.; Dietrich, W.D. Long-term consequences of traumatic brain injury: Current status of potential mechanisms of injury and neurological outcomes. J. Neurotrauma 2015, 32, 1834–1848. [Google Scholar] [CrossRef] [PubMed]
- Mira, R.G.; Lira, M.; Cerpa, W. Traumatic Brain Injury: Mechanisms of Glial Response. Front. Physiol. 2021, 12, 740939. [Google Scholar] [CrossRef]
- Frati, A.; Cerretani, D.; Fiaschi, A.I.; Frati, P.; Gatto, V.; La Russa, R.; Pesce, A.; Pinchi, E.; Santurro, A.; Fraschetti, F.; et al. Diffuse Axonal Injury and Oxidative Stress: A Comprehensive Review. Int. J. Mol. Sci. 2017, 18, 2600. [Google Scholar] [CrossRef]
- Meythaler, J.M.; Pedussi, J.D.; Eleftheriou, E.; Novack, T.A. Current concepts: Diffuse axonal injury–associated traumatic brain injury. Arch. Phys. Med. Rehabil. 2001, 82, 1461–1471. [Google Scholar] [CrossRef]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef]
- Palmer, A.M.; Marion, D.W.; Botscheller, M.L.; Swedlow, P.E.; Styren, S.D.; DeKosky, S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993, 61, 2015–2024. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Rancan, M.; Staherl, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef]
- Pun, P.B.L.; Lu, J.; Moochhala, S. Involvement of ROS in BBB dysfunction. Free Radic. Res. 2009, 43, 348–364. [Google Scholar] [CrossRef]
- Marmarou, A. Traumatic brain edema: An overview. In Brain Edema IX, Proceedings of the Ninth International Symposium, Tokyo, Japan, 16–19 May 1993; Springer: Vienna, Austria, 1994; Volume 60, pp. 421–424. [Google Scholar] [CrossRef]
- Buki, A.; Okonkwo, D.O.; Wang, K.K.; Povlishock, J.T. Cytochrome c release and caspase activation in traumatic axonal injury. J. Neurosci. 2000, 20, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Lee, B.E.; Sohn, M.; Song, H.K.; Suh, S.W. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012, 1481, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Gu, Q.; Peterson, P.L.; Muizelaar, J.P.; Lee, C.P. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma 1997, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Nola, A.; Becker, M.; Picard, K.; Vernoux, N.; Frias, E.S.; Feng, X.; Tremblay, M.E.; Rosi, S. Novel microglia-mediated mechanisms underlying synaptic loss and cognitive impairment after traumatic brain injury. Brain Behav. Immun. 2021, 98, 122–135. [Google Scholar] [CrossRef]
- Wu, L.; Chung, J.Y.; Saith, S.; Tozzi, L.; Buckley, E.M.; Sanders, B.; Franceschini, M.A.; Lule, S.; Izzy, S.; Lok, J.; et al. Repetitive head injury in adolescent mice: A role for vascular inflammation. J. Cereb. Blood Flow. Metab. 2019, 39, 2196–2209. [Google Scholar] [CrossRef]
- Foda, M.A.; Marmarou, A. A new model of diffuse brain injury in rats. Part II: Morphological characterization. J. Neurosurg. 1994, 80, 301–313. [Google Scholar] [CrossRef]
- Povlishock, J.T.; Marmarou, A.; McIntosh, T.; Trojanowski, J.Q.; Moroi, J. Impact acceleration injury in the rat: Evidence for focal axolemmal change and related neurofilament sidearm alteration. J. Neuropathol. Exp. Neurol. 1997, 56, 347–359. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Iishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef]
- Bye, N.; Carron, S.; Han, X.; Agyapomaa, D.; Ng, S.Y.; Yan, E.; Rosenfeld, J.V.; Morganti-Kossmann, M.C. Neurogenesis and glial proliferation are stimulated following diffuse traumatic brain injury in adult rats. J. Neurosci. Res. 2011, 89, 986–1000. [Google Scholar] [CrossRef]
- Ozen, I.; Arkan, S.; Clausen, F.; Ruscher, K.; Marklund, N. Diffuse Traumatic Injury in the Mouse Disrupts Axon-Myelin Integrity in the Cerebellum. J. Neurotrauma 2022, 39, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kilbourne, M.; Kuehn, R.; Tosun, C.; Caridi, J.; Keledjian, K.; Bochicchio, G.; Scalea, T.; Gerzanich, V.; Simard, J.M. Novel model of frontal impact closed head injury in the rat. J. Neurotrauma 2009, 26, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.; Abebe, Z.A.; McInnes, K.A.; Button, E.B.; Tatarnikov, I.; Cheng, W.H.; Haber, M.; Wilkinson, A.; Barron, C.; Diaz-Arrastia, R.; et al. Increased severity of the CHIMERA model induces acute vascular injury, sub-acute deficits in memory recall, and chronic white matter gliosis. Exp. Neurol. 2020, 324, 113116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Gu, J.W.; Li, B.C.; Gao, F.B.; Liao, X.M.; Cui, S.J. Establishment of a novel rat model of blast-related diffuse axonal injury. Exp. Ther. Med. 2018, 16, 93–102. [Google Scholar] [CrossRef]
- Bugay, V.; Bozdemir, E.; Vigil, F.A.; Chun, S.H.; Holstein, D.M.; Elliot, W.R.; Sprague, C.J.; Cavazos, J.E.; Zamora, D.O.; Rule, G.; et al. A Mouse Model of Repetitive Blast Traumatic Brain Injury Reveals Post-Trauma Seizures and Increased Neuronal Excitability. J. Neurotrauma 2020, 37, 248–261. [Google Scholar] [CrossRef]
- Konar, S.K.; Shukla, D.; Amit, A. Posttraumatic Brain Edema: Pathophysiology, Management, and Current Concept. Apollo Med. 2019, 16, 2–7. [Google Scholar] [CrossRef]
- Zusman, B.E.; Kochanek, P.M.; Jha, R.M. Cerebral Edema in Traumatic Brain Injury: A Historical Framework for Current Therapy. Curr. Treat. Options Neurol. 2020, 22, 9. [Google Scholar] [CrossRef]
- Winkler, E.A.; Minter, D.; Yue, J.K.; Manley, G.T. Cerebral Edema in Traumatic Brain Injury Pathophysiology and Prospective Therapeutic Targets. Neurosurg. Clin. N. Am. 2016, 27, 473–488. [Google Scholar] [CrossRef]
- Mishinaga, S.; Koyama, Y. Pathogenesis of Brain Edema and Investigation into Anti-Edema Drugs. Int. J. Mol. Sci. 2015, 16, 9949–9975. [Google Scholar] [CrossRef]
- Dalby, T.; Wohl, E.; Dinsmore, M.; Unger, Z.; Chowdhury, T.; Venkatraghavan, L. Pathophysiology of Cerebral Edema—A Comprehensive Review. J. Neuroanaesth. Crit. Care 2021, 8, 163–172. [Google Scholar] [CrossRef]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J. Neuroinflamm. 2016, 13, 264. [Google Scholar] [CrossRef]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.L.; Rojas, R.; Eisenberg, R.L. Cerebral Edema. Am. J. Roentgenol. 2012, 199, W258–W273. [Google Scholar] [CrossRef] [PubMed]
- Halstead, M.R.; Geocadin, R.G. The Medical Management of Cerebral Edema: Past, Present, and Future Therapies. Neurotherapeutics 2019, 16, 1133–1148. [Google Scholar] [CrossRef]
- Stokum, J.A.; Gerzanich, V.; Simard, J.M. Molecular pathophysiology of cerebral edema. J. Cereb. Blood Flow Metab. 2016, 36, 513–538. [Google Scholar] [CrossRef] [PubMed]
- Obrenovitch, T.P. Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol. Rev. 2008, 88, 211–247. [Google Scholar] [CrossRef]
- Liang, D.; Bhatta, S.; Gerzanich, V.; Simard, J.M. Cytotoxic edema: Mechanisms of pathological cell swelling. Neurosurg. Focus 2007, 22, E2. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, J.; Lu, H. Expression of aquaporin-4 and pathological characteristics of brain injury in a rat model of traumatic brain injury. Mol. Med. Rep. 2015, 12, 7351–7357. [Google Scholar] [CrossRef]
- Fukuda, A.M.; Adami, A.; Pop, V.; Bellone, J.A.; Coats, J.S.; Hartman, R.E.; Ashwal, S.; Obenaus, A.; Badaut, J. Posttraumatic Reduction of Edema with Aquaporin-4 RNA Interference Improves Acute and Chronic Functional Recovery. J. Cereb. Blood Flow Metab. 2013, 33, 1621–1632. [Google Scholar] [CrossRef]
- He, Z.; Lu, H. Aquaporin-4 gene silencing protects injured neurons after early cerebral infarction. Neural Regen. Res. 2015, 10, 1082–1087. [Google Scholar] [CrossRef]
- Higashida, T.; Kreipke, C.W.; Rafols, J.A.; Peng, C.; Schafer, S.; Schafer, P.; Ding, J.Y.; Dornbos, D., 3rd; Li, X.; Guthikonda, M.; et al. The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury: Laboratory investigation. J. Neurosurg. 2011, 114, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Marmarou, A.; Barzó, P.; Fatouros, P.; Corwin, F. Characterization of edema by diffusion-weighted imaging in experimental traumatic brain injury. J. Neurosurg. 1996, 84, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Marmarou, C.R.; Liang, X.; Abidi, N.H.; Parveen, S.; Taya, K.; Henderson, S.C.; Young, H.F.; Filippidis, A.S.; Baumgarten, C.M. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014, 1581, 89–102. [Google Scholar] [CrossRef]
- Filippidis, A.S.; Carozza, R.B.; Rekate, H.L. Aquaporins in Brain Edema and Neuropathological Conditions. Int. J. Mol. Sci. 2017, 18, 55. [Google Scholar] [CrossRef]
- Gross, B.A.; Jankowitz, B.T.; Friedlander, R.M. Cerebral Intraparenchymal Hemorrhage A Review. JAMA 2019, 321, 1295–1303. [Google Scholar] [CrossRef]
- Naumenko, Y.; Yuryshinetz, I.; Zabenko, Y.; Pivneva, T. Mild traumatic brain injury as a pathological process. Heliyon 2023, 9, e18342. [Google Scholar] [CrossRef]
- Qureshi, A.L.; Mendelow, A.D.; Hanley, D.F. Intracerebral haemorrhage. Lancet 2009, 373, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Aromatario, M.; Torsello, A.; D’Errico, S.; Bertozzi, G.; Sessa, F.; Cipolloni, L.; Baldari, B. Traumatic Epidural and Subdural Hematoma: Epidemiology, Outcome, and Dating. Medicina 2021, 57, 125. [Google Scholar] [CrossRef]
- Caceres, J.A.; Goldstein, J.N. Intracranial hemorrhage. Emerg. Med. Clin. N. Am. 2012, 30, 771–794. [Google Scholar] [CrossRef]
- Kushner, D. Mild traumatic brain injury: Toward understanding manifestations and treatment. Arch. Intern. Med. 1998, 158, 1617–1624. [Google Scholar] [CrossRef]
- Hardman, J.M.; Manoukian, A. Pathology of head trauma. Neuroimag. Clin. N. Am. 2002, 12, 175–187. [Google Scholar] [CrossRef]
- Angelova, P.; Kehayov, I.; Davarski, A.; Kitov, B. Contemporary insight into diffuse axonal injury. Folia Med. Plovdiv. 2021, 63, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Hahnel, S. Value of Advanced MR Imaging Techniques in Mild Traumatic Brain Injury. AJNR Am. J. Neuroradiol. 2020, 41, 1269–1270. [Google Scholar] [CrossRef] [PubMed]
- Bigler, E.D. The lesion(s) in traumatic brain injury: Implications for clinical neuropsychology. Arch. Clin. Neuropsychol. 2001, 16, 95–131. [Google Scholar] [CrossRef]
- Andriessen, T.M.J.C.; Horn, J.; Franschman, G.; van der Naalt, J.; Haitsma, I.; Jacobs, B.; Steyerberg, E.W.; Vos, P.E. Epidemiology, severity classification, and outcome of moderate and severe traumatic brain injury: A prospective multicenter study. J. Neurotrauma 2011, 28, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, J.G.; Paton, J.F.R. Brainstem: Neural networks vital for life. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2447–2451. [Google Scholar] [CrossRef]
- Benghanem, S.; Mazeraud, A.; Azabou, E.; Chhor, V.; Shinotsuka, C.R.; Claassen, J.; Rohaut, B.; Sharshar, T. Brainstem dysfunction in critically ill patients. Crit. Care 2020, 24, 5. [Google Scholar] [CrossRef]
- Bolandzadeh, N.; Davis, J.C.; Tam, R.; Handy, T.C.; Liu-Ambrose, T. The association between cognitive function and white matter lesion location in older adults: A systematic review. BMC Neurol. 2012, 12, 126. [Google Scholar] [CrossRef]
- Xu, M.; Qian, L.; Wang, S.; Cai, H.; Sun, Y.; Thakor, N.; Qi, X.; Sun, Y. Brain network analysis reveals convergent and divergent aberrations between mild stroke patients with cortical and subcortical infarcts during cognitive task performing. Front. Aging Neurosci. 2023, 15, 1193292. [Google Scholar] [CrossRef]
- Karakasi, M.V.; Nikova, A.S.; Valsamidou, C.; Pavlidis, P.; Birbilis, T.A. Anatomical Localization of Traumatic Brain Injury Cases in Eastern Macedonia and Thrace, Greece: A 10-year Retrospective Observational Study. Korean J. Neurotrauma 2020, 16, 38–48. [Google Scholar] [CrossRef]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Leal, W.; Martins, L.C.; Diniz, J.A.P.; Dos Santos, Z.A.; Borges, J.A.; Macedo, C.A.C.; Medeiros, A.C.; De Paula, L.S.; Guimaraes, J.S.; Freire, M.A.M.; et al. Neurotropism and neuropathological effects of selected rhabdoviruses on intranasally-infected newborn mice. Acta Trop. 2006, 97, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Souza-Rodrigues, R.D.; Costa, A.M.; Lima, R.R.; Dos Santos, C.D.; Picanço-Diniz, C.W.; Gomes-Leal, W. Inflammatory response and white matter damage after microinjections of endothelin-1 into the rat striatum. Brain Res. 2008, 1200, 78–88. [Google Scholar] [CrossRef]
- Freire, M.A.M. Pathophysiology of neurodegeneration following traumatic brain injury. West Indian Med. J. 2012, 61, 751–755. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Postolache, T.T.; Wadhawan, A.; Can, A.; Lowry, C.A.; Woodbury, M.; Makkar, H.; Hoisington, A.J.; Scott, A.J.; Potocki, E.; Benros, M.E.; et al. Inflammation in Traumatic Brain Injury. J. Alheimers Dis. 2020, 74, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Abudukelimu, A.; Barberis, M.; Redegeld, F.A.; Sahin, N.; Westerhoff, H.V. Predictable Irreversible Switching Between Acute and Chronic Inflammation. Front. Immunol. 2018, 9, 1596. [Google Scholar] [CrossRef]
- Gonzalez, A.C.O.; Costa, T.F.; Andrade, Z.A.; Medrado, A.R.A.P. Wound healing—A literature review. Bras. Dermatol. 2016, 91, 614–620. [Google Scholar] [CrossRef]
- Aruselvan, P.; Fard, M.T.; Tan, W.S.; Gothai, S.; Fakurazi, S.; Norhaizan, M.E.; Kumar, S.S. Role of Antioxidants and Natural Products in Inflammation. Oxid. Med. Cell Longev. 2016, 2016, 5276130. [Google Scholar] [CrossRef]
- Ladak, A.A.; Enam, S.A.; Ibrahim, M.T. A review of the molecular mechanisms of traumatic brain injury. World Neurosurg. 2019, 131, 126–132. [Google Scholar] [CrossRef]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 2016, 173, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Zhang, J.; Dong, J.F.; Shi, F.D. Dissemination of brain inflammation in traumatic brain injury. Cell Mol. Immunol. 2019, 16, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.R.; Santana, L.N.; Fernandes, R.M.; Nascimento, E.M.; Oliveira, A.C.; Fernandes, L.M.; Dos Santos, E.M.; Tavares, P.A.; Dos Santos, I.R.; Gimarães-Santos, A.; et al. Neurodegeneration and Glial Response after Acute Striatal Stroke: Histological Basis for Neuroprotective Studies. Oxid. Med. Cell. Longev. 2016, 2016, 3173564. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Aschner, M. Immune and inflammatory responses in the CNS: Modulation by astrocytes. Toxicol. Lett. 1998, 102–103, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 2014, 20, 160–172. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.; Quintana, F.J. Astrocyte crosstalk in CNS inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275, 305–315. [Google Scholar] [CrossRef]
- Guimaraes, J.S.; Lemos, N.A.M.; Freire, M.A.M.; Pereira, A.; Ribeiro, S. Microelectrode implants, inflammatory response and long-lasting effects on NADPH diaphorase neurons in the rat frontal cortex. Exp. Brain Res. 2022, 240, 2569–2580. [Google Scholar] [CrossRef]
- Okada, S.; Hara, M.; Kobayakawa, K.; Matsumoto, Y.; Nakashima, Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci. Res. 2018, 126, 39–43. [Google Scholar] [CrossRef]
- Streit, W.J. Microglial response to brain injury: A brief synopsis. Toxicol. Pathol. 2000, 28, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Freire, M.A.M.; Lima, R.R.; Bittencourt, L.O.; Guimaraes, J.S.; Falcao, D.; Gomes-Leal, W. Astrocytosis, Inflammation, Axonal Damage and Myelin Impairment in the Internal Capsule following Striatal Ischemic Injury. Cells 2023, 12, 457. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S232–S240. [Google Scholar] [CrossRef]
- Akira, S.; Hirano, T.; Taga, T.; Kishimoto, T. Biology of multifunctional cytokines: IL 6 and related molecules (IL 1 and TNF). FASEB J. 1990, 4, 2860–2867. [Google Scholar] [CrossRef] [PubMed]
- Feghali, C.A.; Wright, T.M. Cytokines in acute and chronic inflammation. Front. Biosci. 1997, 2, d12–d26. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef]
- Olney, J.W. Excitotoxicity: An overview. Can. Dis. Wkly. Rep. 1990, 16 (Suppl. S1E), 47–57; discussion 57–58. [Google Scholar]
- McGinn, M.J.; Povlishock, J.T. Pathophysiology of Traumatic Brain Injury. Neurosurg. Clin. N. Am. 2016, 27, 397–407. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate receptors and the induction of excitotoxic neuronal death. Prog. Brain Res. 1994, 100, 47–51. [Google Scholar] [CrossRef]
- Tantral, L.; Malathi, K.; Kohyama, S.; Silane, M.; Berenstein, A.; Jayaraman, T. Intracellular calcium release is required for caspase-3 and -9 activation. Cell Biochem. Funct. 2004, 22, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Baracaldo-Santamaría, D.; Ariza-Salamanca, D.F.; Corrales-Hernández, M.G.; Pachón-Londoño, M.J.; Hernandez-Duarte, I.; Calderon-Ospina, C.A. Revisiting Excitotoxicity in Traumatic Brain Injury: From Bench to Bedside. Pharmaceutics 2022, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Caspase function in programmed cell death. Cell Death Differ. 2007, 14, 32–43. [Google Scholar] [CrossRef]
- Clark, R.S.; Kochanek, P.M.; Watkins, S.C.; Chen, M.; Dixon, C.E.; Seidberg, N.A.; Melick, J.; Loeffert, J.E.; Nathaniel, P.D.; Jin, K.L.; et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J. Neurochem. 2000, 74, 740–753. [Google Scholar] [CrossRef]
- Lifshitz, J.; Sullivan, P.G.; Hovda, D.A.; Wieloch, T.; McIntosh, T.K. Mitochondrial damage and dysfunction in traumatic brain injury. Mitochondrion 2004, 4, 705–713. [Google Scholar] [CrossRef]
- Hirsch, T.; Marzo, I.; Kroemer, G. Role of the mitochondrial permeability transition pore in apoptosis. Biosci. Rep. 1997, 17, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, J.S.; Freire, M.A.M.; Lima, R.R.; Souza-Rodrigues, R.D.; Costa, A.M.R.; Dos Santos, C.D.; Picanco-Diniz, C.W.; Gomes-Leal, W. Mechanisms of secondary degeneration in the central nervous system during acute neural disorders and white matter damage. Rev. Neurol. 2009, 48, 304–310. [Google Scholar] [CrossRef]
- Knoblach, S.M.; Nikolaeva, M.; Huang, X.; Fan, L.; Krajewski, S.; Reed, J.C.; Faden, A.I. Multiple caspases are activated after traumatic brain injury: Evidence for involvement in functional outcome. J. Neurotrauma 2002, 19, 1155–1170. [Google Scholar] [CrossRef]
- Siman, R.; Bozyczko-Coyne, D.; Savage, M.J.; Roberts-Lewis, J.M. The calcium-activated protease Calpain I and ischemia-induced neurodegeneration. Adv. Neurol. 1996, 71, 167–174. [Google Scholar]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal cell death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Leslie, C.C. Cytosolic phospholipase A₂: Physiological function and role in disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.K.; Xu, X.M. Phospholipase A2 and its molecular mechanism after spinal cord injury. Mol. Neurobiol. 2010, 41, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Liu, Y.; Lin, C.; Xu, X.; Li, Z.; Bao, Z.; Fan, L.; Tao, C.; Zhao, L.; Liu, Y.; et al. Activation of bradykinin B2 receptor induced the inflammatory responses of cytosolic phospholipase A2 after the early traumatic brain injury. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2957–2971. [Google Scholar] [CrossRef] [PubMed]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Arancibia-Hernández, Y.L.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. RONS and Oxidative Stress: An Overview of Basic Concepts. Oxygen 2022, 2, 437–478. [Google Scholar] [CrossRef]
- Waring, W.S. Uric acid: An important antioxidant in acute ischaemic stroke. QJM 2002, 95, 691–693. [Google Scholar] [CrossRef]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.M.; Chandra, N.; Haorah, J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. J. Mol. Neurobiol. 2015, 51, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.K.; Rich, W.; Reilly, M.A. Oxidative stress in the brain and retina after traumatic injury. Front. Neurosci. 2023, 17, 1021152. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Gois, A.M.; Mendonça, D.M.F.; Freire, M.A.M.; Santos, J.R. In vitro and in vivo models of Amyotrophic lateral sclerosis: An updated review. Brain Res. Bull. 2020, 159, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Fesharaki-Zadeh, A. Oxidative Stress in Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 13000. [Google Scholar] [CrossRef]
- Sahel, D.K.; Kaira, M.; Raj, K.; Sharma, S.; Singh, S. Mitochondrial dysfunctioning and neuroinflammation: Recent highlights on the possible mechanisms involved in Traumatic Brain Injury. Neurosci. Lett. 2019, 710, 134347. [Google Scholar] [CrossRef]
- LeBel, C.P.; Bondi, S.C. Oxygen radicals: Common mediators of neurotoxicity. Neurotoxicol. Teratol. 1991, 13, 341–346. [Google Scholar] [CrossRef]
- Lutton, E.M.; Farney, S.K.; Andrews, A.M.; Shuvaev, V.V.; Chuang, G.Y.; Muzykantov, V.R.; Ramirez, S.H. Endothelial targeted strategies to combat oxidative stress: Improving outcomes in traumatic brain injury. Front. Neurol. 2019, 10, 582. [Google Scholar] [CrossRef]
- Atlante, A.; Gagliardi, S.; Minervini, G.M.; Ciotti, M.T.; Marra, E.; Calissano, P. Glutamate neurotoxicity in rat cerebellar granule cells: A major role for xanthine oxidase in oxygen radical formation. J. Neurochem. 1997, 68, 2038–2045. [Google Scholar] [CrossRef]
- Luo, D.; Knezevich, S.; Vincent, S.R. N-methyl-D-aspartate-induced nitric oxide release: An in vivo microdialysis study. Neuroscience 1993, 57, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed]
- Nanda, B.L.; Nataraju, A.; Rajesh, R.; Rangappa, K.S.; Shekar, M.A.; Vishwanath, B.S. PLA2 mediated arachidonate free radicals: PLA2 inhibition and neutralization of free radicals by anti-oxidants—A new role as anti-inflammatory molecule. Curr. Top. Med. Chem. 2007, 7, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar] [CrossRef]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative stress and novel anti-oxidants such as mitoquinone and edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef]
- McGovern, A.J.; Barreto, G.E. Mitochondria dysfunction and inflammation in traumatic brain injury: Androgens to the battlefront. Androg. Clin. Res. Ther. 2021, 2, 304–315. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Rabchevsky, A.G.; Waldmeier, P.C.; Springer, J.E. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J. Neurosci. Res. 2005, 79, 231–239. [Google Scholar] [CrossRef]
- Shokolenko, A.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Jendrach, M.; Mai, S.; Pohl, S.; Voth, M.; Bereiter-Hahn, J. Short-and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion 2008, 8, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tian, F.; Shao, Z.; Shen, X.; Qi, X.; Li, H.; Wang, Z.; Chen, G. Expression and clinical significance of non-phagocytic cell oxidase 2 and 4 after human traumatic brain injury. Neurol. Sci. 2015, 36, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.W.; Wang, J.; Dhandapani, K.M.; Wang, R.; Brann, D. NADPH oxidases in traumatic brain injury—Promising therapeutic targets? Redox Biol. 2018, 16, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Wang, H.D.; Cong, Z.X.; Zhou, X.M.; Xu, J.G.; Jia, Y.; Ding, Y. Puerarin ameliorates oxidative stress in a rodent model of traumatic brain injury. J. Surg. Res. 2014, 186, 328–337. [Google Scholar] [CrossRef]
- Freire, M.A.M.; Rocha, G.S.; Costa, I.M.; Oliveira, L.C.; Guzen, F.P.; Cavalcanti, J.R.L.P. Roles of terpenoid Astragaloside IV in altered states of the nervous system: An updated review. Res. Soc. Dev. 2022, 11, e11711628861. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, H.D.; Zhou, X.M.; Fang, J.; Zhu, L.; Ding, K. N-acetylcysteine amide provides neuroprotection via Nrf2-ARE pathway in a mouse model of traumatic brain injury. Drug Des. Devel. Ther. 2018, 12, 4117–4127. [Google Scholar] [CrossRef]
- Ates, O.; Cayli, S.; Altinoz, E.; Gurses, I.; Yucel, N.; Sener, M.; Kocak, A.; Yologlu, S. Neuroprotection by resveratrol against traumatic brain injury in rats. Mol. Cell Biochem. 2007, 294, 137–144. [Google Scholar] [CrossRef]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell Neurosci. 2019, 13, 528. [Google Scholar] [CrossRef]
- Pingitore, A.; Lima, G.P.P.; Mastorci, F.; Quinones, A.; Iervasi, G.; Vassalle, C. Exercise and oxidative stress: Potential effects of antioxidant dietary strategies in sports. Nutrition 2015, 31, 916–922. [Google Scholar] [CrossRef]
- Rodrigues, A.N.; da Silva, D.C.B.; Baia-da-Silva, D.C.; Mendes, P.F.S.; Ferreira, M.K.M.; Rocha, G.S.; Freire, M.A.M.; Fernandes, L.M.P.; Maia, C.D.S.F.; Gomes-Leal, W.; et al. Aerobic physical training attenuates oxidative stress in the spinal cord of adult rats induced by binge-like ethanol intake. Antioxidants 2023, 12, 1051. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.Q.; Shi, Y.C.; Lin, S.; Chen, X.R. Metabolic disorders on cognitive dysfunction after traumatic brain injury. Trends Endocrinol. Metab. 2022, 33, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, C.; Prabhakar, H.; Bilotta, F. Endocrine Dysfunction After Traumatic Brain Injury: An Ignored Clinical Syndrome? Neurocrit. Care 2023, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Komura, A.; Kawasaki, T.; Yamada, Y.; Uzuyama, S.; Asano, Y.; Shinoda, J. Cerebral Glucose Metabolism in Patients with Chronic Mental and Cognitive Sequelae after a Single Blunt Mild Traumatic Brain Injury without Visible Brain Lesions. J. Neurotrauma 2019, 36, 641–649. [Google Scholar] [CrossRef]
- Franklin, W.; Krishnan, B.; Taglialatela, G. Chronic synaptic insulin resistance after traumatic brain injury abolishes insulin protection from amyloid beta and tau oligomer-induced synaptic dysfunction. Sci. Rep. 2019, 9, 8228. [Google Scholar] [CrossRef]
- Wang, G.H.; Yan, Y.; Shen, H.P.; Chu, Z. The Clinical Characteristics of Electrolyte Disturbance in Patients with Moderate and Severe Traumatic Brain Injury Who Underwent Craniotomy and Its Influence on Prognosis. J. Korean Neurosurg. Soc. 2023, 66, 332–339. [Google Scholar] [CrossRef]
- Vinas-Rios, J.M.; Sanchez-Aguilar, M.; Sanchez-Roriguez, J.J.; Gonzalez-Aguirre, D.; Heinen, C.; Meyer, F.; Kretschmer, T. Hypocalcaemia as a prognostic factor of early mortality in moderate and severe traumatic brain injury. Neurol. Res. 2014, 36, 102–106. [Google Scholar] [CrossRef]
- Vedantam, A.; Robertson, C.S.; Gopinath, S.P. Morbidity and mortality associated with hypernatremia in patients with severe traumatic brain injury. Neurosurg. Focus 2017, 43, E2. [Google Scholar] [CrossRef]
- Wu, X.; Lu, X.; Lu, X.; Yu, J.; Sun, Y.; Du, Z.; Wu, X.; Mao, Y.; Zhou, L.; Wu, S.; et al. Prevalence of severe hypokalaemia in patients with traumatic brain injury. Injury 2015, 46, 35–41. [Google Scholar] [CrossRef]
- Schlogl, M.; Kach, I.; Beeler, P.E.; Pape, H.C.; Neuhaus, V. Trauma patients with hypokalemia have an increased risk of morbidity and mortality. SIPAS 2021, 7, 100041. [Google Scholar] [CrossRef]
- Chen, Y.H.; Huang, E.Y.K.; Kou, T.T.; Miller, J.; Chiang, Y.H.; Hoffer, B.J. Impact of Traumatic Brain Injury on Dopaminergic Transmission. Cell Transpl. 2017, 26, 1156–1168. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Shimada, R.; Okada, Y.; Kibayashi, K. Traumatic brain injury decreases serotonin transporter expression in the rat cerebrum. Neurol. Res. 2016, 38, 358–363. [Google Scholar] [CrossRef]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Krishna, G.; Beitchman, J.A.; Bromberg, C.E.; Thomas, T.C. Approaches to Monitor Circuit Disruption after Traumatic Brain Injury: Frontiers in Preclinical Research. Int. J. Mol. Sci. 2020, 21, 588. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, P.; Rocha, E.E.M. Nutrition Therapy, Glucose Control, and Brain Metabolism in Traumatic Brain Injury: A Multimodal Monitoring Approach. Front. Neurosci. 2020, 14, 190. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Malik, R.; Singh, G.; Bhatia, S.; Al-Harrasi, A.; Mohan, S.; Albratty, M.; Albarrati, A.; Tambuwala, M.M. Pathogenesis and management of traumatic brain injury (TBI): Role of neuroinflammation and anti-inflammatory drugs. Inflammopharmacol 2022, 30, 1153–1166. [Google Scholar] [CrossRef]
- Singh, S.; Singh, T.G. Role of nuclear factor kappa B (NF-κB) signalling in neurodegenerative diseases: An mechanistic approach. Curr. Neuropharmacol. 2020, 18, 918–935. [Google Scholar] [CrossRef]
- Cente, M.; Matyasova, K.; Csicsatkova, N.; Tomikova, A.; Porubska, S.; Niu, Y.; Madjan, M.; Filipcik, P.; Jurisica, I. Traumatic MicroRNAs: Deconvolving the Signal After Severe Traumatic Brain Injury. Cell Mol. Neurobiol. 2023, 43, 1061–1075. [Google Scholar] [CrossRef]
- Gustafsson, D.; Klang, A.; Thams, S.; Rostami, E. The Role of BDNF in Experimental and Clinical Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 3582. [Google Scholar] [CrossRef]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF signaling in context: From synaptic regulation to psychiatric disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef]
- Wang, Y.; Hameed, M.Q.; Rakhade, S.N.; Iglesias, A.H.; Muller, P.A.; Mou, D.-L.; Rotenberg, A. Hippocampal immediate early gene transcription in the rat fluid percussion traumatic brain injury model. NeuroReport 2014, 25, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Failla, M.D.; Conley, Y.P.; Wagner, A.K. Brain-Derived Neurotrophic Factor (BDNF) in Traumatic Brain Injury–Related Mortality: Interrelationships Between Genetics and Acute Systemic and Central Nervous System BDNF Profiles. Neurorehabilit. Neural Repair 2016, 30, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Movahedpour, A.; Vakili, O.; Khalifeh, M.; Mousavi, P.; Mahmoodzadeh, A.; Taheri-Anganeh, M.; Razmeh, S.; Shabaninejad, Z.; Yousefi, F.; Behrouj, H.; et al. Mammalian target of rapamycin (mTOR) signaling pathway and traumatic brain injury: A novel insight into targeted therapy. Cell Biochem. Funct. 2022, 40, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. Targeting molecules to medicine with mTOR, autophagy and neurodegenerative disorders. Br. J. Clin. Pharmacol. 2016, 82, 1245–1266. [Google Scholar] [CrossRef]
- Chen, S.; Atkins, C.M.; Liu, C.L.; Alonso, O.F.; Dietrich, W.D.; Hu, B.R. Alterations in Mammalian Target of Rapamycin Signaling Pathways after Traumatic Brain Injury. J. Cereb. Blood Flow Metab. 2007, 27, 939–949. [Google Scholar] [CrossRef]
- Golarai, G.; Greenwood, A.C.; Feeney, D.M.; Connor, J.A. Physiological and Structural Evidence for Hippocampal Involvement in Persistent Seizure Susceptibility after Traumatic Brain Injury. J. Neurosci. 2001, 21, 8523–8537. [Google Scholar] [CrossRef] [PubMed]
- Arachchige Don, A.S.; Tsang, C.K.; Kazdoba, T.M.; D’Arcangelo, G.; Young, W.; Steven Zheng, X.F. Targeting mTOR as a novel therapeutic strategy for traumatic CNS injuries. Drug Discov. Today 2012, 17, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Cordaro, M.; Ardizzone, A.; Scuderi, S.A.; Cuzzocrea, S.; Esposito, E.; Paterniti, I. The inhibition of mammalian target of rapamycin (mTOR) in improving inflammatory response after traumatic brain injury. J. Cell Mol. Med. 2021, 25, 7855–7866. [Google Scholar] [CrossRef]
- Nikolaeva, I.; Crowell, B.; Valenziano, J.; Meaney, D.; D’Arcangelo, G. Beneficial Effects of Early mTORC1 Inhibition after Traumatic Brain Injury. J. Neurotrauma 2016, 33, 183–193. [Google Scholar] [CrossRef]
- Kumar, S.; Mehan, S.; Narula, A.S. Therapeutic modulation of JAK-STAT, mTOR, and PPAR-γ signaling in neurological dysfunctions. J. Mol. Med. 2023, 101, 9–49. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, Y.; Li, G.; Su, X.; Hang, C. Activation of JAK2/STAT pathway in cerebral cortex after experimental traumatic brain injury of rats. Neurosci. Lett. 2011, 498, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.A.; Kang, Y.; Sanchez-Molano, J.; Furones, C.; Atkins, C.M. STAT3 signaling after traumatic brain injury. J. Neurochem. 2012, 120, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Raible, D.J.; Frey, L.C.; Del Angel, Y.C.; Carlsen, J.; Hund, D.; Russek, S.J.; Smith, B.; Brooks-Kayal, A.R. JAK/STAT pathway regulation of GABAA receptor expression after differing severities of experimental TBI. Exp. Neurol. 2015, 271, 445–456. [Google Scholar] [CrossRef]
- Di Pietro, V.; Amin, D.; Pernagallo, S.; Lazzarino, G.; Tavazzi, B.; Vagnozzi, R.; Pringle, A.; Belli, A. Transcriptomics of traumatic brain injury: Gene expression and molecular pathways of different grades of insult in a rat organotypic hippocampal culture model. J. Neurotrauma 2010, 27, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Muneer, P.M. Nrf2 as a Potential Therapeutic Target for Traumatic Brain Injury. JIN 2023, 22, 81. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Wang, H.-D.; Hu, Z.-G.; Wang, Q.-F.; Yin, H.-X. Activation of Nrf2–ARE pathway in brain after traumatic brain injury. Neurosci. Lett. 2008, 431, 150–154. [Google Scholar] [CrossRef]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Abdul-Muneer, P.M. Traumatic brain injury-induced downregulation of Nrf2 activates inflammatory response and apoptotic cell death. J. Mol. Med. 2019, 97, 1627–1641. [Google Scholar] [CrossRef]
- Huie, J.R.; Mondello, S.; Lindsell, C.J.; Antiga, L.; Yuh, E.L.; Zanier, E.R.; Masson, S.; Rosario, B.L.; Ferguson, A.R. Biomarkers for Traumatic Brain Injury: Data Standards and Statistical Considerations. J. Neurotrauma 2021, 38, 2514–2529. [Google Scholar] [CrossRef]
- Dadas, A.; Washington, J.; Dias-Arrastia, R.; Janigro, D. Biomarkers in traumatic brain injury (TBI): A review. Neuropsychiatr. Dis. Treat. 2018, 14, 2989–3000. [Google Scholar] [CrossRef]
- Ghaith, H.S.; Nawar, A.A.; Gabra, M.D.; Abdelrahman, M.E.; Nafady, M.H.; Bahbah, E.I. Ebada, M.A.; Ashraf, G.M.; Negida, A.; Barreto, G.E. A Literature Review of Traumatic Brain Injury Biomarkers. Mol. Neurobiol. 2022, 59, 4141–4158. [Google Scholar] [CrossRef]
- Deshetty, U.M.; Periyasamy, P. Potential Biomarkers in Experimental Animal Models for Traumatic Brain Injury. J. Clin. Med. 2023, 12, 3923. [Google Scholar] [CrossRef] [PubMed]
- Rothermundt, M.; Peters, M.; Prehn, J.H.; Arolt, V. S100B in brain damage and neurodegeneration. Microsc. Res. Tech. 2003, 60, 614–632. [Google Scholar] [CrossRef] [PubMed]
- Rocha, G.S.; Freire, M.A.M.; Paiva, K.M.; Oliveira, R.F.; Norrara, B.; Morais, P.L.A.G.; Oliveira, L.C.; Engelberth, R.C.G.J.; Cavalcante, J.S.; Cavalcanti, J.R.L.P. Effect of senescence on the tyrosine hydroxylase and S100B immunoreactivity in the nigrostriatal pathway of the rat. J. Chem. Neuroanat. 2022, 124, 102136. [Google Scholar] [CrossRef]
- Arrais, A.C.; Melo, L.H.M.F.; Norrara, B.; Almeida, M.A.B.; Freire, K.F.; Melo, A.M.M.F.; Oliveira, L.C.; Lima, F.O.V.; Engelberth, R.C.G.J.; Cavalcante, J.S.; et al. S100B protein: General characteristics and pathophysiological implications in the Central Nervous System. Int. J. Neurosci. 2022, 132, 313–321. [Google Scholar] [CrossRef]
- Michetti, F.; Clementi, M.E.; Di Liddo, R.; Valeriani, F.; Ria, F.; Rende, M.; Di Sante, G.; Spica, V.R. The S100B Protein: A Multifaceted Pathogenic Factor More Than a Biomarker. Int. J. Mol. Sci. 2023, 24, 9605. [Google Scholar] [CrossRef] [PubMed]
- Michetti, F.; D’Ambrosi, N.; Toesca, A.; Puglisi, M.A.; Serrano, A.; Marchese, E.; Corvino, V.; Geloso, M.C. The S100B story: From biomarker to active factor in neural injury. J. Neurochem. 2019, 148, 168–187. [Google Scholar] [CrossRef] [PubMed]
- Michetti, F.; Di Sante, G.; Clementi, M.E.; Sampaolese, B.; Casalbore, P.; Volonté, C.; Spica, V.R.; Parnigotto, P.P.; Di Liddo, R.; Amadio, S.; et al. Growing role of S100B protein as a putative therapeutic target for neurological- and nonneurological-disorders. Neurosci. Biobehav. Rev. 2021, 127, 446–458. [Google Scholar] [CrossRef]
- Corne, R.; Besson, V.; Slimane, S.A.S.; Coutan, M.; Palhas, M.L.C.; Shen, F.X.; Marchand-Leroux, C.; Ogier, M.; Mongeau, R. Insulin-like Growth Factors may be Markers of both Traumatic Brain Injury and Fear-Related Stress. Neuroscience 2021, 466, 205–221. [Google Scholar] [CrossRef]
- Kou, Z.; Wu, Z.; Tong, K.A.; Holshouser, B.; Benson, R.R.; Hu, J.; Haacke, E.M. The role of advanced MR imaging findings as biomarkers of traumatic brain injury. J. Head. Trauma. Rehabil. 2010, 25, 267–282. [Google Scholar] [CrossRef]
- Diaz-Arrastia, R.; Wang, K.K.W.; Papa, L.; Sorani, M.D.; Yue, J.K.; Puccio, A.M.; McMahon, P.J.; Inoue, T.; Yuh, E.L.; Lingsma, H.F.; et al. Acute biomarkers of traumatic brain injury: Relationship between plasma levels of ubiquitin C-terminal hydrolase-L1 and glial fibrillary acidic protein. J. Neurotrauma 2014, 31, 19–25. [Google Scholar] [CrossRef]
- Tewari, B.P.; Chaunsali, L.; Prim, C.E.; Sontheimer, H. A glial perspective on the extracellular matrix and perineuronal net remodeling in the central nervous system. Front. Cell. Neurosci. 2022, 16, 1022754. [Google Scholar] [CrossRef]
- Minta, K.; Brinkmalm, G.; Nimer, F.A.; Thelin, E.P.; Piehl, F.; Tullberg, M. Jeppsson, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; et al. Dynamics of cerebrospinal fluid levels of matrix metalloproteinases in human traumatic brain injury. Sci. Rep. 2020, 10, 18075. [Google Scholar] [CrossRef]
- Nagalakshmi, B.; Sagarkar, S.; Sakharkar, A.J. Epigenetic Mechanisms of Traumatic Brain Injuries. Prog. Mol. Biol. Transl. Sci. 2018, 157, 263–298. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, C.; Wang, J.; Ye, Y.; Li, Y.; Xu, Q.; Kang, X.; Gu, L. Neuroinflammatory Biomarkers in the Brain, Cerebrospinal Fluid, and Blood After Ischemic Stroke. Mol. Neurobiol. 2023, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Stanzione, R.; Cotugno, M.; Bianch, F.; Marchitti, S.; Forte, M.; Volpe, M.; Rubattu, S. Pathogenesis of Ischemic Stroke: Role of Epigenetic Mechanisms. Genes 2020, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Mortezaee, K.; Khanlarkhani, N.; Beyer, C.; Zendedel, A. Inflammasome: Its role in traumatic brain and spinal cord injury. J. Cell. Physiol. 2018, 233, 5160–5169. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, P.; Faure, E.; Kipnis, E. Inflammasomes in Tissue Damages and Immune Disorders After Trauma. Front. Immunol. 2018, 9, 1900. [Google Scholar] [CrossRef]
- Lenart, N.; Brough, D.; Den-es, A. Inflammasomes link vascular disease with neuroinflammation and brain disorders. J. Cereb. Blood Flow Metab. 2016, 36, 1668–1685. [Google Scholar] [CrossRef]
- Chakraborty, R.; Tabassum, H.; Parvez, S. NLRP3 inflammasome in traumatic brain injury: Its implication in the disease pathophysiology and potential as a therapeutic target. Life Sci. 2023, 314, 121352. [Google Scholar] [CrossRef]
- Freeman, L.C.; Ting, J.P.Y. The pathogenic role of the inflammasome in neurodegenerative diseases. J. Neurochem. 2016, 136 (Suppl. S1), 29–38. [Google Scholar] [CrossRef]
- Tsuchiya, K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol. Immunol. 2020, 64, 252–269. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; de Rivero Vaccari, J.P.; Truettner, J.S.; Dietrich, W.D.; Keane, R.W. The role of microglial inflammasome activation in pyroptotic cell death following penetrating traumatic brain injury. J. Neuroinflamm. 2019, 16, 27. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, W.T.; Pham, L.; Symons, G.F.; Monif, M.; Shultz, S.R.; McDonald, S.J. The NLRP3 inflammasome in traumatic brain injury: Potential as a biomarker and therapeutic target. J. Neuroinflamm. 2020, 17, 104. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Ahmed, H.A.; Adris, T.; Parveen, K.; Thakor, P.; Ishrat, T. The NLRP3 inflammasome: A potential therapeutic target for traumatic brain injury. Neural Regen. Res. 2021, 16, 49–57. [Google Scholar] [CrossRef]
- Ma, X.; Aravind, A.; Pfister, B.J.; Chandra, N.; Haorah, J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol. Neurobiol. 2019, 56, 5332–5345. [Google Scholar] [CrossRef]
- Sorby-Adams, A.J.; Vink, R.; Turner, R.J. Large animal models of stroke and traumatic brain injury as translational tools. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R165–R190. [Google Scholar] [CrossRef]
- Risling, M.; Smith, D.; Stein, T.D.; Thelin, E.P.; Zanier, E.R.; Ankarcrona, M.; Nilsson, P. Modelling human pathology of traumatic brain injury in animal models. J. Intern. Med. 2019, 285, 594–607. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, J.; Li, H.; Li, H.; Xie, F. Models of traumatic brain injury-highlights and drawbacks. Front. Neurol. 2023, 14, 1151660. [Google Scholar] [CrossRef]
- Vink, R. Large animal models of traumatic brain injury. J. Neurosci. Res. 2018, 96, 527–535. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.X.; Ma, Y.B.; Le, N.Y.; Cao, J.; Wang, Y. Large animal models of traumatic brain injury. Int. J. Neurosci. 2018, 128, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Jarrahi, A.; Braun, M.; Ahluwalia, M.; Gupta, R.V.; Wilson, M.; Munie, S.; Ahluwalia, P.; Vender, J.R.; Vale, F.L.; Dhandapani, K.M.; et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 2020, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.R.; Meno, D.K.; Manley, G.T.; Abrams, M.; Akerlund, C.; Andelic, C.; Aries, M.; Bashford, T.; Bell, M.J.; Bodien, Y.G.; et al. Traumatic brain injury: Progress and challenges in prevention, clinical care, and research. Lancet Neurol. 2022, 21, 1004–1060. [Google Scholar] [CrossRef]
- Nishimura, K.; Cordeiro, J.G.; Ahmed, A.I.; Yokobori, S.; Gajavelli, S. Advances in Traumatic Brain Injury Biomarkers. Cureus 2022, 14, e23804. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Model | Animal Model | Type of Injury | Pathway/Structure Evaluated | Outcomes | Reference |
|---|---|---|---|---|---|
| Marmarou weight drop model | Rat | Focal | Apoptotic pathways | Cyto c released, activating caspase 3 apoptotic pathway | Buki et al. [23] |
| Weight drop TBI model | Rat | Focal | Oxidative stress, Inflammation | Oxidative injury, blood-brain barrier disruption, microglial activation | Choi et al. [24] |
| Controlled cortical impact injury model | Rat | Focal | Energetic metabolism | Mitochondrial dysfunction | Xiong et al. [25] |
| Controlled cortical impact injury model | Rat | Focal | Excitotoxicity | Excitotoxic injury caused by higher concentrations of aspartate and glutamate in the brain parenchyma | Palmer et al. [19] |
| Controlled cortical impact model | Mouse | Focal | Microglia | Microglial activation, synaptic loss | Krukowski et al. [26] |
| Closed-head injury model | Mouse | Diffuse | Proinflammatory pathways | Increase in Interleukin 1-expression following repetitive brain lesion | Wu et al. [27] |
| Marmarou weight drop model | Rat | Diffuse | Structural organization | Axonal injury, brain edema | Foda and Marmarou [28] |
| Marmarou weight drop model | Focal | Altered axolemmal permeability, cytoskeletal disturbances | Povlishock et al. [29] | ||
| Dorsal column crush injury model | Rat | Focal/diffuse | Tissue swelling (brain edema) | Water channel aquaporin-4 cell surface increases in response to hypoxia-induced cell swelling | Kitchen et al. [30] |
| Marmarou weight drop model | Rat | Focal | Inflammatory response | Proliferation and increase of reactive astrocytes and microglia | Bye et al. [31] |
| Central fluid percussion injury mode | Mouse | Diffuse | Structural organization | Neurofilament phosphorylation, myelin impairment | Ozen et al. [32] |
| Maryland closed-head injury model | Rat | Diffuse | Axonal structure, apoptotic pathways | Petechial hemorrhage, axonal damage, caspase 3 activation | Kilbourne et al. [33] |
| Closed-head impact model of engineered rotational acceleration | Mouse | Diffuse | Axonal structure, glial structure | White matter gliosis, axonal damage | Bashir et al. [34] |
| Blast TBI model | Rat | Diffuse | Axonal structure | Axonal injury | Zhang et al. [35] |
| Repetitive blast TBI model | Mouse | Diffuse | Structural organization, glial structure | Tau phosphorylation, microglial activation | Bugay et al. [36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freire, M.A.M.; Rocha, G.S.; Bittencourt, L.O.; Falcao, D.; Lima, R.R.; Cavalcanti, J.R.L.P. Cellular and Molecular Pathophysiology of Traumatic Brain Injury: What Have We Learned So Far? Biology 2023, 12, 1139. https://doi.org/10.3390/biology12081139
Freire MAM, Rocha GS, Bittencourt LO, Falcao D, Lima RR, Cavalcanti JRLP. Cellular and Molecular Pathophysiology of Traumatic Brain Injury: What Have We Learned So Far? Biology. 2023; 12(8):1139. https://doi.org/10.3390/biology12081139
Chicago/Turabian StyleFreire, Marco Aurelio M., Gabriel Sousa Rocha, Leonardo Oliveira Bittencourt, Daniel Falcao, Rafael Rodrigues Lima, and Jose Rodolfo Lopes P. Cavalcanti. 2023. "Cellular and Molecular Pathophysiology of Traumatic Brain Injury: What Have We Learned So Far?" Biology 12, no. 8: 1139. https://doi.org/10.3390/biology12081139
APA StyleFreire, M. A. M., Rocha, G. S., Bittencourt, L. O., Falcao, D., Lima, R. R., & Cavalcanti, J. R. L. P. (2023). Cellular and Molecular Pathophysiology of Traumatic Brain Injury: What Have We Learned So Far? Biology, 12(8), 1139. https://doi.org/10.3390/biology12081139