
Inter-Individual Variation in DNA Methylation Patterns across Two Tissues and Leukocytes in Mature Brahman Cattle
 ,
,  , and
, and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods & Materials
2.1. Animal Procedures
2.2. Sample Preparation & DNA Extraction
2.3. DNA Methylation Analysis
2.4. Statistical Analysis
2.4.1. Genome-Wide Inter-Individual Methylation Variation
2.4.2. Genomic Feature Inter-Individual Methylation Variation
3. Results
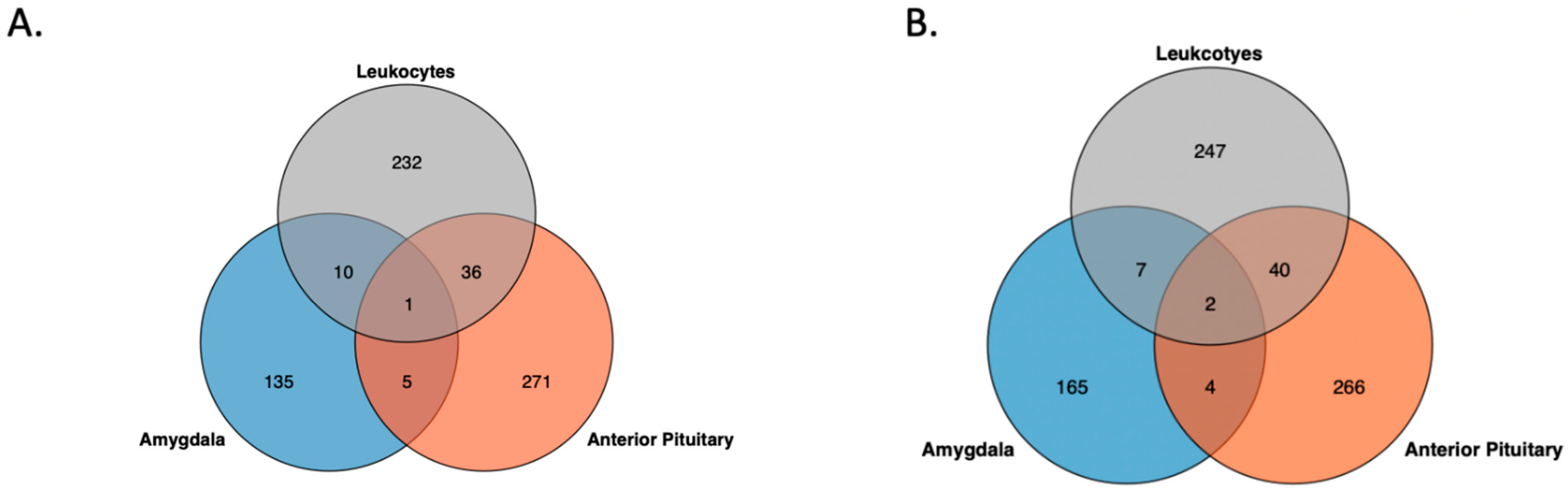
3.1. Genome-Wide Inter-Individual Methylation Variation
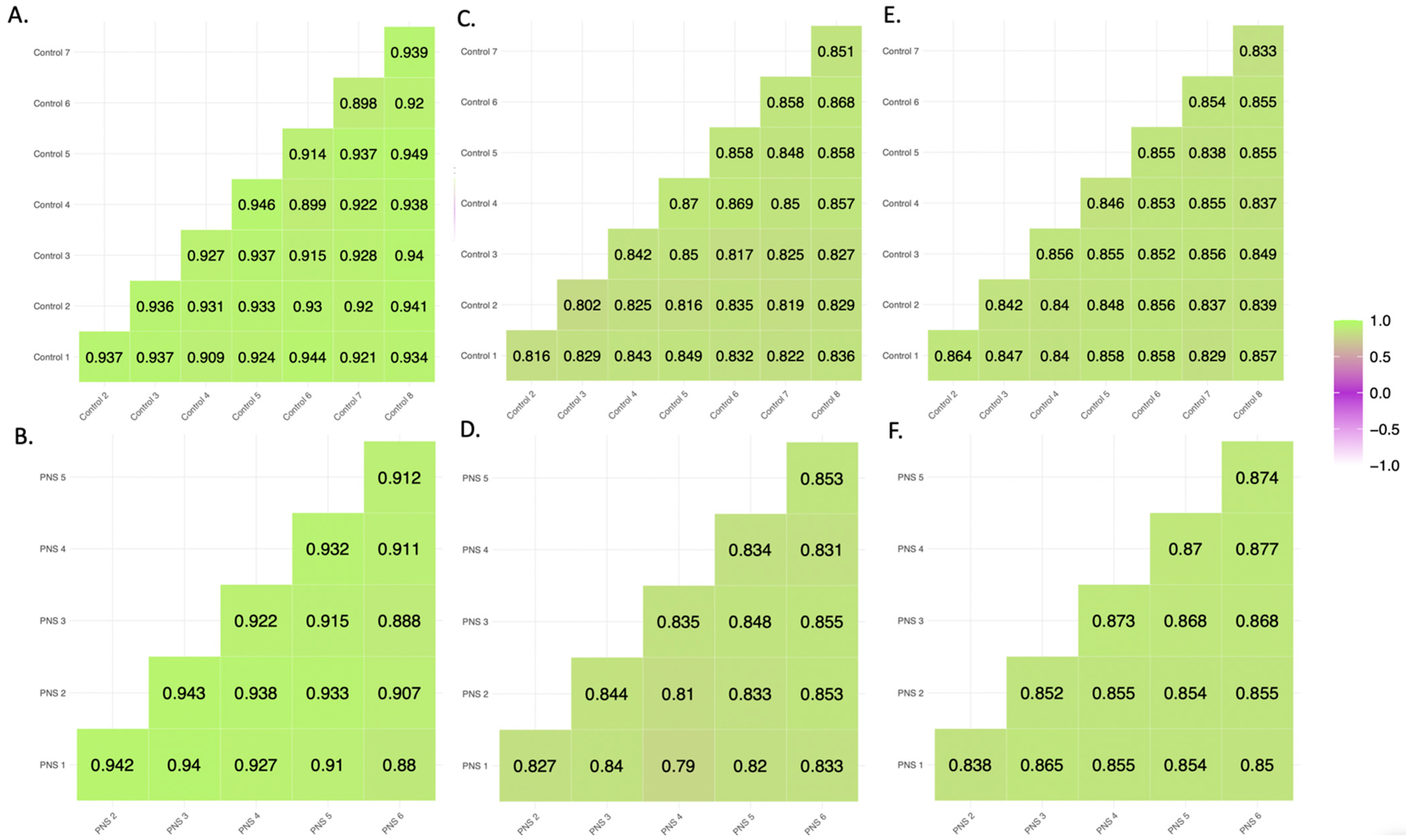
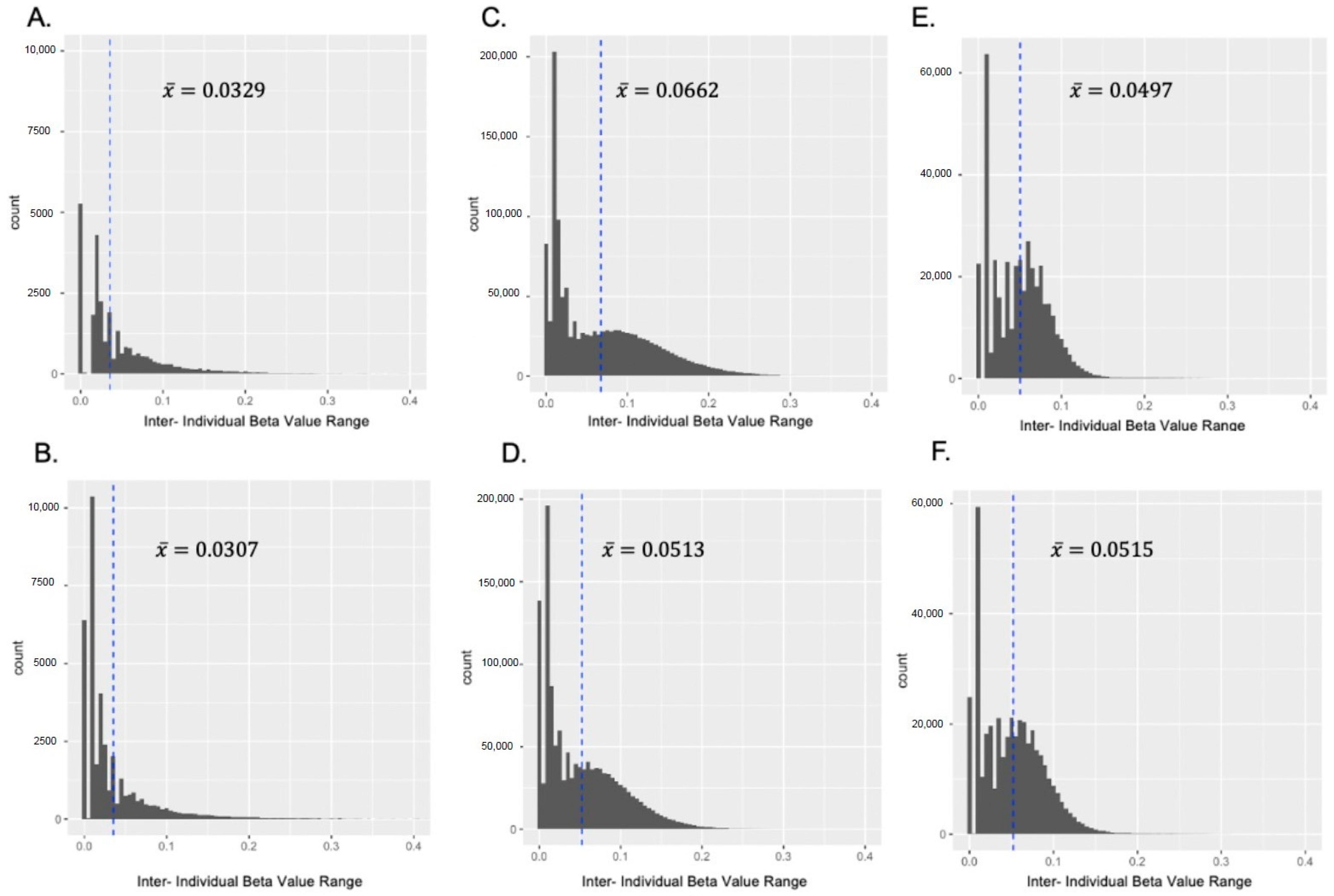
3.1.1. Pearson Correlation and Inter-Individual β Value Range
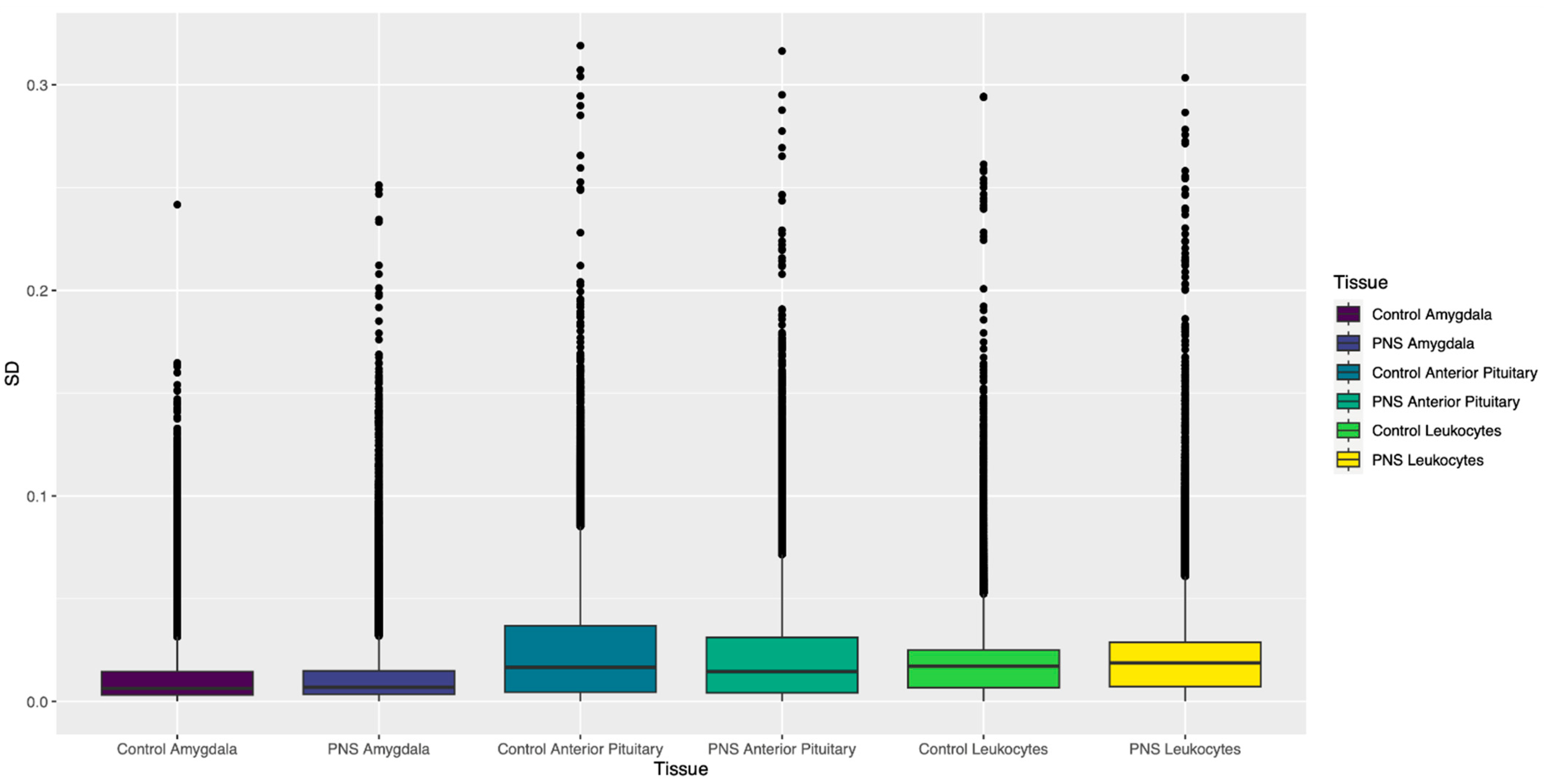
3.1.2. Standard Deviation of β Values
3.1.3. Chi-Square Test for the Variance
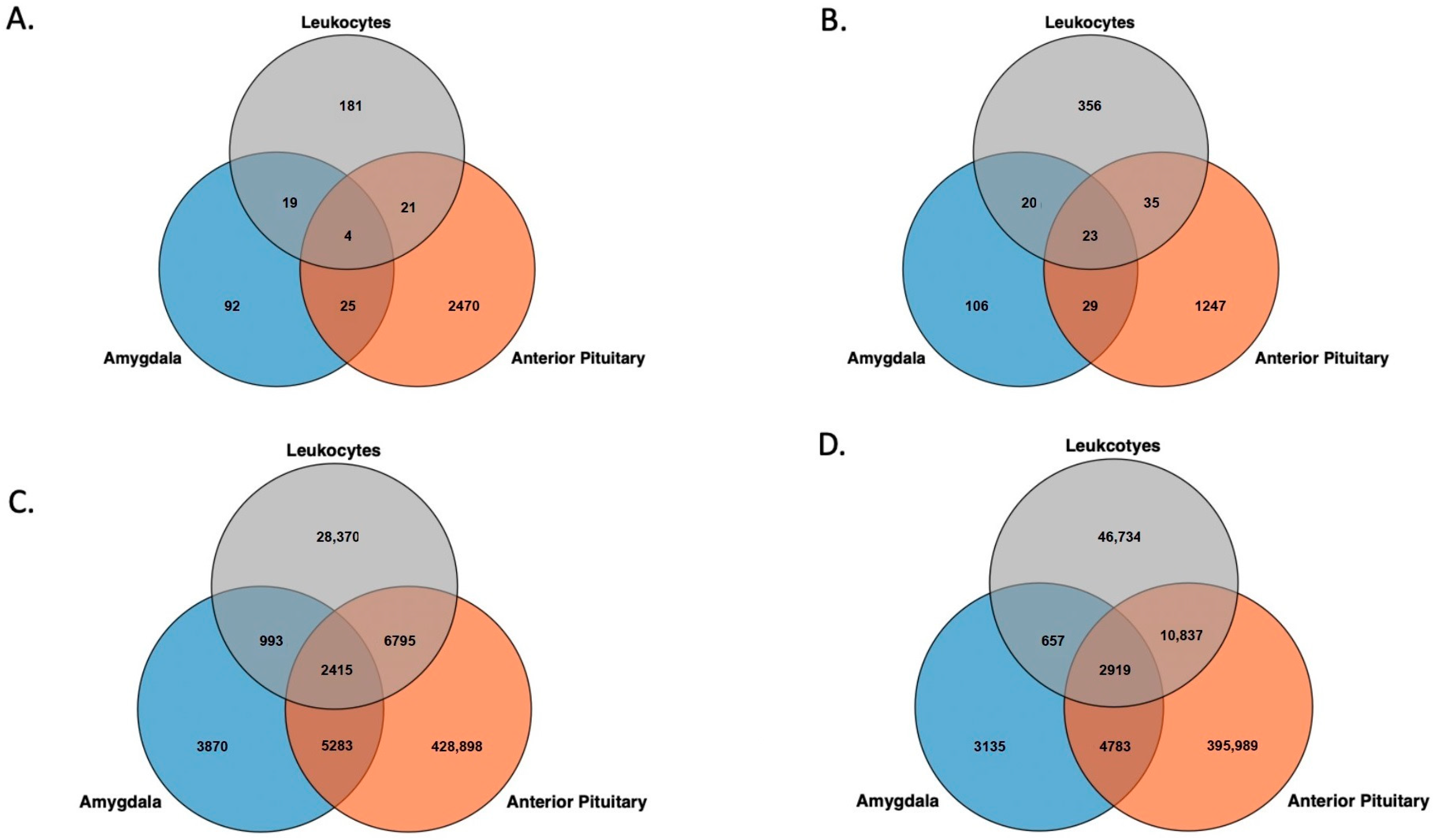
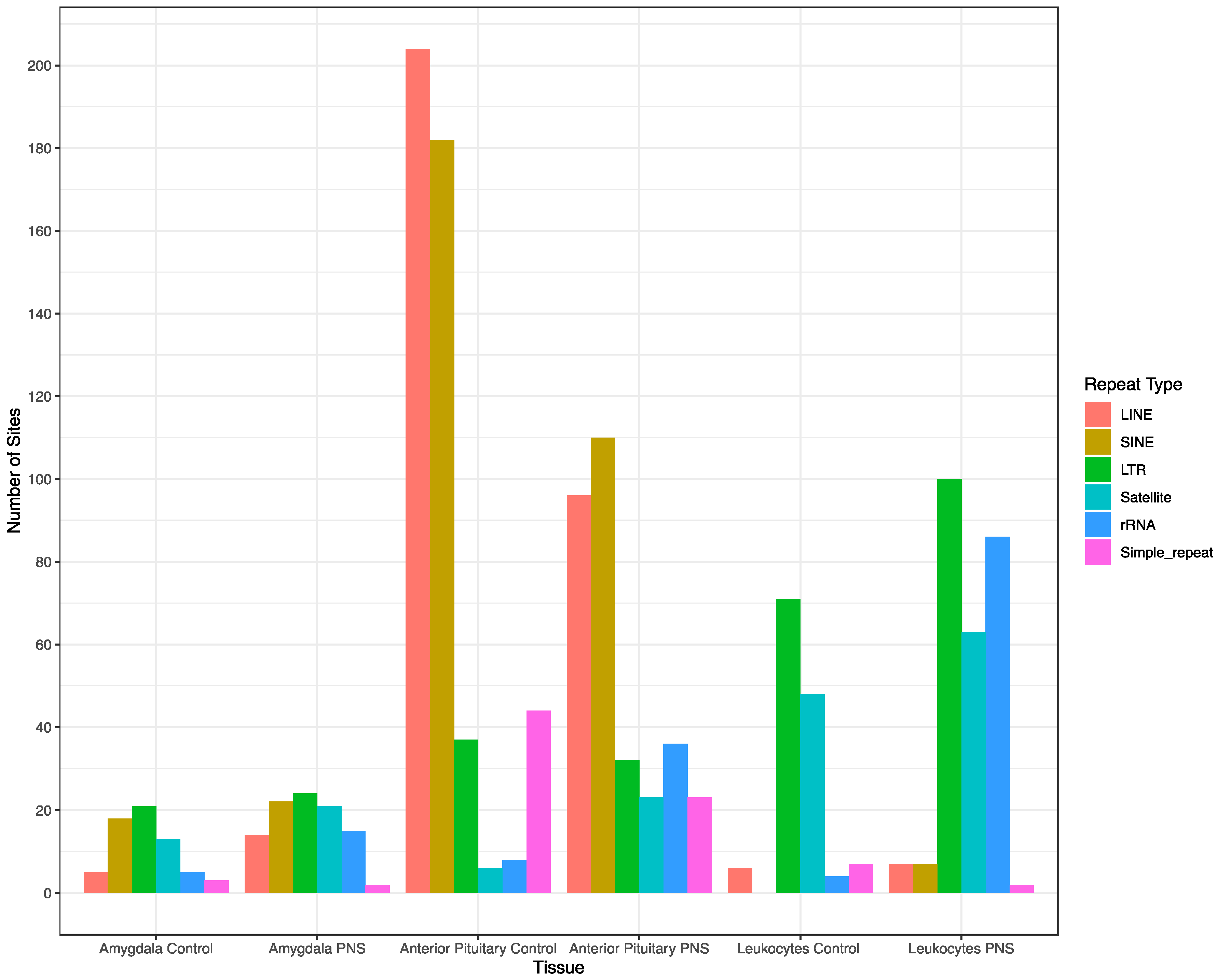
3.2. Genomic Feature Inter-Individual Methylation Variation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P. Epigenetic variation and human disease. J. Nutr. 2002, 132, 2388S–2392S. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Popendikyte, V.; Pozdniakovaite, N.; Sobolev, M.; Assadzadeh, A.; Schumacher, A.; Zangeneh, M.; Lau, L.; Virtanen, C.; Wang, S.-C.; et al. Intra- and interindividual epigenetic variation in human germ cells. Am. J. Hum. Genet. 2006, 79, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Moran, S.; Hernando-Herraez, I.; Sayols, S.; Gomez, A.; Sandoval, J.; Monk, D.; Hata, K.; Marques-Bonet, T.; Wang, L.; et al. DNA methylation contributes to natural human variation. Genome Res. 2013, 23, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Duncan, E.J.; Parry, M.F.; Weeks, R.J.; Morison, I.M. Genome-wide DNA methylation map of human neutrophils reveals widespread inter-individual epigenetic variation. Sci. Rep. 2015, 5, 17328. [Google Scholar] [CrossRef]
- Shen, H.; Qiu, C.; Li, J.; Tian, Q.; Deng, H.-W. Characterization of the DNA methylome and its interindividual variation in human peripheral blood monocytes. Epigenomics 2013, 5, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011, 12, R10. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Gruenewald-Schneider, U.; De Sousa, D.; Webb, S.; Merusi, C.; Kerr, A.R.W.; James, K.D.; Smith, C.; Walker, R.; Andrews, R.; et al. Inter-individual variability contrasts with regional homogeneity in the human brain DNA methylome. Nucleic Acids Res. 2015, 43, 732–744. [Google Scholar] [CrossRef]
- Gutierrez-Arcelus, M.; Lappalainen, T.; Montgomery, S.B.; Buil, A.; Ongen, H.; Yurovsky, A.; Bryois, J.; Giger, T.; Romano, L.; Planchon, A.; et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife 2013, 2, e00523. [Google Scholar] [CrossRef]
- Gonseth, S.; de Smith, A.J.; Roy, R.; Zhou, M.; Lee, S.-T.; Shao, X.; Ohja, J.; Wrensch, M.R.; Walsh, K.M.; Metayer, C.; et al. Genetic contribution to variation in DNA methylation at maternal smoking-sensitive loci in exposed neonates. Epigenetics 2016, 11, 664–673. [Google Scholar] [CrossRef]
- Iwamoto, K.; Bundo, M.; Ueda, J.; Oldham, M.C.; Ukai, W.; Hashimoto, E.; Saito, T.; Geschwind, D.H.; Kato, T. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011, 21, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Martino, D.J.; Tulic, M.K.; Gordon, L.; Hodder, M.; Richman, T.R.; Metcalfe, J.; Prescott, S.L.; Saffery, R. Evidence for age-related and individual-specific changes in DNA methylation profile of mononuclear cells during early immune development in humans. Epigenetics 2011, 6, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Mulder, R.H.; Neumann, A.; Cecil, C.A.M.; Walton, E.; Houtepen, L.C.; Simpkin, A.J.; Rijlaarsdam, J.; Heijmans, B.T.; Gaunt, T.R.; Felix, J.F.; et al. Epigenome-wide change and variation in DNA methylation in childhood: Trajectories from birth to late adolescence. Hum. Mol. Genet. 2021, 30, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Hannon, E.; Knox, O.; Sugden, K.; Burrage, J.; Wong, C.C.Y.; Belsky, D.W.; Corcoran, D.L.; Arseneault, L.; Moffitt, T.E.; Caspi, A.; et al. Characterizing genetic and environmental influences on variable DNA methylation using monozygotic and dizygotic twins. PLoS Genet. 2018, 14, e1007544. [Google Scholar] [CrossRef] [PubMed]
- Teh, A.L.; Pan, H.; Chen, L.; Ong, M.L.; Dogra, S.; Wong, J.; MacIsaac, J.L.; Mah, S.M.; McEwen, L.M.; Saw, S.M.; et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 2014, 24, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, B.P.; Price, D.M.; Neuendorff, D.A.; Carroll, J.A.; Vann, R.C.; Riggs, P.K.; Riley, D.G.; Long, C.R.; Welsh, T.H., Jr.; Randel, R.D. Prenatal transportation stress alters genome-wide DNA methylation in suckling Brahman bull calves. J. Anim. Sci. 2018, 96, 5075–5099. [Google Scholar] [CrossRef]
- Baker, E.C.; Cilkiz, K.Z.; Riggs, P.K.; Littlejohn, B.P.; Long, C.R.; Welsh, T.H.; Randel, R.D.; Riley, D.G. Effect of prenatal transportation stress on DNA methylation in Brahman heifers. Livest. Sci. 2020, 240, 104116. [Google Scholar] [CrossRef]
- Cilkiz, K.Z.; Baker, E.C.; Riggs, P.K.; Littlejohn, B.P.; Long, C.R.; Welsh, T.H.; Randel, R.D.; Riley, D.G. Genome-wide DNA methylation alteration in prenatally stressed Brahman heifer calves with the advancement of age. Epigenetics 2021, 16, 519–536. [Google Scholar] [CrossRef]
- Vukojevic, V.; Kolassa, I.-T.; Fastenrath, M.; Gschwind, L.; Spalek, K.; Milnik, A.; Heck, A.; Vogler, C.; Wilker, S.; Demougin, P.; et al. Epigenetic modification of the glucocorticoid receptor gene is linked to traumatic memory and post-traumatic stress disorder risk in genocide survivors. J. Neurosci. Res. 2014, 34, 10274–10284. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Relton, C.L. Statistical and integrative system-level analysis of DNA methylation data. Nat. Rev. Genet. 2018, 19, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Fang, L.; Zhou, Y.; Santos, D.J.A.; Xiang, R.; Daetwyler, H.D.; Chamberlain, A.J.; Cole, J.B.; Li, C.-j.; Yu, Y.; et al. Analyses of inter-individual variations of sperm DNA methylation and their potential implications in cattle. BMC Genom. 2019, 20, 888. [Google Scholar] [CrossRef] [PubMed]
- Federation of Animal Science Societies (FASS). Guide for the Care and Use of Agricultural Animals in Research and Teaching, 4th ed.; FASS: Champaign, IL, USA, 2020. [Google Scholar]
- Price, D.M.; Lewis, A.W.; Neuendorff, D.A.; Carroll, J.A.; Burdick Sanchez, N.C.; Vann, R.C.; Welsh, T.H.; Randel, R.D. Physiological and metabolic responses of gestating Brahman cows to repeated transportation. J. Anim. Sci. 2015, 93, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Jiang, R.; Jones, M.J.; Chen, E.; Neumann, S.M.; Fraser, H.B.; Miller, G.E.; Kobor, M.S. Discordance of DNA methylation variance between two accessible human tissues. Sci. Rep. 2015, 5, 8257. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Navarro Gonzalez, J.; Zweig, A.S.; Speir, M.L.; Schmelter, D.; Rosenbloom, K.R.; Raney, B.J.; Powell, C.C.; Nassar, L.R.; Maulding, N.D.; Lee, C.M.; et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021, 49, D1046–D1057. [Google Scholar] [CrossRef]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov Andrey, G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2021, 50, D988–D995. [Google Scholar] [CrossRef]
- Jaffe, A.E.; Irizarry, R.A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014, 15, R31. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, B.T.; Gudnason, H.; Aspelund, T.; Harris, T.B.; Launer, L.J.; Eiriksdottir, G.; Smith, A.V.; Gudnason, V. Heterogeneity in white blood cells has potential to confound DNA methylation measurements. PLoS ONE 2012, 7, e46705. [Google Scholar] [CrossRef] [PubMed]
- Tosevski, J.; Malikovic, A.; Mojsilovic-Petrovic, J.; Lackovic, V.; Peulic, M.; Sazdanovic, P.; Alexopulos, C. Types of neurons and some dendritic patterns of basolateral amygdala in humans—A Golgi study. Ann. Anat. 2002, 184, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Le Tissier, P.R.; Hodson, D.J.; Lafont, C.; Fontanaud, P.; Schaeffer, M.; Mollard, P. Anterior pituitary cell networks. Front. Neuroendocrinol. 2012, 33, 252–266. [Google Scholar] [CrossRef]
- Zhang, D.; Cheng, L.; Badner, J.A.; Chen, C.; Chen, Q.; Luo, W.; Craig, D.W.; Redman, M.; Gershon, E.S.; Liu, C. Genetic control of individual differences in gene-specific methylation in human brain. J. Hum. Genet. 2010, 86, 411–419. [Google Scholar] [CrossRef]
- Wang, D.; Liu, X.; Zhou, Y.; Xie, H.; Hong, X.; Tsai, H.-J.; Wang, G.; Liu, R.; Wang, X. Individual variation and longitudinal pattern of genome-wide DNA methylation from birth to the first two years of life. Epigenetics 2012, 7, 594–605. [Google Scholar] [CrossRef]
- Jeziorska, D.M.; Murray, R.J.S.; De Gobbi, M.; Gaentzsch, R.; Garrick, D.; Ayyub, H.; Chen, T.; Li, E.; Telenius, J.; Lynch, M.; et al. DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E7526–E7535. [Google Scholar] [CrossRef]
- Wagner, J.R.; Busche, S.; Ge, B.; Kwan, T.; Pastinen, T.; Blanchette, M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014, 15, R37. [Google Scholar] [CrossRef]
- Verhulst, E.C.; Mateman, A.C.; Zwier, M.V.; Caro, S.P.; Verhoeven, K.J.F.; van Oers, K. Evidence from pyrosequencing indicates that natural variation in animal personality is associated with DRD4 DNA methylation. Mol. Ecol. 2016, 25, 1801–1811. [Google Scholar] [CrossRef]
- Kühnen, P.; Handke, D.; Waterland, R.A.; Hennig, B.J.; Silver, M.; Fulford, A.J.; Dominguez-Salas, P.; Moore, S.E.; Prentice, A.M.; Spranger, J.; et al. Interindividual variation in DNA methylation at a putative POMC metastable epiallele is associated with obesity. Cell Metab. 2016, 24, 502–509. [Google Scholar] [CrossRef]
- Kim, T.-K.; Han, P.-L. Functional connectivity of basolateral amygdala neurons carrying orexin receptors and melanin-concentrating hormone receptors in regulating sociability and mood-related behaviors. Exp. Neurobiol. 2016, 25, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Schang, A.L.; Quérat, B.; Simon, V.; Garrel, G.; Bleux, C.; Counis, R.; Cohen-Tannoudji, J.; Laverrière, J.N. Mechanisms underlying the tissue-specific and regulated activity of the GNRH promoter in mammals. Front. Endocrinol. 2012, 3, 162. [Google Scholar] [CrossRef] [PubMed]
- Mendez, J.A.; Bourque, M.-J.; Fasano, C.; Kortleven, C.; Trudeau, L.-E. Somatodendritic dopamine release requires Synaptotagmin 4 and 7 and the participation of voltage-gated calcium channels. J. Biol. Chem. 2011, 286, 23928–23937. [Google Scholar] [CrossRef]
- Yang, M.-F.; Long, X.-X.; Hu, H.-S.; Bin, Y.-L.; Chen, X.-M.; Wu, B.-H.; Peng, Q.-Z.; Wang, L.-S.; Yao, J.; Li, D.-F. Comprehensive analysis on the expression profile and prognostic values of Synaptotagmins (SYTs) family members and their methylation levels in gastric cancer. Bioengineered 2021, 12, 3550–3565. [Google Scholar] [CrossRef] [PubMed]
- Córdova-Palomera, A.; Fatjó-Vilas, M.; Gastó, C.; Navarro, V.; Krebs, M.O.; Fañanás, L. Genome-wide methylation study on depression: Differential methylation and variable methylation in monozygotic twins. Transl. Psychiatry 2015, 5, e557. [Google Scholar] [CrossRef]
- Tejedor, J.R.; Fraga, M.F. Interindividual epigenetic variability: Sound or noise? BioEssays 2017, 39, 1700055. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.L.; Emberly, E.; Fraser, H.B.; Neumann, S.M.; Chen, E.; Miller, G.E.; Kobor, M.S. Factors underlying variable DNA methylation in a human community cohort. Proc. Natl. Acad. Sci. USA 2012, 109, 17253–17260. [Google Scholar] [CrossRef] [PubMed]
- Michels, K.B.; Harris, H.R.; Barault, L. Birthweight, maternal weight trajectories and global DNA methylation of LINE-1 repetitive elements. PLoS ONE 2011, 6, e25254. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Algarra, F.; Seaborne, R.A.E.; Danson, A.F.; Yildizoglu, S.; Yoshikawa, H.; Law, P.P.; Ahmad, Z.; Maudsley, V.A.; Brew, A.; Holmes, N.; et al. Genetic variation at mouse and human ribosomal DNA influences associated epigenetic states. Genome Biol. 2022, 23, 54. [Google Scholar] [CrossRef]
- Hannon, E.; Lunnon, K.; Schalkwyk, L.; Mill, J. Interindividual methylomic variation across blood, cortex, and cerebellum: Implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 2015, 10, 1024–1032. [Google Scholar] [CrossRef]
- Hannon, E.; Mansell, G.; Walker, E.; Nabais, M.F.; Burrage, J.; Kepa, A.; Best-Lane, J.; Rose, A.; Heck, S.; Moffitt, T.E.; et al. Assessing the co-variability of DNA methylation across peripheral cells and tissues: Implications for the interpretation of findings in epigenetic epidemiology. PLoS Genet. 2021, 17, e1009443. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, R43. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Ouidir, M.; Tekola-Ayele, F. Genetic and in utero environmental contributions to DNA methylation variation in placenta. Hum. Mol. Genet. 2021, 30, 1968–1976. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SD ≥ 0.1 1 | Mean Variance 2 | p ≤ 0.001 3 | |
|---|---|---|---|
| Amygdala | |||
| Control | 140 | 2.76 × 10−4 | 12,471 |
| Prenatally Stressed | 178 | 4.81 × 10−5 | 11,494 |
| Anterior Pituitary | |||
| Control | 2520 | 4.11 × 10−5 | 443,391 |
| Prenatally Stressed | 1334 | 2.19 × 10−4 | 414,528 |
| Leukocytes | |||
| Control | 225 | 2.95 × 10−4 | 38,573 |
| Prenatally Stressed | 434 | 3.26 × 10−4 | 61,147 |
| Total Features Tested 1 | Significant 2 | % of Total Features Tested 3 | |
|---|---|---|---|
| Amygdala | |||
| Promoter 4 | 13,053 | 52 | 0.40% |
| Gene | 16,828 | 151 | 0.90% |
| CpG 5 islands | 16,808 | 51 | 0.50% |
| CpG shores 6 | 18,754 | 81 | 0.43% |
| Leukocytes | |||
| Promoter | 15,602 | 168 | 0.94% |
| Gene | 18,829 | 281 | 1.49% |
| CpG islands | 20,016 | 216 | 0.01% |
| CpG shores | 17,417 | 149 | 0.61% |
| Anterior Pituitary | |||
| Promoter | 20,688 | 226 | 1.09% |
| Gene | 19,083 | 314 | 1.65% |
| CpG islands | 20,605 | 307 | 1.49% |
| CpG shores | 30,252 | 446 | 1.47% |
| Total Tested 1 | Significant 2 | % of Total Features Tested 3 | |
|---|---|---|---|
| Amygdala | |||
| Promoter 4 | 13,177 | 52 | 0.40% |
| Gene | 17,021 | 178 | 1.05% |
| CpG islands 5 | 16,700 | 51 | 0.31% |
| CpG shores 6 | 19,244 | 7 | 0.04% |
| Leukocytes | |||
| Promoter | 16,255 | 168 | 1.03% |
| Gene | 19,290 | 298 | 1.54% |
| CpG islands | 20,400 | 216 | 1.06% |
| CpG shores | 29,987 | 149 | 0.50% |
| Pituitary Gland | |||
| Promoter | 20148 | 372 | 1.85% |
| Gene | 18,965 | 313 | 1.65% |
| CpG islands | 20,594 | 313 | 1.52% |
| CpG shores | 29,675 | 399 | 1.34% |
| Feature | Anterior Pituitary | Amygdala | Leukocytes |
|---|---|---|---|
| Promoter 1 | 92 | 1 | 14 |
| Gene | 81 | 6 | 54 |
| CpG 2 islands | 73 | 0 | 26 |
| CpG shores 3 | 96 | 0 | 3 |
| Feature Name | Feature Type | Standard Deviation 3 |
|---|---|---|
| Prenatally Stressed group | ||
| ENSBTAG00000020813 | Promoter 4 | 32.97 |
| WAP four-disulfide core domain 2 | Promoter | 27.23 |
| ENSBTAG00000014329 | Gene | 27.19 |
| ENSBTAG00000052841 | Gene | 26.61 |
| ENSBTAG00000036102 | Gene | 26.02 |
| Interleukin-34 | Promoter | 24.99 |
| Secretory leukocyte peptidase inhibitor | Promoter | 24.6 |
| Bovine major histocompatibility complex | Promoter | 24.29 |
| Interleukin 9 receptor | Promoter | 23.86 |
| Myelin oligodendrocyte glycoprotein | Promoter | 22.72 |
| Transmembrane protein 176B | Gene | 21.44 |
| ENSBTAG00000048980 | Gene | 20.35 |
| Interleukin 36 alpha | Gene | 18.73 |
| ENSBTAG00000023563 | Gene | 18.26 |
| C-X-C chemokine receptor type 2 | Gene | 18.13 |
| Triggering receptor expressed on myeloid cells 1 | Gene | 17.94 |
| Testicular cell adhesion molecule 1 | Gene | 17.32 |
| Interleukin-4 | Gene | 16.9 |
| ENSBTAG00000006864 | Gene | 16.48 |
| Control group | ||
| Myelin oligodendrocyte glycoprotein | Promoter | 37.67 |
| ENSBTAG00000045810 | Gene | 33.86 |
| ENSBTAG00000020813 | Promoter | 33.04 |
| ENSBTAG00000055111 | Promoter | 30.69 |
| ENSBTAG00000006864 | Gene | 29.81 |
| Peptidase inhibitor 3 | Gene | 28.62 |
| Interleukin-34 | Promoter | 26.04 |
| ENSBTAG00000051008 | Promoter | 25.91 |
| Calcium-dependent phospholipase A2 | Gene | 24.7 |
| ENSBTAG00000050878 | Gene | 24.28 |
| Interleukin 2 receptor subunit beta | Promoter | 23.78 |
| CCAAT enhancer binding protein epsilon | Gene | 22.53 |
| ENSBTAG00000053521 | Gene | 22.38 |
| Major histocompatibility complex, class I-related | Gene | 19.45 |
| 5,-aminolevulinate synthase 2 | Gene | 19.27 |
| C-C motif chemokine ligand 25 | Gene | 18.83 |
| C-C motif chemokine ligand 1 | Gene | 17.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baker, E.C.; San, A.E.; Cilkiz, K.Z.; Littlejohn, B.P.; Cardoso, R.C.; Ghaffari, N.; Long, C.R.; Riggs, P.K.; Randel, R.D.; Welsh, T.H., Jr.; et al. Inter-Individual Variation in DNA Methylation Patterns across Two Tissues and Leukocytes in Mature Brahman Cattle. Biology 2023, 12, 252. https://doi.org/10.3390/biology12020252
Baker EC, San AE, Cilkiz KZ, Littlejohn BP, Cardoso RC, Ghaffari N, Long CR, Riggs PK, Randel RD, Welsh TH Jr., et al. Inter-Individual Variation in DNA Methylation Patterns across Two Tissues and Leukocytes in Mature Brahman Cattle. Biology. 2023; 12(2):252. https://doi.org/10.3390/biology12020252
Chicago/Turabian StyleBaker, Emilie C., Audrey E. San, Kubra Z. Cilkiz, Brittni P. Littlejohn, Rodolfo C. Cardoso, Noushin Ghaffari, Charles R. Long, Penny K. Riggs, Ronald D. Randel, Thomas H. Welsh, Jr., and et al. 2023. "Inter-Individual Variation in DNA Methylation Patterns across Two Tissues and Leukocytes in Mature Brahman Cattle" Biology 12, no. 2: 252. https://doi.org/10.3390/biology12020252
APA StyleBaker, E. C., San, A. E., Cilkiz, K. Z., Littlejohn, B. P., Cardoso, R. C., Ghaffari, N., Long, C. R., Riggs, P. K., Randel, R. D., Welsh, T. H., Jr., & Riley, D. G. (2023). Inter-Individual Variation in DNA Methylation Patterns across Two Tissues and Leukocytes in Mature Brahman Cattle. Biology, 12(2), 252. https://doi.org/10.3390/biology12020252