
Identification of Two Subsets of Subcompartment A1 Associated with High Transcriptional Activity and Frequent Loop Extrusion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experiments
2.1.1. Cell Culture
2.1.2. Cleavage under Targets and Tagmentation (CUT&Tag)
2.1.3. Chromatin Immunoprecipitation Sequencing (ChIP-Seq)
2.2. Data Analysis
2.2.1. Hi-C Data Processing
2.2.2. Assignments of Hi-C Subcompartments
2.2.3. Calculation of Interchromosomal A/B Ratio
2.2.4. RNA-Seq Data Analysis
2.2.5. Calculation of Domain Strength
2.2.6. Three-Dimensional Simulation
2.2.7. ChIP-Seq Data Analysis
2.2.8. CUT&Tag Data Analysis
2.2.9. Data Availability
3. Results
3.1. Subcompartment Relocations Frequently Occur and These Transitions Preferentially Correlate with Changes in Gene Expression Levels
3.1.1. Identification of Subcompartments in Five Distinct Cell Types
3.1.2. Frequent Occurrences of Subcompartment Relocations during Cell Differentiation
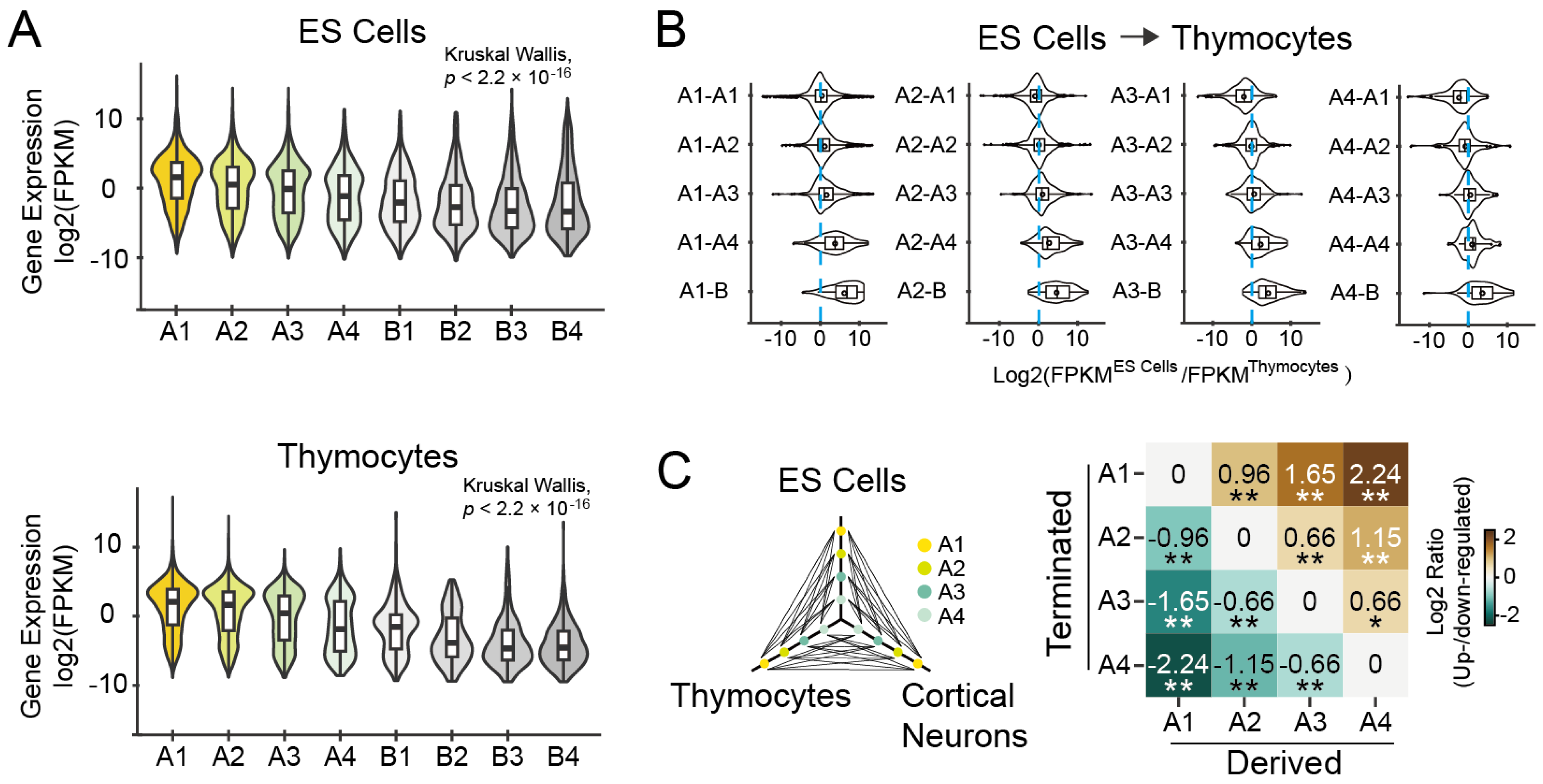
3.1.3. Expression Levels of Genes Are Associated with Their Subcompartment Positioning
3.2. Differential Spatial Distribution Characteristics between Housekeeping and Cell Type-Specific Genes
3.2.1. Identification of Common A1 Regions That Are Highly Enriched in Active Genes and Highly Expressed Genes
3.2.2. Different Spatial Positioning of Housekeeping and Cell Type-Specific Genes in Distinct Cell Types
3.3. Subcompartments A1 Containing More Housekeeping and Cell Type-Specific Genes Harbor Most Strongly Looped Domains
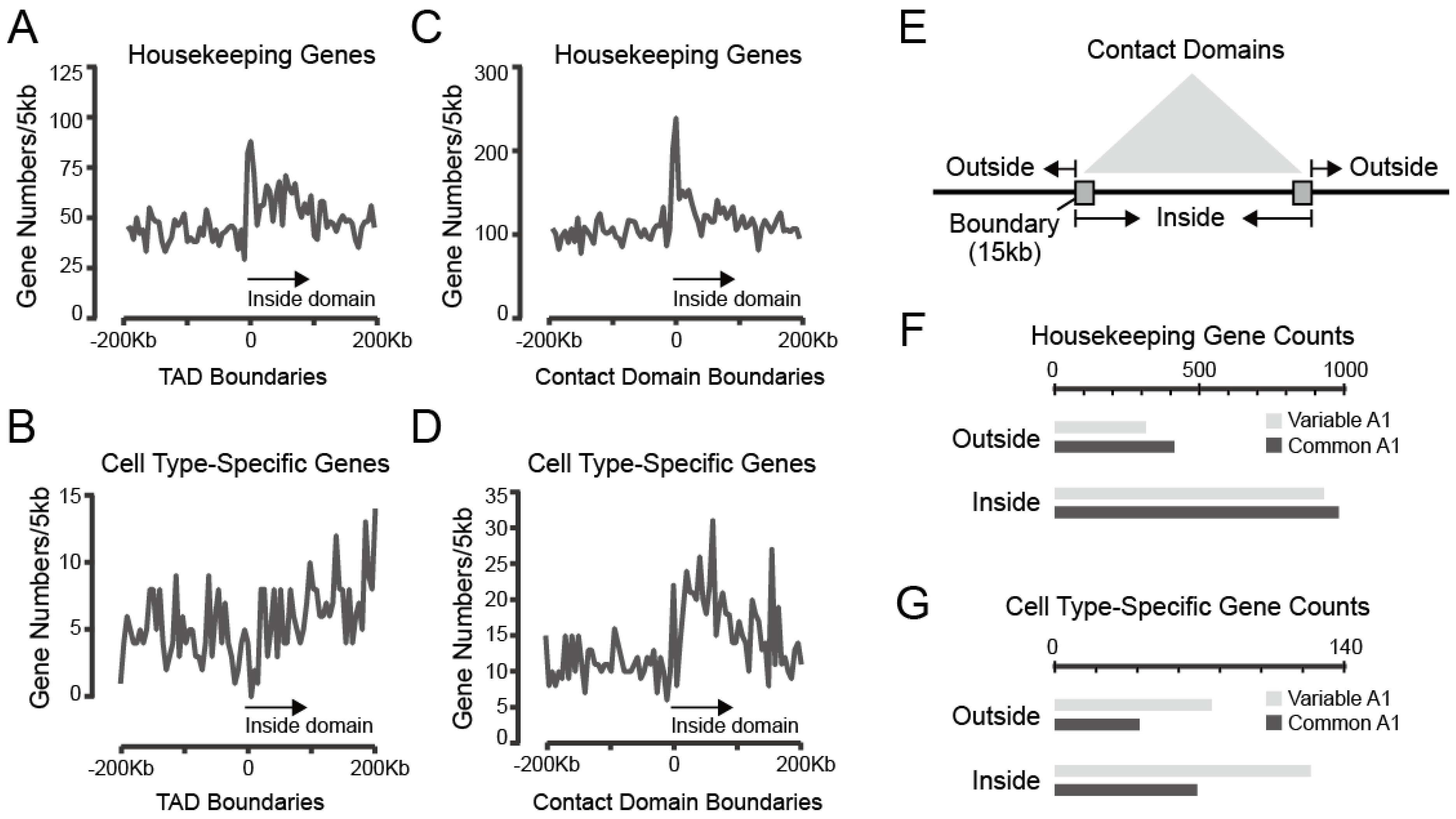
3.3.1. Both Housekeeping and Cell Type-Specific Genes Exhibit a Tendency to Reside within Contact Domains
3.3.2. Strongly Looped Domains Are Most Frequently Located in Subcompartments A1
3.3.3. Chromatin Interaction Strength within Contact Domains Is Associated with Their Subcompartment Positioning
4. Discussion

5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Misteli, T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Kempfer, R.; Pombo, A. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 2020, 21, 207–226. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Meaburn, K.J.; Misteli, T. Cell biology: Chromosome territories. Nature 2007, 445, 379–781. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Liu, Y.; Nanni, L.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Mina, M.; Ceri, S.; Oricchio, E.; Ciriello, G. Systematic inference and comparison of multi-scale chromatin sub-compartments connects spatial organization to cell phenotypes. Nat. Commun. 2021, 12, 2439. [Google Scholar] [CrossRef]
- Bickmore, W.A. The spatial organization of the human genome. Annu. Rev. Genomics. Hum. Genet. 2013, 14, 67–84. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Xiong, K.; Ma, J. Revealing Hi-C subcompartments by imputing inter-chromosomal chromatin interactions. Nat. Commun. 2019, 10, 5069. [Google Scholar] [CrossRef]
- Ashoor, H.; Chen, X.; Rosikiewicz, W.; Wang, J.; Cheng, A.; Wang, P.; Ruan, Y.; Li, S. Graph embedding and unsupervised learning predict genomic sub-compartments from HiC chromatin interaction data. Nat. Commun. 2020, 11, 1173. [Google Scholar] [CrossRef] [PubMed]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Zhang, H.; Emerson, D.J.; Gilgenast, T.G.; Titus, K.R.; Lan, Y.; Huang, P.; Zhang, D.; Wang, H.; Keller, C.A.; Giardine, B.; et al. Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature 2019, 576, 158–162. [Google Scholar] [CrossRef]
- Guo, Y.; Al-Jibury, E.; Garcia-Millan, R.; Ntagiantas, K.; King, J.W.D.; Nash, A.J.; Galjart, N.; Lenhard, B.; Rueckert, D.; Fisher, A.G.; et al. Chromatin jets define the properties of cohesin-driven in vivo loop extrusion. Mol. Cell 2022, 82, 3769–3780. [Google Scholar] [CrossRef] [PubMed]
- Nasmyth, K.; Haering, C.H. Cohesin: Its roles and mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef] [PubMed]
- Losada, A. Cohesin in cancer: Chromosome segregation and beyond. Nat. Rev. Cancer 2014, 14, 389–393. [Google Scholar] [CrossRef]
- Dorsett, D. Cohesin: Genomic insights into controlling gene transcription and development. Curr. Opin. Genet. Dev 2011, 21, 199–206. [Google Scholar] [CrossRef]
- Merkenschlager, M.; Nora, E.P. CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu. Rev. Genom. Hum. Genet. 2016, 17, 17–43. [Google Scholar] [CrossRef]
- Seitan, V.C.; Faure, A.J.; Zhan, Y.; McCord, R.P.; Lajoie, B.R.; Ing-Simmons, E.; Lenhard, B.; Giorgetti, L.; Heard, E.; Fisher, A.G.; et al. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Res. 2013, 23, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Krijger, P.H.; Di Stefano, B.; de Wit, E.; Limone, F.; van Oevelen, C.; de Laat, W.; Graf, T. Cell-of-Origin-Specific 3D Genome Structure Acquired during Somatic Cell Reprogramming. Cell Stem Cell 2016, 18, 597–610. [Google Scholar] [CrossRef] [PubMed]
- El Khattabi, L.; Zhao, H.; Kalchschmidt, J.; Young, N.; Jung, S.; Van Blerkom, P.; Kieffer-Kwon, P.; Kieffer-Kwon, K.R.; Park, S.; Wang, X.; et al. A Pliable Mediator Acts as a Functional Rather Than an Architectural Bridge between Promoters and Enhancers. Cell 2019, 178, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, F.; Chen, Q.; Wan, X.; Shi, M.; Chen, A.K.; Ma, Z.; Li, G.; Wang, M.; Ying, Y.; et al. MyoD is a 3D genome structure organizer for muscle cell identity. Nat. Commun. 2022, 13, 205. [Google Scholar] [CrossRef]
- Haarhuis, J.H.I.; van der Weide, R.H.; Blomen, V.A.; Flach, K.D.; Teunissen, H.; Willems, L.; Brummelkamp, T.R.; Rowland, B.D.; de Wit, E. A Mediator-cohesin axis controls heterochromatin domain formation. Nat. Commun. 2022, 13, 754. [Google Scholar] [CrossRef]
- Xu, J.; Song, F.; Lyu, H.; Kobayashi, M.; Zhang, B.; Zhao, Z.; Hou, Y.; Wang, X.; Luan, Y.; Jia, B.; et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature 2022, 611, 387–398. [Google Scholar] [CrossRef]
- Emerson, D.J.; Zhao, P.A.; Cook, A.L.; Barnett, R.J.; Klein, K.N.; Saulebekova, D.; Ge, C.; Zhou, L.; Simandi, Z.; Minsk, M.K.; et al. Cohesin-mediated loop anchors confine the locations of human replication origins. Nature 2022, 606, 812–819. [Google Scholar] [CrossRef]
- Piazza, A.; Bordelet, H.; Dumont, A.; Thierry, A.; Savocco, J.; Girard, F.; Koszul, R. Cohesin regulates homology search during recombinational DNA repair. Nat. Cell Biol. 2021, 23, 1176–1186. [Google Scholar] [CrossRef]
- Sanders, J.T.; Freeman, T.F.; Xu, Y.; Golloshi, R.; Stallard, M.A.; Hill, A.M.; San Martin, R.; Balajee, A.S.; McCord, R.P. Radiation-induced DNA damage and repair effects on 3D genome organization. Nat. Commun. 2020, 11, 6178. [Google Scholar] [CrossRef] [PubMed]
- Arnould, C.; Rocher, V.; Finoux, A.L.; Clouaire, T.; Li, K.; Zhou, F.; Caron, P.; Mangeot, P.E.; Ricci, E.P.; Mourad, R.; et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 2021, 590, 660–665. [Google Scholar] [CrossRef]
- Abramo, K.; Valton, A.L.; Venev, S.V.; Ozadam, H.; Fox, A.N.; Dekker, J. A chromosome folding intermediate at the condensin-to-cohesin transition during telophase. Nat. Cell Biol. 2019, 21, 1393–1402. [Google Scholar] [CrossRef]
- Naumova, N.; Imakaev, M.; Fudenberg, G.; Zhan, Y.; Lajoie, B.R.; Mirny, L.A.; Dekker, J. Organization of the mitotic chromosome. Science 2013, 342, 948–953. [Google Scholar] [CrossRef]
- Valton, A.L.; Venev, S.V.; Mair, B.; Khokhar, E.S.; Tong, A.H.Y.; Usaj, M.; Chan, K.; Pai, A.A.; Moffat, J.; Dekker, J. A cohesin traffic pattern genetically linked to gene regulation. Nat. Struct. Mol. Biol. 2022, 29, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, Q.; Canzio, D.; Shou, J.; Li, J.; Gorkin, D.U.; Jung, I.; Wu, H.; Zhai, Y.; Tang, Y.; et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 2015, 162, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.H.; Corces, V.G. A CTCF Code for 3D Genome Architecture. Cell 2015, 162, 703–705. [Google Scholar] [CrossRef]
- Ghirlando, R.; Felsenfeld, G. CTCF: Making the right connections. Genes. Dev. 2016, 30, 881–891. [Google Scholar] [CrossRef]
- de Wit, E.; Vos, E.S.; Holwerda, S.J.; Valdes-Quezada, C.; Verstegen, M.J.; Teunissen, H.; Splinter, E.; Wijchers, P.J.; Krijger, P.H.; de Laat, W. CTCF Binding Polarity Determines Chromatin Looping. Mol. Cell 2015, 60, 676–684. [Google Scholar] [CrossRef]
- Vietri Rudan, M.; Barrington, C.; Henderson, S.; Ernst, C.; Odom, D.T.; Tanay, A.; Hadjur, S. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015, 10, 1297–1309. [Google Scholar] [CrossRef]
- Tang, Z.; Luo, O.J.; Li, X.; Zheng, M.; Zhu, J.J.; Szalaj, P.; Trzaskoma, P.; Magalska, A.; Wlodarczyk, J.; Ruszczycki, B.; et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 2015, 163, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Schwalie, P.C.; Ross-Innes, C.S.; Hurtado, A.; Brown, G.D.; Carroll, J.S.; Flicek, P.; Odom, D.T. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 2010, 20, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Faure, A.J.; Schmidt, D.; Watt, S.; Schwalie, P.C.; Wilson, M.D.; Xu, H.; Ramsay, R.G.; Odom, D.T.; Flicek, P. Cohesin regulates tissue-specific expression by stabilizing highly occupied cis-regulatory modules. Genome Res. 2012, 22, 2163–2175. [Google Scholar] [CrossRef] [PubMed]
- Cuartero, S.; Weiss, F.D.; Dharmalingam, G.; Guo, Y.; Ing-Simmons, E.; Masella, S.; Robles-Rebollo, I.; Xiao, X.; Wang, Y.F.; Barozzi, I.; et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat. Immunol. 2018, 19, 932–941. [Google Scholar] [CrossRef]
- Banigan, E.J.; Tang, W.; van den Berg, A.A.; Stocsits, R.R.; Wutz, G.; Brandao, H.B.; Busslinger, G.A.; Peters, J.M.; Mirny, L.A. Transcription shapes 3D chromatin organization by interacting with loop extrusion. Proc. Natl. Acad. Sci. USA 2023, 120, e2210480120. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.J.; Vert, J.P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Thirumalai, D. A maximum-entropy model to predict 3D structural ensembles of chromatin from pairwise distances with applications to interphase chromosomes and structural variants. Nat. Commun. 2023, 14, 1150. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Purushotham, D.; Harrison, J.K.; Hsu, S.; Zhuo, X.; Fan, C.; Liu, S.; Xu, V.; Chen, S.; Xu, J.; et al. WashU Epigenome Browser update 2022. Nucleic Acids Res. 2022, 50, W774–W781. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M. NGmerge: Merging paired-end reads via novel empirically-derived models of sequencing errors. BMC Bioinform. 2018, 19, 536. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef]
- Li, B.; Qing, T.; Zhu, J.; Wen, Z.; Yu, Y.; Fukumura, R.; Zheng, Y.; Gondo, Y.; Shi, L. A Comprehensive Mouse Transcriptomic BodyMap across 17 Tissues by RNA-seq. Sci. Rep. 2017, 7, 4200. [Google Scholar] [CrossRef]
- Saksouk, N.; Simboeck, E.; Dejardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 2015, 8, 3. [Google Scholar] [CrossRef]
- Wang, L.; Gao, Y.; Zheng, X.; Liu, C.; Dong, S.; Li, R.; Zhang, G.; Wei, Y.; Qu, H.; Li, Y.; et al. Histone Modifications Regulate Chromatin Compartmentalization by Contributing to a Phase Separation Mechanism. Mol. Cell 2019, 76, 646–659. [Google Scholar] [CrossRef]
- Sanulli, S.; Trnka, M.J.; Dharmarajan, V.; Tibble, R.W.; Pascal, B.D.; Burlingame, A.L.; Griffin, P.R.; Gross, J.D.; Narlikar, G.J. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 2019, 575, 390–394. [Google Scholar] [CrossRef]
- Machida, S.; Takizawa, Y.; Ishimaru, M.; Sugita, Y.; Sekine, S.; Nakayama, J.I.; Wolf, M.; Kurumizaka, H. Structural Basis of Heterochromatin Formation by Human HP1. Mol. Cell 2018, 69, 385–397. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Young, R.A. Biomolecular Condensates in the Nucleus. Trends Biochem. Sci. 2020, 45, 961–977. [Google Scholar] [CrossRef]
- Lamond, A.I.; Spector, D.L. Nuclear speckles: A model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 2003, 4, 605–612. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Wang, Y.; Zhang, L.; Brinkman, E.K.; Adam, S.A.; Goldman, R.; van Steensel, B.; Ma, J.; Belmont, A.S. Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J. Cell Biol. 2018, 217, 4025–4048. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. Beyond the sequence: Cellular organization of genome function. Cell 2007, 128, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Miron, E.; Oldenkamp, R.; Brown, J.M.; Pinto, D.M.S.; Xu, C.S.; Faria, A.R.; Shaban, H.A.; Rhodes, J.D.P.; Innocent, C.; de Ornellas, S.; et al. Chromatin arranges in chains of mesoscale domains with nanoscale functional topography independent of cohesin. Sci. Adv. 2020, 6, eaba8811. [Google Scholar] [CrossRef] [PubMed]
- van Steensel, B.; Furlong, E.E.M. The role of transcription in shaping the spatial organization of the genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 327–337. [Google Scholar] [CrossRef]
- Gilbert, N.; Boyle, S.; Fiegler, H.; Woodfine, K.; Carter, N.P.; Bickmore, W.A. Chromatin architecture of the human genome: Gene-rich domains are enriched in open chromatin fibers. Cell 2004, 118, 555–566. [Google Scholar] [CrossRef]
- Croft, J.A.; Bridger, J.M.; Boyle, S.; Perry, P.; Teague, P.; Bickmore, W.A. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 1999, 145, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef]
- Hou, C.; Li, L.; Qin, Z.S.; Corces, V.G. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell 2012, 48, 471–484. [Google Scholar] [CrossRef]
- Banigan, E.J.; Mirny, L.A. Loop extrusion: Theory meets single-molecule experiments. Curr. Opin Cell Biol. 2020, 64, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Davidson, I.F.; Peters, J.M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Hoencamp, C.; Rowland, B.D. Genome control by SMC complexes. Nat. Rev. Mol. Cell Biol. 2023, 1–18. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, Z.; Cui, S.; Xue, S.; Xie, Y.; Wang, Y.; Zhao, C.; Zhang, Z.; Wu, T.; Hou, G.; Wang, W.; et al. Identification of Two Subsets of Subcompartment A1 Associated with High Transcriptional Activity and Frequent Loop Extrusion. Biology 2023, 12, 1058. https://doi.org/10.3390/biology12081058
Yin Z, Cui S, Xue S, Xie Y, Wang Y, Zhao C, Zhang Z, Wu T, Hou G, Wang W, et al. Identification of Two Subsets of Subcompartment A1 Associated with High Transcriptional Activity and Frequent Loop Extrusion. Biology. 2023; 12(8):1058. https://doi.org/10.3390/biology12081058
Chicago/Turabian StyleYin, Zihang, Shuang Cui, Song Xue, Yufan Xie, Yefan Wang, Chengling Zhao, Zhiyu Zhang, Tao Wu, Guojun Hou, Wuming Wang, and et al. 2023. "Identification of Two Subsets of Subcompartment A1 Associated with High Transcriptional Activity and Frequent Loop Extrusion" Biology 12, no. 8: 1058. https://doi.org/10.3390/biology12081058
APA StyleYin, Z., Cui, S., Xue, S., Xie, Y., Wang, Y., Zhao, C., Zhang, Z., Wu, T., Hou, G., Wang, W., Xie, S. Q., Wu, Y., & Guo, Y. (2023). Identification of Two Subsets of Subcompartment A1 Associated with High Transcriptional Activity and Frequent Loop Extrusion. Biology, 12(8), 1058. https://doi.org/10.3390/biology12081058